Blood-Brain Barrier Dysfunction and the Pathogenesis of Alzheimer’s Disease


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
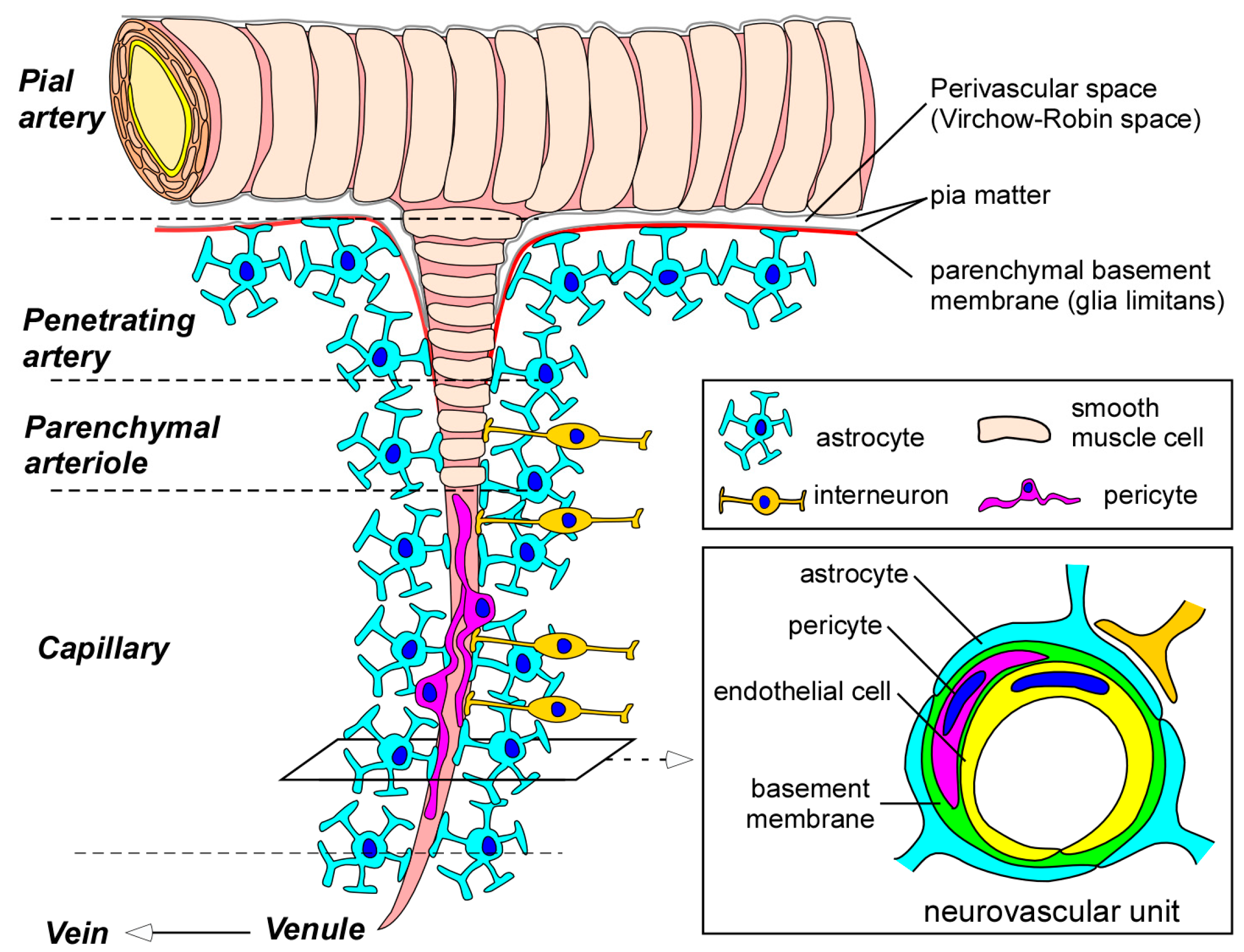
2. Blood-Brain Barrier (BBB) in the Neurovascular Unit
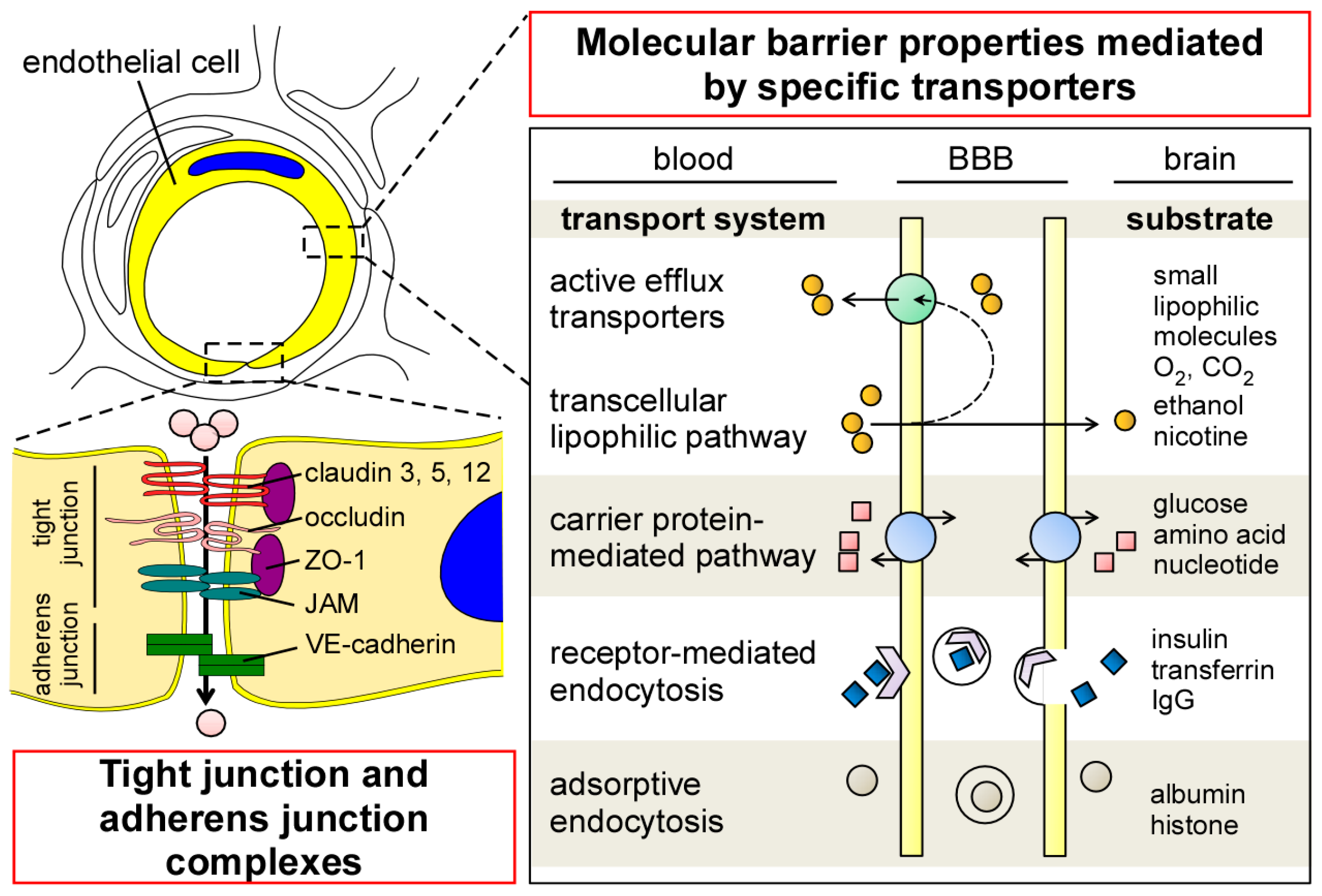
2.1. Endothelial Cells
2.2. Pericytes
2.3. Astrocytes
2.4. Basement Membranes
3. BBB Alteration in Alzheimer’s Disease (AD)
3.1. Leakages of Blood-Derived Molecules in Postmortem AD Brains
3.2. Cerebrospinal Fluid (CSF)/Blood Albumin Ratio in AD Patients
3.3. Evaluation of BBB Function through Brain Imaging in AD Patients
4. Neurovascular Unit Dysregulation and AD
4.1. Endothelial Cell Alternation in AD
4.2. Cerebrovascular Pericyte Degeneration in AD
4.3. Altered Perivascular Astrocytic End-Feet in AD
4.4. Cerebrovascular Basement Membrane Pathology in AD
5. Transport of Aβ across the BBB
6. Summary and Perspective
Acknowledgments
Conflicts of Interest
Abbreviations and Acronyms
| 5XFAD | 5 familial AD mutations |
| ABCG2 | ATP-binding cassette sub-family G member 2 |
| ABCG4 | ATP-binding cassette sub-family G member 4 |
| AD | Alzheimer’s disease |
| AQP4 | Aquaporin 4 |
| Aβ | Amyloid-β |
| BBB | Blood-brain barrier |
| CA1 | Cornu ammonis 1 |
| CAA | Cerebral amyloid angiopathy |
| CNS | Central nervous system |
| CSF | Cerebrospinal fluid |
| CT | Computed tomography |
| ELISA | Enzyme-linked immunosorbent assay |
| GFAP | Glial fibrillary acidic protein |
| GLUT1 | Glucose transporter 1 |
| HPLC | High performance liquid chromatography |
| HUVEC | Human umbilical vein endothelial cell |
| ISF | Interstitial fluid |
| JAM | Junctional adhesion molecule |
| LAM | Leukocyte adhesion molecule |
| LRP1 | Low-density lipoprotein receptor-related protein 1 |
| MAGUK | Membrane-associated guanylate kinases |
| MCI | Mild cognitive impairment |
| MRI | Magnetic resonance imaging |
| mRNA | Messenger RNA |
| PDGFRβ | Platelet-derived growth factor receptor β |
| PET | Positron emission tomography |
| P-gp | P-glycoprotein |
| RAGE | Receptor for advanced glycation end products |
| TJ | Tight junction |
| VE-cadherin | Vascular endothelial-cadherin |
| ZO-1 | Zonula occludens-1 |
References
- Cines, D.B.; Pollak, E.S.; Buck, C.A.; Loscalzo, J.; Zimmerman, G.A.; McEver, R.P.; Pober, J.S.; Wick, T.M.; Konkle, B.A.; Schwartz, B.S.; et al. Endothelial cells in physiology and in the pathophysiology of vascular disorders. Blood 1998, 91, 3527–3561. [Google Scholar] [PubMed]
- Pittman, R.N. Regulation of Tissue Oxygenation. In Colloquium Series on Integrated Systems Physiology: From Molecule to Function; Biota Publishing: Princeton, NJ, USA, 2011. [Google Scholar]
- Schaeffer, M.; Hodson, D.J.; Lafont, C.; Mollard, P. Endocrine cells and blood vessels work in tandem to generate hormone pulses. J. Mol. Endocrinol. 2011, 47, R59–R66. [Google Scholar] [CrossRef] [PubMed]
- Abbas, A.K.; Lichtman, A.H.; Pillai, S. Cellular and Molecular Immunology, 8th ed.; Elsevier Health Sciences: Amsterdam, The Netherlands, 2014; 535p. [Google Scholar]
- Daneman, R.; Prat, A. The blood-brain barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef] [PubMed]
- Daneman, R. The blood-brain barrier in health and disease. Ann. Neurol. 2012, 72, 648–672. [Google Scholar] [CrossRef] [PubMed]
- Zlokovic, B.V. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron 2008, 57, 178–201. [Google Scholar] [CrossRef] [PubMed]
- Erickson, M.A.; Banks, W.A. Blood-brain barrier dysfunction as a cause and consequence of Alzheimer’s disease. J. Cereb. Blood Flow Metab. 2013, 33, 1500–1513. [Google Scholar] [CrossRef] [PubMed]
- Zenaro, E.; Piacentino, G.; Constantin, G. The blood-brain barrier in Alzheimer’s disease. Neurobiol. Dis. 2016, in press. [Google Scholar]
- Zlokovic, B.V. Neurovascular mechanisms of Alzheimer’s neurodegeneration. Trends Neurosci. 2005, 28, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Hebert, L.E.; Weuve, J.; Scherr, P.A.; Evans, D.A. Alzheimer disease in the United States (2010–2050) estimated using the 2010 census. Neurology 2013, 80, 1778–1783. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer's Association. 2016 Alzheimer’s disease facts and figures. Alzheimers Dement. 2016, 12, 459–509.
- Ellis, R.J.; Olichney, J.M.; Thal, L.J.; Mirra, S.S.; Morris, J.C.; Beekly, D.; Heyman, A. Cerebral amyloid angiopathy in the brains of patients with Alzheimer’s disease: The CERAD experience, Part XV. Neurology 1996, 46, 1592–1596. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, M.; Murray, M.E.; Frank, R.D.; DeTure, M.; Yamazaki, Y.; Tachibana, M.; Atagi, Y.; Davis, M.D.; Liu, C.C.; Zhao, N.; et al. Impact of sex and APOE4 on cerebral amyloid angiopathy in Alzheimer’s disease. Acta Neuropathol. 2016, 132, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef]
- Spires-Jones, T.L.; Hyman, B.T. The intersection of amyloid beta and tau at synapses in Alzheimer’s disease. Neuron 2014, 82, 756–771. [Google Scholar] [CrossRef] [PubMed]
- Kapasi, A.; Schneider, J.A. Vascular contributions to cognitive impairment, clinical Alzheimer’s disease, and dementia in older persons. Biochim. Biophys. Acta 2016, 1862, 878–886. [Google Scholar] [CrossRef] [PubMed]
- Yarchoan, M.; Xie, S.X.; Kling, M.A.; Toledo, J.B.; Wolk, D.A.; Lee, E.B.; van Deerlin, V.; Lee, V.M.; Trojanowski, J.Q.; Arnold, S.E. Cerebrovascular atherosclerosis correlates with Alzheimer pathology in neurodegenerative dementias. Brain J. Neurol. 2012, 135, 3749–3756. [Google Scholar] [CrossRef] [PubMed]
- Deeken, J.F.; Loscher, W. The blood-brain barrier and cancer: Transporters, treatment, and Trojan horses. Clin. Cancer Res. 2007, 13, 1663–1674. [Google Scholar] [CrossRef] [PubMed]
- Gunzel, D.; Yu, A.S. Claudins and the modulation of tight junction permeability. Physiol. Rev. 2013, 93, 525–569. [Google Scholar] [CrossRef] [PubMed]
- Furuse, M.; Sasaki, H.; Fujimoto, K.; Tsukita, S. A single gene product, claudin-1 or -2, reconstitutes tight junction strands and recruits occludin in fibroblasts. J. Cell Biol. 1998, 143, 391–401. [Google Scholar] [CrossRef] [PubMed]
- Furuse, M.; Furuse, K.; Sasaki, H.; Tsukita, S. Conversion of zonulae occludentes from tight to leaky strand type by introducing claudin-2 into Madin-Darby canine kidney I cells. J. Cell Biol. 2001, 153, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Van Itallie, C.; Rahner, C.; Anderson, J.M. Regulated expression of claudin-4 decreases paracellular conductance through a selective decrease in sodium permeability. J. Clin. Investig. 2001, 107, 1319–1327. [Google Scholar] [CrossRef] [PubMed]
- Amasheh, S.; Schmidt, T.; Mahn, M.; Florian, P.; Mankertz, J.; Tavalali, S.; Gitter, A.H.; Schulzke, J.D.; Fromm, M. Contribution of claudin-5 to barrier properties in tight junctions of epithelial cells. Cell Tissue Res. 2005, 321, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Gomes, A.S.; Paul, D.L.; Goodenough, D.A. Study of claudin function by RNA interference. J. Biol. Chem. 2006, 281, 36117–36123. [Google Scholar] [CrossRef] [PubMed]
- Tsukita, S.; Furuse, M. Occludin and claudins in tight-junction strands: Leading or supporting players? Trends Cell Biol. 1999, 9, 268–273. [Google Scholar] [CrossRef]
- Morita, K.; Sasaki, H.; Furuse, M.; Tsukita, S. Endothelial claudin: Claudin-5/TMVCF constitutes tight junction strands in endothelial cells. J. Cell Biol. 1999, 147, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Liebner, S.; Fischmann, A.; Rascher, G.; Duffner, F.; Grote, E.H.; Kalbacher, H.; Wolburg, H. Claudin-1 and claudin-5 expression and tight junction morphology are altered in blood vessels of human glioblastoma multiforme. Acta Neuropathol. 2000, 100, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Wolburg, H.; Wolburg-Buchholz, K.; Kraus, J.; Rascher-Eggstein, G.; Liebner, S.; Hamm, S.; Duffner, F.; Grote, E.H.; Risau, W.; Engelhardt, B. Localization of claudin-3 in tight junctions of the blood-brain barrier is selectively lost during experimental autoimmune encephalomyelitis and human glioblastoma multiforme. Acta Neuropathol. 2003, 105, 586–592. [Google Scholar] [PubMed]
- Nitta, T.; Hata, M.; Gotoh, S.; Seo, Y.; Sasaki, H.; Hashimoto, N.; Furuse, M.; Tsukita, S. Size-selective loosening of the blood-brain barrier in claudin-5-deficient mice. J. Cell Biol. 2003, 161, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Furuse, M.; Hirase, T.; Itoh, M.; Nagafuchi, A.; Yonemura, S.; Tsukita, S. Occludin: A novel integral membrane protein localizing at tight junctions. J. Cell Biol. 1993, 123, 1777–1788. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, K.M.; Skare, I.B.; Stankewich, M.C.; Furuse, M.; Tsukita, S.; Rogers, R.A.; Lynch, R.D.; Schneeberger, E.E. Occludin is a functional component of the tight junction. J. Cell Sci. 1996, 109, 2287–2298. [Google Scholar] [PubMed]
- Wong, V.; Gumbiner, B.M. A synthetic peptide corresponding to the extracellular domain of occludin perturbs the tight junction permeability barrier. J. Cell Biol. 1997, 136, 399–409. [Google Scholar] [CrossRef] [PubMed]
- Saitou, M.; Furuse, M.; Sasaki, H.; Schulzke, J.D.; Fromm, M.; Takano, H.; Noda, T.; Tsukita, S. Complex phenotype of mice lacking occludin, a component of tight junction strands. Mol. Biol. Cell 2000, 11, 4131–4142. [Google Scholar] [CrossRef] [PubMed]
- Fanning, A.S.; Jameson, B.J.; Jesaitis, L.A.; Anderson, J.M. The tight junction protein ZO-1 establishes a link between the transmembrane protein occludin and the actin cytoskeleton. J. Biol. Chem. 1998, 273, 29745–29753. [Google Scholar] [CrossRef] [PubMed]
- Itoh, M.; Furuse, M.; Morita, K.; Kubota, K.; Saitou, M.; Tsukita, S. Direct binding of three tight junction-associated MAGUKs, ZO-1, ZO-2, and ZO-3, with the COOH termini of claudins. J. Cell Biol. 1999, 147, 1351–1363. [Google Scholar] [CrossRef] [PubMed]
- Tornavaca, O.; Chia, M.; Dufton, N.; Almagro, L.O.; Conway, D.E.; Randi, A.M.; Schwartz, M.A.; Matter, K.; Balda, M.S. ZO-1 controls endothelial adherens junctions, cell-cell tension, angiogenesis, and barrier formation. J. Cell Biol. 2015, 208, 821–838. [Google Scholar] [CrossRef] [PubMed]
- Dejana, E.; Orsenigo, F.; Lampugnani, M.G. The role of adherens junctions and VE-cadherin in the control of vascular permeability. J. Cell Sci. 2008, 121, 2115–2122. [Google Scholar] [CrossRef] [PubMed]
- Vestweber, D. VE-cadherin: The major endothelial adhesion molecule controlling cellular junctions and blood vessel formation. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Coomber, B.L.; Stewart, P.A. Morphometric analysis of CNS microvascular endothelium. Microvasc. Res. 1985, 30, 99–115. [Google Scholar] [CrossRef]
- Tuma, P.; Hubbard, A.L. Transcytosis: Crossing cellular barriers. Physiol. Rev. 2003, 83, 871–932. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.D.; Ye, M.; Levy, A.F.; Rothstein, J.D.; Bergles, D.E.; Searson, P.C. The blood-brain barrier: An engineering perspective. Front. Neuroeng. 2013, 6, 7. [Google Scholar] [CrossRef] [PubMed]
- Chow, B.W.; Gu, C. The molecular constituents of the blood-brain barrier. Trends Neurosci. 2015, 38, 598–608. [Google Scholar] [CrossRef] [PubMed]
- Smith, Q.R. A review of blood-brain barrier transport techniques. Methods Mol. Med. 2003, 89, 193–208. [Google Scholar] [PubMed]
- Enerson, B.E.; Drewes, L.R. The rat blood-brain barrier transcriptome. J. Cereb. Blood Flow Metab. 2006, 26, 959–973. [Google Scholar] [CrossRef] [PubMed]
- Henninger, D.D.; Panes, J.; Eppihimer, M.; Russell, J.; Gerritsen, M.; Anderson, D.C.; Granger, D.N. Cytokine-induced VCAM-1 and ICAM-1 expression in different organs of the mouse. J. Immunol. 1997, 158, 1825–1832. [Google Scholar] [PubMed]
- Aird, W.C. Phenotypic heterogeneity of the endothelium: II. Representative vascular beds. Circ. Res. 2007, 100, 174–190. [Google Scholar] [PubMed]
- Daneman, R.; Zhou, L.; Agalliu, D.; Cahoy, J.D.; Kaushal, A.; Barres, B.A. The mouse blood-brain barrier transcriptome: A new resource for understanding the development and function of brain endothelial cells. PLoS ONE 2010, 5, e13741. [Google Scholar] [CrossRef] [PubMed]
- Muldoon, L.L.; Alvarez, J.I.; Begley, D.J.; Boado, R.J.; Del Zoppo, G.J.; Doolittle, N.D.; Engelhardt, B.; Hallenbeck, J.M.; Lonser, R.R.; Ohlfest, J.R.; et al. Immunologic privilege in the central nervous system and the blood-brain barrier. J. Cereb. Blood Flow Metab. 2013, 33, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Bonkowski, D.; Katyshev, V.; Balabanov, R.D.; Borisov, A.; Dore-Duffy, P. The CNS microvascular pericyte: Pericyte-astrocyte crosstalk in the regulation of tissue survival. Fluids Barriers CNS 2011, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- Winkler, E.A.; Bell, R.D.; Zlokovic, B.V. Central nervous system pericytes in health and disease. Nat. Neurosci. 2011, 14, 1398–1405. [Google Scholar] [CrossRef] [PubMed]
- Daneman, R.; Agalliu, D.; Zhou, L.; Kuhnert, F.; Kuo, C.J.; Barres, B.A. Wnt/β-catenin signaling is required for CNS, but not non-CNS, angiogenesis (vol 106, pg 641, 2009). Proc. Natl. Acad. Sci. USA 2009, 106, 641–646. [Google Scholar] [CrossRef] [PubMed]
- Daneman, R.; Zhou, L.; Kebede, A.A.; Barres, B.A. Pericytes are required for blood-brain barrier integrity during embryogenesis. Nature 2010, 468, 562–566. [Google Scholar] [CrossRef] [PubMed]
- Armulik, A.; Abramsson, A.; Betsholtz, C. Endothelial/pericyte interactions. Circ. Res. 2005, 97, 512–523. [Google Scholar] [CrossRef] [PubMed]
- Armulik, A.; Genove, G.; Mae, M.; Nisancioglu, M.H.; Wallgard, E.; Niaudet, C.; He, L.; Norlin, J.; Lindblom, P.; Strittmatter, K.; et al. Pericytes regulate the blood-brain barrier. Nature 2010, 468, 557–561. [Google Scholar] [CrossRef] [PubMed]
- Bell, R.D.; Winkler, E.A.; Sagare, A.P.; Singh, I.; LaRue, B.; Deane, R.; Zlokovic, B.V. Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron 2010, 68, 409–427. [Google Scholar] [CrossRef] [PubMed]
- Peppiatt, C.M.; Howarth, C.; Mobbs, P.; Attwell, D. Bidirectional control of CNS capillary diameter by pericytes. Nature 2006, 443, 700–704. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, N.B.; Attwell, D.; Hall, C.N. Pericyte-mediated regulation of capillary diameter: A component of neurovascular coupling in health and disease. Front. Neuroenerg. 2010, 2, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Shepro, D.; Morel, N.M. Pericyte physiology. FASEB J. 1993, 7, 1031–1038. [Google Scholar] [PubMed]
- Oberheim, N.A.; Takano, T.; Han, X.; He, W.; Lin, J.H.; Wang, F.; Xu, Q.; Wyatt, J.D.; Pilcher, W.; Ojemann, J.G.; et al. Uniquely hominid features of adult human astrocytes. J. Neurosci. 2009, 29, 3276–3287. [Google Scholar] [CrossRef] [PubMed]
- Abbott, N.J.; Ronnback, L.; Hansson, E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Haydon, P.G.; Carmignoto, G. Astrocyte control of synaptic transmission and neurovascular coupling. Physiol. Rev. 2006, 86, 1009–1031. [Google Scholar] [CrossRef] [PubMed]
- Markiewicz, I.; Lukomska, B. The role of astrocytes in the physiology and pathology of the central nervous system. Acta Neurobiol. Exp. 2006, 66, 343–358. [Google Scholar]
- Sidoryk-Wegrzynowicz, M.; Wegrzynowicz, M.; Lee, E.; Bowman, A.B.; Aschner, M. Role of astrocytes in brain function and disease. Toxicol. Pathol. 2011, 39, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Attwell, D.; Buchan, A.M.; Charpak, S.; Lauritzen, M.; Macvicar, B.A.; Newman, E.A. Glial and neuronal control of brain blood flow. Nature 2010, 468, 232–243. [Google Scholar] [CrossRef] [PubMed]
- Gordon, G.R.; Howarth, C.; MacVicar, B.A. Bidirectional control of arteriole diameter by astrocytes. Exp. Physiol. 2011, 96, 393–399. [Google Scholar] [CrossRef] [PubMed]
- DeBault, L.E.; Cancilla, P.A. γ-Glutamyl transpeptidase in isolated brain endothelial cells: Induction by glial cells in vitro. Science 1980, 207, 653–655. [Google Scholar] [CrossRef] [PubMed]
- Janzer, R.C.; Raff, M.C. Astrocytes induce blood-brain barrier properties in endothelial cells. Nature 1987, 325, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.W.; Kim, W.J.; Choi, Y.K.; Song, H.S.; Son, M.J.; Gelman, I.H.; Kim, Y.J.; Kim, K.W. SSeCKS regulates angiogenesis and tight junction formation in blood-brain barrier. Nat. Med. 2003, 9, 900–906. [Google Scholar] [CrossRef] [PubMed]
- Yousif, L.F.; Di Russo, J.; Sorokin, L. Laminin isoforms in endothelial and perivascular basement membranes. Cell Adhes. Migr. 2013, 7, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Morris, A.W.; Carare, R.O.; Schreiber, S.; Hawkes, C.A. The cerebrovascular basement membrane: Role in the clearance of β-amyloid and cerebral amyloid angiopathy. Front. Aging Neurosci. 2014, 6, 251. [Google Scholar] [CrossRef] [PubMed]
- Sixt, M.; Engelhardt, B.; Pausch, F.; Hallmann, R.; Wendler, O.; Sorokin, L.M. Endothelial cell laminin isoforms, laminins 8 and 10, play decisive roles in T cell recruitment across the blood-brain barrier in experimental autoimmune encephalomyelitis. J. Cell Biol. 2001, 153, 933–946. [Google Scholar] [CrossRef] [PubMed]
- Owens, T.; Bechmann, I.; Engelhardt, B. Perivascular spaces and the two steps to neuroinflammation. J. Neuropathol. Exp. Neurol. 2008, 67, 1113–1121. [Google Scholar] [CrossRef] [PubMed]
- Baeten, K.M.; Akassoglou, K. Extracellular matrix and matrix receptors in blood-brain barrier formation and stroke. Dev. Neurobiol. 2011, 71, 1018–1039. [Google Scholar] [CrossRef] [PubMed]
- Sorokin, L. The impact of the extracellular matrix on inflammation. Nat. Rev. Immunol. 2010, 10, 712–723. [Google Scholar] [CrossRef] [PubMed]
- Van Horssen, J.; Bo, L.; Vos, C.M.; Virtanen, I.; de Vries, H.E. Basement membrane proteins in multiple sclerosis-associated inflammatory cuffs: Potential role in influx and transport of leukocytes. Neuropathol. Exp. Neurol. 2005, 64, 722–729. [Google Scholar] [CrossRef]
- Savettieri, G.; Di Liegro, I.; Catania, C.; Licata, L.; Pitarresi, G.L.; D’Agostino, S.; Schiera, G.; De Caro, V.; Giandalia, G.; Giannola, L.I.; Cestelli, A. Neurons and ECM regulate occludin localization in brain endothelial cells. Neuroreport 2000, 11, 1081–1084. [Google Scholar] [CrossRef] [PubMed]
- Tilling, T.; Korte, D.; Hoheisel, D.; Galla, H.J. Basement membrane proteins influence brain capillary endothelial barrier function in vitro. J. Neurochem. 1998, 71, 1151–1157. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, H.M.; Kozlowski, P.B. Evidence for blood-brain barrier changes in senile dementia of the Alzheimer type (SDAT). Ann. N. Y. Acad. Sci. 1982, 396, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, H.M.; Vorbrodt, A.W.; Wegiel, J. Amyloid angiopathy and blood-brain barrier changes in Alzheimer’s disease. Ann. N. Y. Acad. Sci. 1997, 826, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Slemmon, J.R.; Hughes, C.M.; Campbell, G.A.; Flood, D.G. Increased levels of hemoglobin-derived and other peptides in Alzheimer’s disease cerebellum. J. Neurosci. 1994, 14, 2225–2235. [Google Scholar] [PubMed]
- Zipser, B.D.; Johanson, C.E.; Gonzalez, L.; Berzin, T.M.; Tavares, R.; Hulette, C.M.; Vitek, M.P.; Hovanesian, V.; Stopa, E.G. Microvascular injury and blood-brain barrier leakage in Alzheimer’s disease. Neurobiol. Aging 2007, 28, 977–986. [Google Scholar] [CrossRef] [PubMed]
- Alafuzoff, I.; Adolfsson, R.; Grundke-Iqbal, I.; Winblad, B. Blood-brain barrier in Alzheimer dementia and in non-demented elderly. An immunocytochemical study. Acta Neuropathol. 1987, 73, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Tomimoto, H.; Akiguchi, I.; Suenaga, T.; Nishimura, M.; Wakita, H.; Nakamura, S.; Kimura, J. Alterations of the blood-brain barrier and glial cells in white-matter lesions in cerebrovascular and Alzheimer’s disease patients. Stroke 1996, 27, 2069–2074. [Google Scholar] [CrossRef] [PubMed]
- Rozemuller, J.M.; Eikelenboom, P.; Kamphorst, W.; Stam, F.C. Lack of evidence for dysfunction of the blood-brain barrier in Alzheimer’s disease: An immunohistochemical study. Neurobiol. Aging 1988, 9, 383–391. [Google Scholar] [CrossRef]
- Elovaara, I.; Icen, A.; Palo, J.; Erkinjuntti, T. CSF in Alzheimer’s disease. Studies on blood-brain barrier function and intrathecal protein synthesis. J. Neurol. Sci. 1985, 70, 73–80. [Google Scholar] [CrossRef]
- Skoog, I.; Wallin, A.; Fredman, P.; Hesse, C.; Aevarsson, O.; Karlsson, I.; Gottfries, C.G.; Blennow, K. A population study on blood-brain barrier function in 85-year-olds: Relation to Alzheimer’s disease and vascular dementia. Neurology 1998, 50, 966–971. [Google Scholar] [CrossRef] [PubMed]
- Alafuzoff, I.; Adolfsson, R.; Bucht, G.; Winblad, B. Albumin and immunoglobulin in plasma and cerebrospinal fluid, and blood-cerebrospinal fluid barrier function in patients with dementia of Alzheimer type and multi-infarct dementia. J. Neurol. Sci. 1983, 60, 465–472. [Google Scholar] [CrossRef]
- Algotsson, A.; Winblad, B. The integrity of the blood-brain barrier in Alzheimer’s disease. Acta Neurol. Scand. 2007, 115, 403–408. [Google Scholar] [CrossRef] [PubMed]
- Blennow, K.; Wallin, A.; Fredman, P.; Karlsson, I.; Gottfries, C.G.; Svennerholm, L. Blood-brain barrier disturbance in patients with Alzheimer’s disease is related to vascular factors. Acta Neurol. Scand. 1990, 81, 323–326. [Google Scholar] [CrossRef] [PubMed]
- Blennow, K.; Wallin, A.; Uhlemann, C.; Gottfries, C.G. White-matter lesions on CT in Alzheimer patients: Relation to clinical symptomatology and vascular factors. Acta Neurol. Scand. 1991, 83, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Wallin, A.; Blennow, K.; Rosengren, L. Cerebrospinal fluid markers of pathogenetic processes in vascular dementia, with special reference to the subcortical subtype. Alzheimer Dis. Assoc. Disord. 1999, 13 (Suppl. S3), S102–S105. [Google Scholar] [PubMed]
- Farrall, A.J.; Wardlaw, J.M. Blood-brain barrier: Ageing and microvascular disease—Systematic review and meta-analysis. Neurobiol. Aging 2009, 30, 337–352. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.L. Is it appropriate to use albumin CSF/plasma ratio to assess blood brain barrier permeability? Neurobiol. Aging 2011, 32, 1338–1339. [Google Scholar] [CrossRef] [PubMed]
- Silverberg, G.D.; Heit, G.; Huhn, S.; Jaffe, R.A.; Chang, S.D.; Bronte-Stewart, H.; Rubenstein, E.; Possin, K.; Saul, T.A. The cerebrospinal fluid production rate is reduced in dementia of the Alzheimer’s type. Neurology 2001, 57, 1763–1766. [Google Scholar] [CrossRef] [PubMed]
- Caserta, M.T.; Caccioppo, D.; Lapin, G.D.; Ragin, A.; Groothuis, D.R. Blood-brain barrier integrity in Alzheimer’s disease patients and elderly control subjects. J. Neuropsychiatry Clin. Neurosci. 1998, 10, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Dysken, M.W.; Nelson, M.J.; Hoover, K.M.; Kuskowski, M.; McGeachie, R. Rapid dynamic CT scanning in primary degenerative dementia and age-matched controls. Biol. Psychiatry 1990, 28, 425–434. [Google Scholar] [CrossRef]
- Schlageter, N.L.; Carson, R.E.; Rapoport, S.I. Examination of blood-brain barrier permeability in dementia of the Alzheimer type with [68Ga] EDTA and positron emission tomography. J. Cereb. Blood Flow Metab. 1987, 7, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Starr, J.M.; Farrall, A.J.; Armitage, P.; McGurn, B.; Wardlaw, J. Blood-brain barrier permeability in Alzheimer’s disease: A case-control MRI study. Psychiatry Res. 2009, 171, 232–241. [Google Scholar] [CrossRef] [PubMed]
- Montagne, A.; Barnes, S.R.; Sweeney, M.D.; Halliday, M.R.; Sagare, A.P.; Zhao, Z.; Toga, A.W.; Jacobs, R.E.; Liu, C.Y.; Amezcua, L.; et al. Blood-brain barrier breakdown in the aging human hippocampus. Neuron 2015, 85, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Carrano, A.; Hoozemans, J.J.; van der Vies, S.M.; van Horssen, J.; de Vries, H.E.; Rozemuller, A.J. Neuroinflammation and blood-brain barrier changes in capillary amyloid angiopathy. Neurodegener. Dis. 2012, 10, 329–331. [Google Scholar] [CrossRef] [PubMed]
- Carrano, A.; Hoozemans, J.J.; van der Vies, S.M.; Rozemuller, A.J.; van Horssen, J.; de Vries, H.E. Amyloid Beta induces oxidative stress-mediated blood-brain barrier changes in capillary amyloid angiopathy. Antioxid. Redox Signal. 2011, 15, 1167–1178. [Google Scholar] [CrossRef] [PubMed]
- Kook, S.Y.; Hong, H.S.; Moon, M.; Ha, C.M.; Chang, S.; Mook-Jung, I. Aβ1-42-RAGE interaction disrupts tight junctions of the blood-brain barrier via Ca2+-calcineurin signaling. J. Neurosci. 2012, 32, 8845–8854. [Google Scholar] [CrossRef] [PubMed]
- Wan, W.; Cao, L.; Liu, L.; Zhang, C.; Kalionis, B.; Tai, X.; Li, Y.; Xia, S. Aβ1–42 oligomer-induced leakage in an in vitro blood-brain barrier model is associated with up-regulation of RAGE and metalloproteinases, and down-regulation of tight junction scaffold proteins. J. Neurochem. 2015, 134, 382–393. [Google Scholar] [CrossRef] [PubMed]
- Tai, L.M.; Holloway, K.A.; Male, D.K.; Loughlin, A.J.; Romero, I.A. Amyloid-beta-induced occludin down-regulation and increased permeability in human brain endothelial cells is mediated by MAPK activation. J. Cell. Mol. Med. 2010, 14, 1101–1112. [Google Scholar] [PubMed]
- Marco, S.; Skaper, S.D. Amyloid β-peptide1–42 alters tight junction protein distribution and expression in brain microvessel endothelial cells. Neurosci. Lett. 2006, 401, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Chao, A.C.; Lee, T.C.; Juo, S.H.; Yang, D.I. Hyperglycemia increases the production of amyloid β-peptide leading to decreased endothelial tight junction. CNS Neurosci. Ther. 2016, 22, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Beese, M.; Wyss, K.; Haubitz, M.; Kirsch, T. Effect of cAMP derivates on assembly and maintenance of tight junctions in human umbilical vein endothelial cells. BMC Cell Biol. 2010, 11, 68. [Google Scholar] [CrossRef] [PubMed]
- Muradashvili, N.; Tyagi, R.; Metreveli, N.; Tyagi, S.C.; Lominadze, D. Ablation of MMP9 gene ameliorates paracellular permeability and fibrinogen-amyloid beta complex formation during hyperhomocysteinemia. J. Cereb. Blood Flow Metab. 2014, 34, 1472–1482. [Google Scholar] [CrossRef] [PubMed]
- Harik, S.I. Changes in the glucose transporter of brain capillaries. Can. J. Physiol. Pharmacol. 1992, 70, S113–S117. [Google Scholar] [CrossRef] [PubMed]
- Horwood, N.; Davies, D.C. Immunolabelling of hippocampal microvessel glucose transporter protein is reduced in Alzheimer’s disease. Virchows Arch. 1994, 425, 69–72. [Google Scholar] [CrossRef] [PubMed]
- Mooradian, A.D.; Chung, H.C.; Shah, G.N. GLUT-1 expression in the cerebra of patients with Alzheimer’s disease. Neurobiol. Aging 1997, 18, 469–474. [Google Scholar] [CrossRef]
- Merlini, M.; Meyer, E.P.; Ulmann-Schuler, A.; Nitsch, R.M. Vascular β-amyloid and early astrocyte alterations impair cerebrovascular function and cerebral metabolism in transgenic arcAbeta mice. Acta Neuropathol. 2011, 122, 293–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hooijmans, C.R.; Graven, C.; Dederen, P.J.; Tanila, H.; van Groen, T.; Kiliaan, A.J. Amyloid beta deposition is related to decreased glucose transporter-1 levels and hippocampal atrophy in brains of aged APP/PS1 mice. Brain Res. 2007, 1181, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Winkler, E.A.; Nishida, Y.; Sagare, A.P.; Rege, S.V.; Bell, R.D.; Perlmutter, D.; Sengillo, J.D.; Hillman, S.; Kong, P.; Nelson, A.R.; et al. GLUT1 reductions exacerbate Alzheimer’s disease vasculo-neuronal dysfunction and degeneration. Nat. Neurosci. 2015, 18, 521–530. [Google Scholar] [CrossRef] [PubMed]
- De Vivo, D.C.; Trifiletti, R.R.; Jacobson, R.I.; Ronen, G.M.; Behmand, R.A.; Harik, S.I. Defective glucose transport across the blood-brain barrier as a cause of persistent hypoglycorrhachia, seizures, and developmental delay. N. Engl. J. Med. 1991, 325, 703–709. [Google Scholar] [CrossRef] [PubMed]
- De Giorgis, V.; Veggiotti, P. GLUT1 deficiency syndrome 2013: Current state of the art. Seizure 2013, 22, 803–811. [Google Scholar] [CrossRef] [PubMed]
- Brockmann, K. The expanding phenotype of GLUT1-deficiency syndrome. Brain Dev. 2009, 31, 545–552. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.D.; Ayyadurai, S.; Zlokovic, B.V. Pericytes of the neurovascular unit: Key functions and signaling pathways. Nat. Neurosci. 2016, 19, 771–783. [Google Scholar] [CrossRef] [PubMed]
- Sengillo, J.D.; Winkler, E.A.; Walker, C.T.; Sullivan, J.S.; Johnson, M.; Zlokovic, B.V. Deficiency in mural vascular cells coincides with blood-brain barrier disruption in Alzheimer’s disease. Brain Pathol. 2013, 23, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Sagare, A.P.; Bell, R.D.; Zhao, Z.; Ma, Q.; Winkler, E.A.; Ramanathan, A.; Zlokovic, B.V. Pericyte loss influences Alzheimer-like neurodegeneration in mice. Nat. Commun. 2013, 4, 2932. [Google Scholar] [CrossRef] [PubMed]
- Hoshi, A.; Yamamoto, T.; Shimizu, K.; Ugawa, Y.; Nishizawa, M.; Takahashi, H.; Kakita, A. Characteristics of aquaporin expression surrounding senile plaques and cerebral amyloid angiopathy in Alzheimer disease. Neuropathol. Exp. Neurol. 2012, 71, 750–759. [Google Scholar] [CrossRef] [PubMed]
- Moftakhar, P.; Lynch, M.D.; Pomakian, J.L.; Vinters, H.V. Aquaporin expression in the brains of patients with or without cerebral amyloid angiopathy. Neuropathol. Exp. Neurol. 2010, 69, 1201–1209. [Google Scholar] [CrossRef] [PubMed]
- Zeppenfeld, D.M.; Simon, M.; Haswell, J.D.; D’Abreo, D.; Murchison, C.; Quinn, J.F.; Grafe, M.R.; Woltjer, R.L.; Kaye, J.; Iliff, J.J. Association of Perivascular Localization of Aquaporin-4 With Cognition and Alzheimer Disease in Aging Brains. JAMA Neurol. 2017, 74, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Wilcock, D.M.; Vitek, M.P.; Colton, C.A. Vascular amyloid alters astrocytic water and potassium channels in mouse models and humans with Alzheimer’s disease. Neuroscience 2009, 159, 1055–1069. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Lunde, L.K.; Nuntagij, P.; Oguchi, T.; Camassa, L.M.; Nilsson, L.N.; Lannfelt, L.; Xu, Y.; Amiry-Moghaddam, M.; Ottersen, O.P.; et al. Loss of astrocyte polarization in the tg-ArcSwe mouse model of Alzheimer’s disease. J. Alzheimer’s Dis. 2011, 27, 711–722. [Google Scholar]
- Iliff, J.J.; Wang, M.; Liao, Y.; Plogg, B.A.; Peng, W.; Gundersen, G.A.; Benveniste, H.; Vates, G.E.; Deane, R.; Goldman, S.A.; Nagelhus, E.A.; Nedergaard, M. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid beta. Sci. Translat. Med. 2012, 4, 147ra111. [Google Scholar] [CrossRef] [PubMed]
- Farkas, E.; Luiten, P.G. Cerebral microvascular pathology in aging and Alzheimer’s disease. Prog. Neurobiol. 2001, 64, 575–611. [Google Scholar] [CrossRef]
- Lepelletier, F.X.; Mann, D.M.; Robinson, A.C.; Pinteaux, E.; Boutin, H. Early changes in extracellular matrix in Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 2017, 43, 167–182. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Shi, J.; Smallman, R.; Iwatsubo, T.; Mann, D.M. Relationships in Alzheimer’s disease between the extent of Abeta deposition in cerebral blood vessel walls, as cerebral amyloid angiopathy, and the amount of cerebrovascular smooth muscle cells and collagen. Neuropathol. Appl. Neurobiol. 2006, 32, 332–340. [Google Scholar] [CrossRef] [PubMed]
- Kalaria, R.N.; Pax, A.B. Increased collagen content of cerebral microvessels in Alzheimer’s disease. Brain Res. 1995, 705, 349–352. [Google Scholar] [CrossRef]
- Christov, A.; Ottman, J.; Hamdheydari, L.; Grammas, P. Structural changes in Alzheimer’s disease brain microvessels. Curr. Alzheimer Res. 2008, 5, 392–395. [Google Scholar] [CrossRef] [PubMed]
- Morris, A.W.; Sharp, M.M.; Albargothy, N.J.; Fernandes, R.; Hawkes, C.A.; Verma, A.; Weller, R.O.; Carare, R.O. Vascular basement membranes as pathways for the passage of fluid into and out of the brain. Acta Neuropathol. 2016, 131, 725–736. [Google Scholar] [CrossRef] [PubMed]
- Hawkes, C.A.; Gatherer, M.; Sharp, M.M.; Dorr, A.; Yuen, H.M.; Kalaria, R.; Weller, R.O.; Carare, R.O. Regional differences in the morphological and functional effects of aging on cerebral basement membranes and perivascular drainage of amyloid-beta from the mouse brain. Aging Cell 2013, 12, 224–236. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein E and Alzheimer disease: Risk, mechanisms and therapy. Nat. Rev. Neurol. 2013, 9, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, Y.; Painter, M.M.; Bu, G.; Kanekiyo, T. Apolipoprotein E as a Therapeutic Target in Alzheimer’s Disease: A Review of Basic Research and Clinical Evidence. CNS Drugs 2016, 30, 773–789. [Google Scholar] [CrossRef] [PubMed]
- Hawkes, C.A.; Sullivan, P.M.; Hands, S.; Weller, R.O.; Nicoll, J.A.; Carare, R.O. Disruption of arterial perivascular drainage of amyloid-beta from the brains of mice expressing the human APOE epsilon4 allele. PLoS ONE 2012, 7, e41636. [Google Scholar] [CrossRef] [PubMed]
- Saido, T.; Leissring, M.A. Proteolytic degradation of amyloid beta-protein. Cold Spring Harb. Perspect. Med. 2012, 2, a006379. [Google Scholar] [CrossRef] [PubMed]
- Kanekiyo, T.; Bu, G. The low-density lipoprotein receptor-related protein 1 and amyloid-beta clearance in Alzheimer’s disease. Front. Aging Neurosci. 2014, 6, 93. [Google Scholar] [CrossRef] [PubMed]
- Shibata, M.; Yamada, S.; Kumar, S.R.; Calero, M.; Bading, J.; Frangione, B.; Holtzman, D.M.; Miller, C.A.; Strickland, D.K.; Ghiso, J.; et al. Clearance of Alzheimer’s amyloid-ss(1–40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J. Clin. Investig. 2000, 106, 1489–1499. [Google Scholar] [CrossRef] [PubMed]
- Deane, R.; Wu, Z.; Sagare, A.; Davis, J.; Du Yan, S.; Hamm, K.; Xu, F.; Parisi, M.; LaRue, B.; Hu, H.W.; et al. LRP/amyloid beta-peptide interaction mediates differential brain efflux of Abeta isoforms. Neuron 2004, 43, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Storck, S.E.; Meister, S.; Nahrath, J.; Meissner, J.N.; Schubert, N.; Di Spiezio, A.; Baches, S.; Vandenbroucke, R.E.; Bouter, Y.; Prikulis, I.; et al. Endothelial LRP1 transports amyloid-beta(1–42) across the blood-brain barrier. J. Clin. Investig. 2016, 126, 123–136. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Ueno, T.; Ohtsuki, S.; Terasaki, T. Lack of brain-to-blood efflux transport activity of low-density lipoprotein receptor-related protein-1 (LRP-1) for amyloid-beta peptide(1–40) in mouse: Involvement of an LRP-1-independent pathway. J. Neurochem. 2010, 113, 1356–1363. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Hashimoto, T.; Yabuki, C.; Nagae, Y.; Tachikawa, M.; Strickland, D.K.; Liu, Q.; Bu, G.; Basak, J.M.; Holtzman, D.M.; et al. The low density lipoprotein receptor-related protein 1 mediates uptake of amyloid beta peptides in an in vitro model of the blood-brain barrier cells. J. Biol. Chem. 2008, 283, 34554–34562. [Google Scholar] [CrossRef] [PubMed]
- Pflanzner, T.; Janko, M.C.; Andre-Dohmen, B.; Reuss, S.; Weggen, S.; Roebroek, A.J.; Kuhlmann, C.R.; Pietrzik, C.U. LRP1 mediates bidirectional transcytosis of amyloid-beta across the blood-brain barrier. Neurobiol. Aging 2011, 32, 2323.e1–2323.e11. [Google Scholar] [CrossRef] [PubMed]
- Nazer, B.; Hong, S.; Selkoe, D.J. LRP promotes endocytosis and degradation, but not transcytosis, of the amyloid-beta peptide in a blood-brain barrier in vitro model. Neurobiol. Dis. 2008, 30, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Candela, P.; Saint-Pol, J.; Kuntz, M.; Boucau, M.C.; Lamartiniere, Y.; Gosselet, F.; Fenart, L. In vitro discrimination of the role of LRP1 at the BBB cellular level: Focus on brain capillary endothelial cells and brain pericytes. Brain Res. 2015, 1594, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Schinkel, A.H. P-Glycoprotein, a gatekeeper in the blood-brain barrier. Adv. Drug Deliv. Rev. 1999, 36, 179–194. [Google Scholar] [CrossRef]
- Cirrito, J.R.; Deane, R.; Fagan, A.M.; Spinner, M.L.; Parsadanian, M.; Finn, M.B.; Jiang, H.; Prior, J.L.; Sagare, A.; Bales, K.R.; et al. P-glycoprotein deficiency at the blood-brain barrier increases amyloid-beta deposition in an Alzheimer disease mouse model. J. Clin. Investig. 2005, 115, 3285–3290. [Google Scholar] [CrossRef] [PubMed]
- Park, R.; Kook, S.Y.; Park, J.C.; Mook-Jung, I. Abeta1–42 reduces P-glycoprotein in the blood-brain barrier through RAGE-NF-kappaB signaling. Cell Death Dis. 2014, 5, e1299. [Google Scholar] [CrossRef] [PubMed]
- Hartz, A.M.; Zhong, Y.; Wolf, A.; LeVine, H.; Miller, D.S.; Bauer, B. Abeta40 Reduces p-glycoprotein at the blood-brain barrier through the ubiquitin-proteasome pathway. J. Neurosci. 2016, 36, 1930–1941. [Google Scholar] [CrossRef] [PubMed]
- Vogelgesang, S.; Cascorbi, I.; Schroeder, E.; Pahnke, J.; Kroemer, H.K.; Siegmund, W.; Kunert-Keil, C.; Walker, L.C.; Warzok, R.W. Deposition of Alzheimer’s beta-amyloid is inversely correlated with P-glycoprotein expression in the brains of elderly non-demented humans. Pharmacogenetics 2002, 12, 535–541. [Google Scholar] [CrossRef] [PubMed]
- Van Assema, D.M.; Lubberink, M.; Bauer, M.; van der Flier, W.M.; Schuit, R.C.; Windhorst, A.D.; Comans, E.F.; Hoetjes, N.J.; Tolboom, N.; Langer, O.; et al. Blood-brain barrier P-glycoprotein function in Alzheimer’s disease. Brain J. Neurol. 2012, 135, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, M.; Sato, N.; Kurinami, H.; Takeuchi, D.; Takeda, S.; Shimamura, M.; Yamashita, T.; Uchiyama, Y.; Rakugi, H.; Morishita, R. Reduction of brain beta-amyloid (Abeta) by fluvastatin, a hydroxymethylglutaryl-CoA reductase inhibitor, through increase in degradation of amyloid precursor protein C-terminal fragments (APP-CTFs) and Abeta clearance. J. Biol. Chem. 2010, 285, 22091–22102. [Google Scholar] [CrossRef] [PubMed]
- Hartz, A.M.; Miller, D.S.; Bauer, B. Restoring blood-brain barrier P-glycoprotein reduces brain amyloid-beta in a mouse model of Alzheimer’s disease. Mol. Pharmacol. 2010, 77, 715–723. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.F.; Ramasamy, R.; Schmidt, A.M. The RAGE axis: A fundamental mechanism signaling danger to the vulnerable vasculature. Circ. Res. 2010, 106, 842–853. [Google Scholar] [CrossRef] [PubMed]
- Kierdorf, K.; Fritz, G. RAGE regulation and signaling in inflammation and beyond. J. Leukoc. Biol. 2013, 94, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.C.; Tavares, R.; Johanson, C.E.; Hovanesian, V.; Donahue, J.E.; Gonzalez, L.; Silverberg, G.D.; Stopa, E.G. Hippocampal RAGE immunoreactivity in early and advanced Alzheimer’s disease. Brain Res. 2008, 1230, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Deane, R.; Singh, I.; Sagare, A.P.; Bell, R.D.; Ross, N.T.; LaRue, B.; Love, R.; Perry, S.; Paquette, N.; Deane, R.J.; et al. A multimodal RAGE-specific inhibitor reduces amyloid beta-mediated brain disorder in a mouse model of Alzheimer disease. J. Clin. Investig. 2012, 122, 1377–1392. [Google Scholar] [CrossRef] [PubMed]
- Deane, R.; Du Yan, S.; Submamaryan, R.K.; LaRue, B.; Jovanovic, S.; Hogg, E.; Welch, D.; Manness, L.; Lin, C.; Yu, J.; et al. RAGE mediates amyloid-beta peptide transport across the blood-brain barrier and accumulation in brain. Nat. Med. 2003, 9, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Xiong, H.Q.; Callaghan, D.; Jones, A.; Bai, J.Y.; Rasquinha, I.; Smith, C.; Pei, K.; Walker, D.; Lue, L.F.; Stanimirovic, D.; et al. ABCG2 Is Upregulated in Alzheimer’s Brain with Cerebral Amyloid Angiopathy and May Act as a Gatekeeper at the Blood-Brain Barrier for A beta(1–40) Peptides. J. Neurosci. 2009, 29, 5463–5475. [Google Scholar] [CrossRef] [PubMed]
- Do, T.M.; Noel-Hudson, M.S.; Ribes, S.; Besengez, C.; Smirnova, M.; Cisternino, S.; Buyse, M.; Calon, F.; Chimini, G.; Chacun, H.; et al. ABCG2-and ABCG4-mediated efflux of amyloid-beta peptide 1–40 at the mouse blood-brain barrier. J. Alzheimer’s Dis. 2012, 30, 155–166. [Google Scholar]
- De Bruijn, R.F.; Ikram, M.A. Cardiovascular risk factors and future risk of Alzheimer’s disease. BMC Med. 2014, 12, 130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, C.Y.; Snyder, P.J.; Wu, W.-C.; Zhang, M.; Echeverria, A.; Alber, J. Pathophysiologic relationship between Alzheimer’s disease, cerebrovascular disease, and cardiovascular risk: A review and synthesis. Alzheimer’s Dement. Diagn. Assess. Dis. Monit. 2017, 7, 69–87. [Google Scholar] [CrossRef] [PubMed]
- Gorelick, P.B. Risk factors for vascular dementia and Alzheimer disease. Stroke 2004, 35, 2620–2622. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M.; Engelhardt, B. The anatomical and cellular basis of immune surveillance in the central nervous system. Nat. Rev. Immunol. 2012, 12, 623–635. [Google Scholar] [CrossRef] [PubMed]
- Prinz, M.; Priller, J. The role of peripheral immune cells in the CNS in steady state and disease. Nat. Neurosci. 2017, 20, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Lampron, A.; Elali, A.; Rivest, S. Innate immunity in the CNS: Redefining the relationship between the CNS and Its environment. Neuron 2013, 78, 214–232. [Google Scholar] [CrossRef] [PubMed]
- Grammas, P. Neurovascular dysfunction, inflammation and endothelial activation: Implications for the pathogenesis of Alzheimer’s disease. J. Neuroinflamm. 2011, 8, 26. [Google Scholar] [CrossRef] [PubMed]
- Rossi, B.; Angiari, S.; Zenaro, E.; Budui, S.L.; Constantin, G. Vascular inflammation in central nervous system diseases: Adhesion receptors controlling leukocyte-endothelial interactions. J. Leukoc. Biol. 2011, 89, 539–556. [Google Scholar] [CrossRef] [PubMed]
- Zenaro, E.; Pietronigro, E.; Della Bianca, V.; Piacentino, G.; Marongiu, L.; Budui, S.; Turano, E.; Rossi, B.; Angiari, S.; Dusi, S.; et al. Neutrophils promote Alzheimer’s disease-like pathology and cognitive decline via LFA-1 integrin. Nat. Med. 2015, 21, 880–886. [Google Scholar] [CrossRef] [PubMed]



© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yamazaki, Y.; Kanekiyo, T. Blood-Brain Barrier Dysfunction and the Pathogenesis of Alzheimer’s Disease. Int. J. Mol. Sci. 2017, 18, 1965. https://doi.org/10.3390/ijms18091965
Yamazaki Y, Kanekiyo T. Blood-Brain Barrier Dysfunction and the Pathogenesis of Alzheimer’s Disease. International Journal of Molecular Sciences. 2017; 18(9):1965. https://doi.org/10.3390/ijms18091965
Chicago/Turabian StyleYamazaki, Yu, and Takahisa Kanekiyo. 2017. "Blood-Brain Barrier Dysfunction and the Pathogenesis of Alzheimer’s Disease" International Journal of Molecular Sciences 18, no. 9: 1965. https://doi.org/10.3390/ijms18091965
APA StyleYamazaki, Y., & Kanekiyo, T. (2017). Blood-Brain Barrier Dysfunction and the Pathogenesis of Alzheimer’s Disease. International Journal of Molecular Sciences, 18(9), 1965. https://doi.org/10.3390/ijms18091965