Light-Up RNA Aptamers and Their Cognate Fluorogens: From Their Development to Their Applications

Abstract
:1. Introduction
2. Development of RNA-Based Fluorogenic Modules
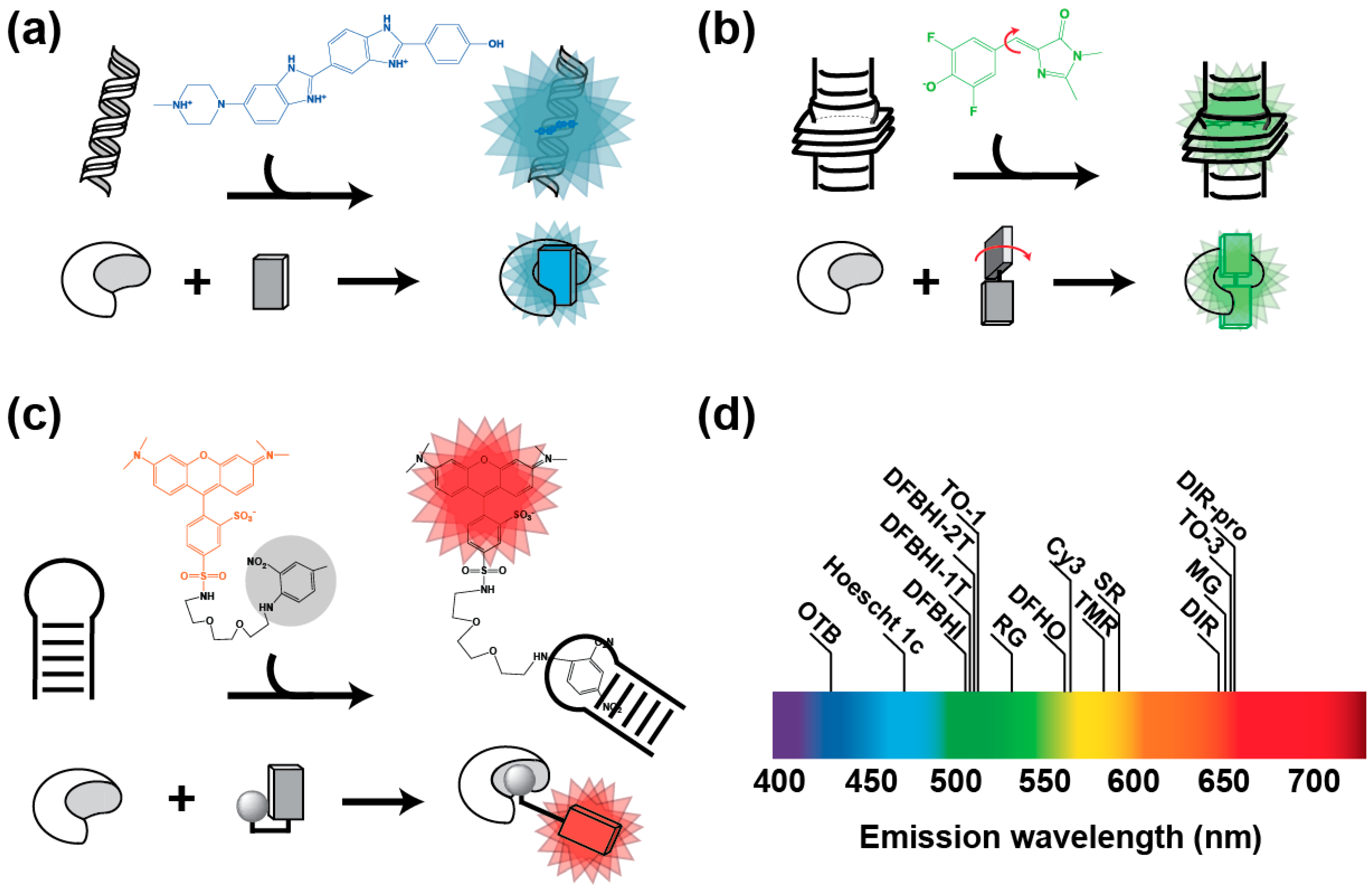
2.1. Fluorogenic Dye Engineering
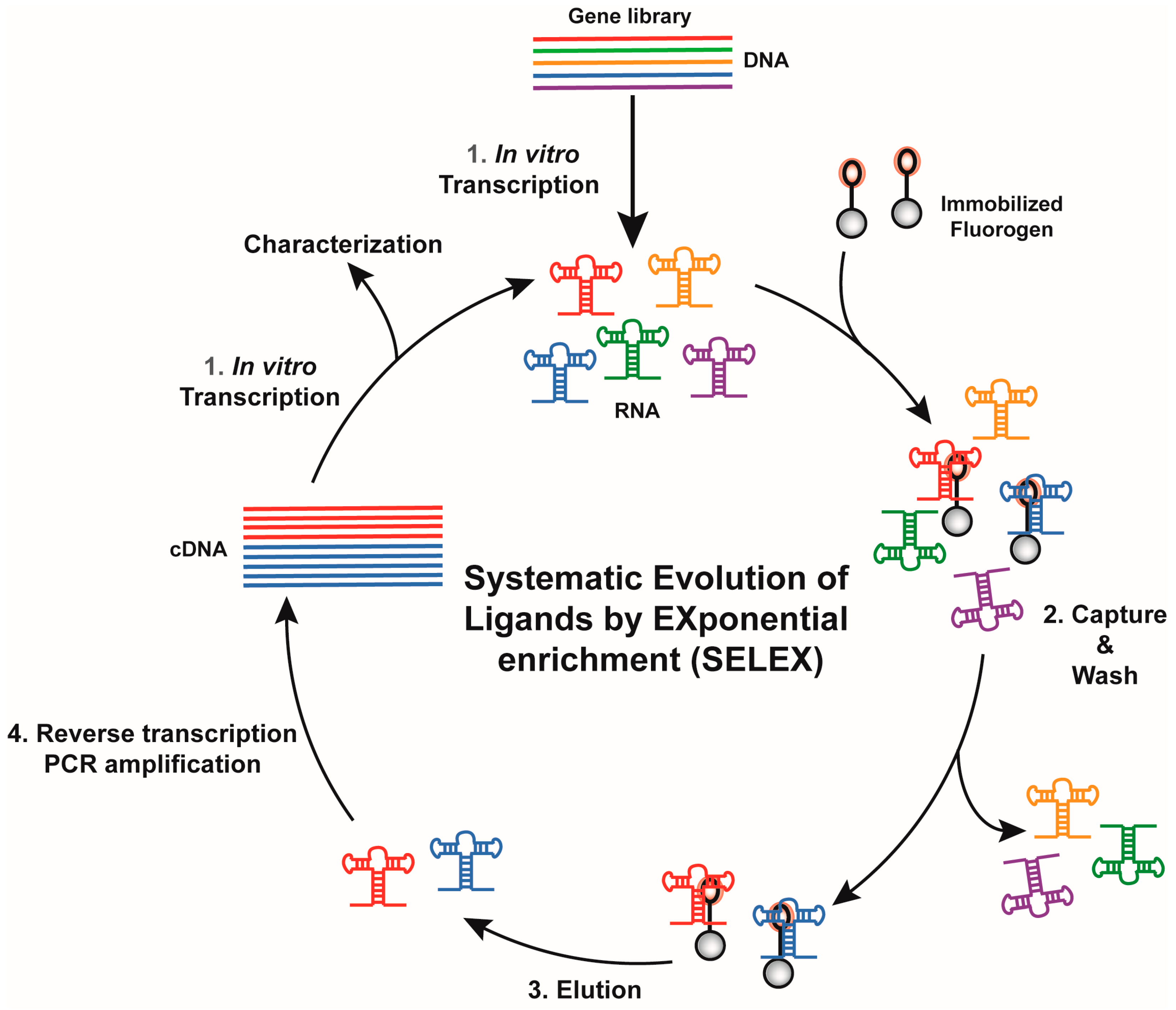
2.2. Isolation of Fluorogenic RNA Aptamers
2.2.1. Aptamers Selection Based on Binding Capacity
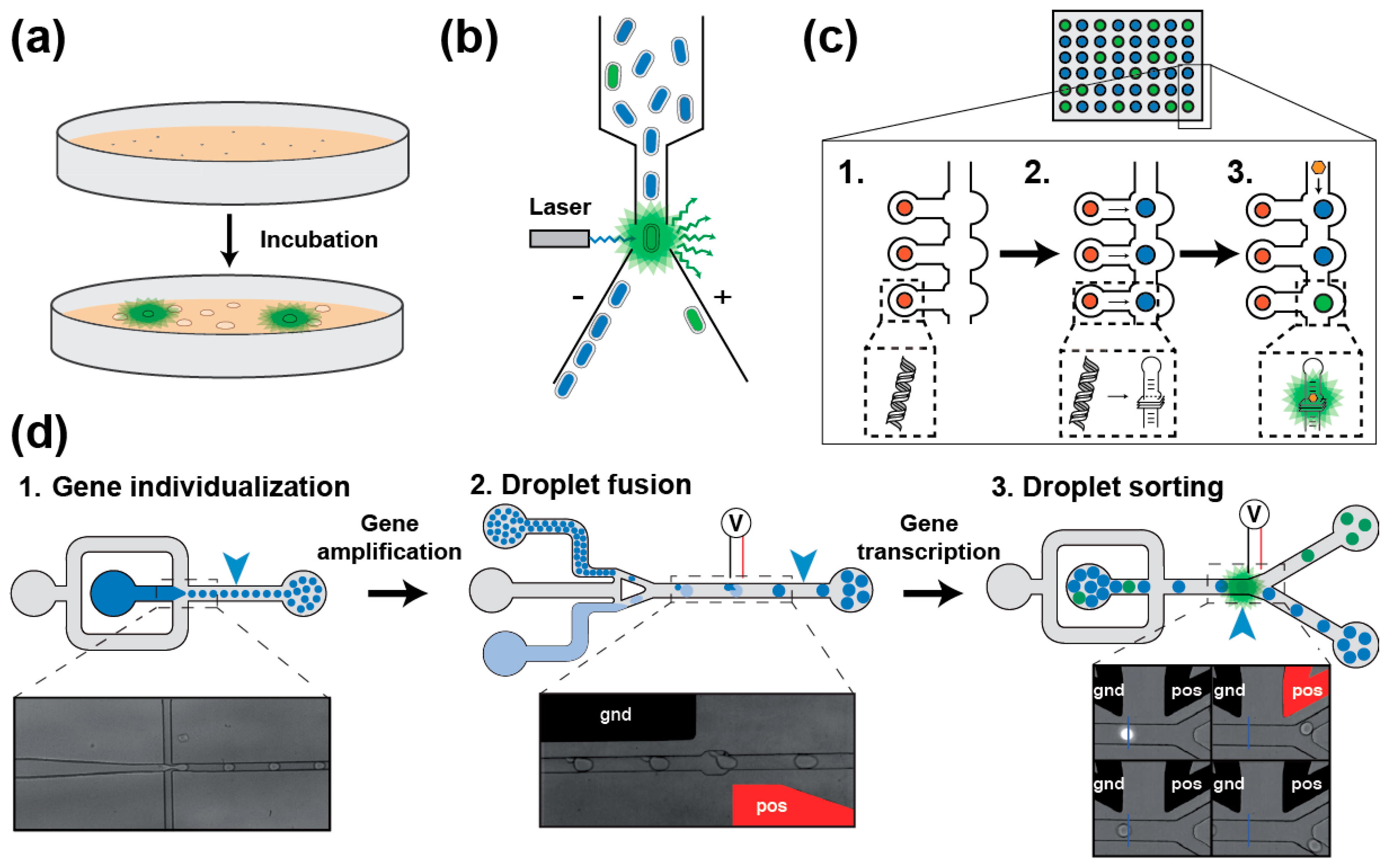
2.2.2. Aptamers Isolation Based on Their Fluorogenic Capacity
2.3. Features of the Best Characterized RNA-Based Fluorogenic Modules
3. Applications of RNA-Based Fluorogenic Modules
3.1. Live-Cell Imaging of Biomolecules
3.1.1. Live-Cell RNA Imaging
3.1.2. Live-Cell Imaging of Metabolites and Proteins
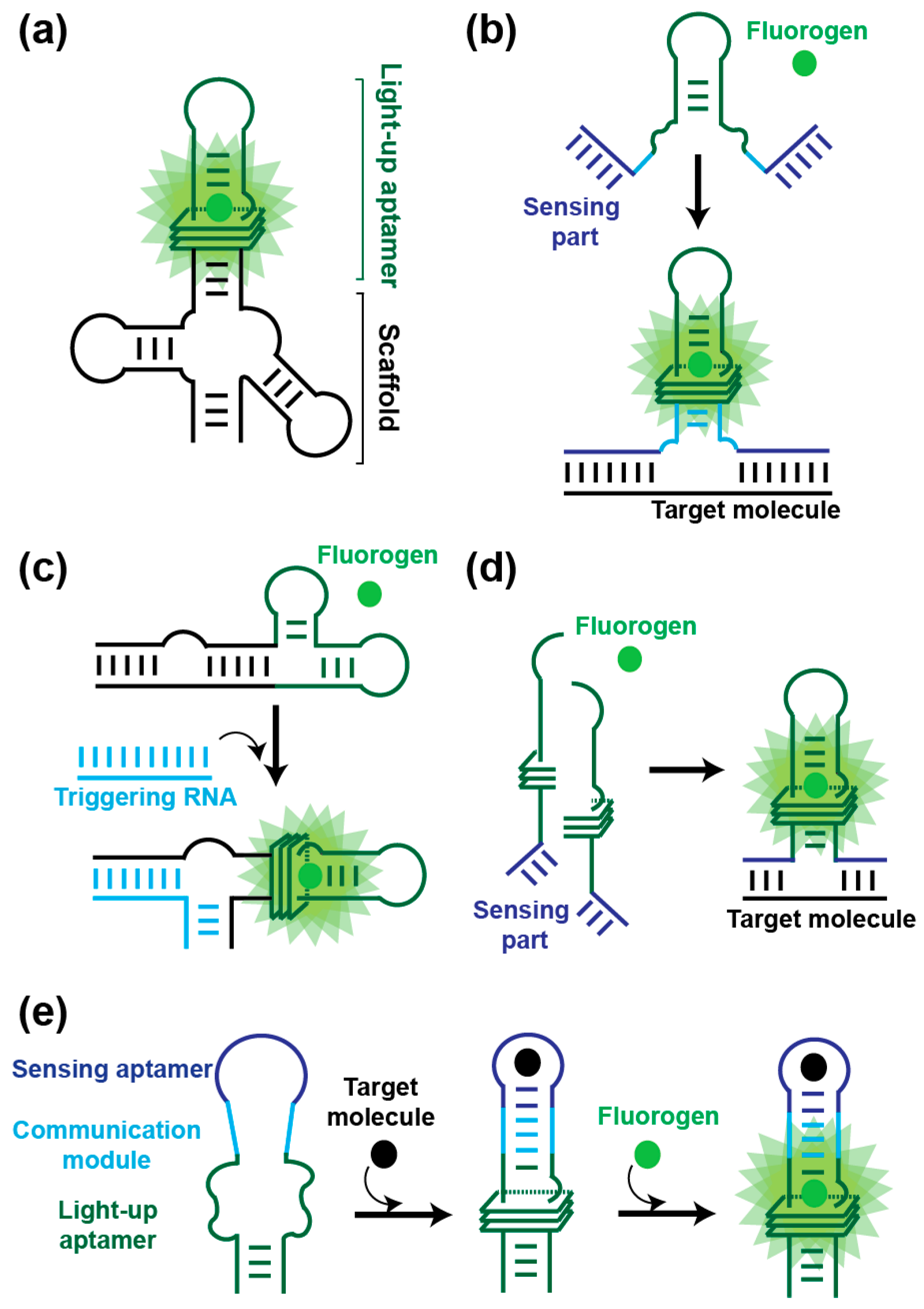
3.2. In Vitro Applications
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| Cy3-BHQ1 | Cyanine 3-Black Hole Quencher 1 |
| DIR | Dimethyl Indole Red |
| DFHBI | 3,5-difluoro-4-hydroxybenzylidene imidazolinone |
| DFHBI-1(2)T | 3,5-difluoro-4-hydroxybenzylidene imidazolinone 1 (2) trifluoroethyl |
| DFHO | 3,5-difluoro-4-hydroxybenzylidene imidazolinone-2-oxime |
| FACS | Fluorescence Activated Cell Sorter |
| GFP | Green Fluorescent Protein |
| MG | Malachite Green |
| OTB | Oxazole Thiazole Blue |
| RG-DN | Rhodamine Green-DiNitroaniline |
| SELEX | Systematic Evolution of Ligands by EXponential enrichment |
| SR-DN | Sulforhodamine-DiNitroaniline |
| TMR-DN | Tertamethyl rhodamine-DiNitroaniline |
| TO-1 (3) | Thiazole Orange 1 (3) |
References
- Schaferling, M. The art of fluorescence imaging with chemical sensors. Angew. Chem. 2012, 51, 3532–3554. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.K.; Todd, M.H.; Rutledge, P.J. Recent advances in macrocyclic fluorescent probes for ion sensing. Molecules 2017, 22, 200. [Google Scholar] [CrossRef] [PubMed]
- Cui, C.; Shu, W.; Li, P. Fluorescence in situ hybridization: Cell-based genetic diagnostic and research applications. Front. Cell Dev. Biol. 2016, 4, 89. [Google Scholar] [CrossRef] [PubMed]
- Shimomura, O.; Johnson, F.H.; Saiga, Y. Extraction, purification and properties of aequorin, a bioluminescent protein from the luminous hydromedusan, Aequorea. J. Cell. Comp. Physiol. 1962, 59, 223–239. [Google Scholar] [CrossRef] [PubMed]
- Cody, C.W.; Prasher, D.C.; Westler, W.M.; Prendergast, F.G.; Ward, W.W. Chemical structure of the hexapeptide chromophore of the Aequorea green-fluorescent protein. Biochemistry 1993, 32, 1212–1218. [Google Scholar] [CrossRef] [PubMed]
- Chalfie, M.; Tu, Y.; Euskirchen, G.; Ward, W.W.; Prasher, D.C. Green fluorescent protein as a marker for gene expression. Science 1994, 263, 802–805. [Google Scholar] [CrossRef] [PubMed]
- Heim, R.; Prasher, D.C.; Tsien, R.Y. Wavelength mutations and posttranslational autoxidation of green fluorescent protein. Proc. Natl. Acad. Sci. USA 1994, 91, 12501–12504. [Google Scholar] [CrossRef] [PubMed]
- Ormo, M.; Cubitt, A.B.; Kallio, K.; Gross, L.A.; Tsien, R.Y.; Remington, S.J. Crystal structure of the Aequorea victoria green fluorescent protein. Science 1996, 273, 1392–1395. [Google Scholar] [CrossRef] [PubMed]
- Chudakov, D.M.; Matz, M.V.; Lukyanov, S.; Lukyanov, K.A. Fluorescent proteins and their applications in imaging living cells and tissues. Physiol. Rev. 2010, 90, 1103–1163. [Google Scholar] [CrossRef] [PubMed]
- Griffin, B.A.; Adams, S.R.; Tsien, R.Y. Specific covalent labeling of recombinant protein molecules inside live cells. Science 1998, 281, 269–272. [Google Scholar] [CrossRef] [PubMed]
- Correa, I.R., Jr. Live-cell reporters for fluorescence imaging. Curr. Opin. Chem. Biol. 2014, 20, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Tebo, A.G.; Gautier, A. Fluorogenic labeling strategies for biological imaging. Int. J. Mol. Sci. 2017, 18, 1473. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, E.; Chartrand, P.; Schaefer, M.; Shenoy, S.M.; Singer, R.H.; Long, R.M. Localization of ASH1 mRNA particles in living yeast. Mol. Cell 1998, 2, 437–445. [Google Scholar] [CrossRef]
- Wu, B.; Chen, J.; Singer, R.H. Background free imaging of single mRNAs in live cells using split fluorescent proteins. Sci. Rep. 2014, 4, 3615. [Google Scholar] [CrossRef] [PubMed]
- Buxbaum, A.R.; Haimovich, G.; Singer, R.H. In the right place at the right time: Visualizing and understanding mRNA localization. Nat. Rev. Mol. Cell Biol. 2015, 16, 95–109. [Google Scholar] [CrossRef] [PubMed]
- Holeman, L.A.; Robinson, S.L.; Szostak, J.W.; Wilson, C. Isolation and characterization of fluorophore-binding RNA aptamers. Fold. Des. 1998, 3, 423–431. [Google Scholar] [CrossRef]
- Shin, I.; Ray, J.; Gupta, V.; Ilgu, M.; Beasley, J.; Bendickson, L.; Mehanovic, S.; Kraus, G.A.; Nilsen-Hamilton, M. Live-cell imaging of pol II promoter activity to monitor gene expression with RNA IMAGEtag reporters. Nucleic Acids Res. 2014, 42, e90. [Google Scholar] [CrossRef] [PubMed]
- Eydeler, K.; Magbanua, E.; Werner, A.; Ziegelmuller, P.; Hahn, U. Fluorophore binding aptamers as a tool for RNA visualization. Biophys. J. 2009, 96, 3703–3707. [Google Scholar] [CrossRef] [PubMed]
- Babendure, J.R.; Adams, S.R.; Tsien, R.Y. Aptamers switch on fluorescence of triphenylmethane dyes. J. Am. Chem. Soc. 2003, 125, 14716–14717. [Google Scholar] [CrossRef] [PubMed]
- Grate, D.; Wilson, C. Laser-mediated, site-specific inactivation of RNA transcripts. Proc. Natl. Acad. Sci. USA 1999, 96, 6131–6136. [Google Scholar] [CrossRef] [PubMed]
- Ouellet, J. RNA fluorescence with light-up aptamers. Front. Chem. 2016, 4, 29. [Google Scholar] [CrossRef] [PubMed]
- Rath, A.K.; Rentmeister, A. Genetically encoded tools for RNA imaging in living cells. Curr. Opin. Biotechnol. 2015, 31, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Pauff, S.; Withers, J.M.; McKean, I.J.; Mackay, S.P.; Burley, G.A. Synthetic biological approaches for RNA labelling and imaging: Design principles and future opportunities. Curr. Opin. Biotechnol. 2017, 48, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Schneider, A.F.; Hackenberger, C.P. Fluorescent labelling in living cells. Curr. Opin. Biotechnol. 2017, 48, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Van Gijtenbeek, L.A.; Kok, J. Illuminating messengers: An update and outlook on RNA visualization in bacteria. Front. Microbiol. 2017, 8, 1161. [Google Scholar] [CrossRef] [PubMed]
- Heim, R.; Cubitt, A.B.; Tsien, R.Y. Improved green fluorescence. Nature 1995, 373, 663–664. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.; Constantin, T.P.; Sloane, K.L.; Waggoner, A.S.; Bruchez, M.P.; Armitage, B.A. Fluoromodules consisting of a promiscuous RNA aptamer and red or blue fluorogenic cyanine dyes: Selection, characterization, and bioimaging. J. Am. Chem. Soc. 2017, 139, 9001–9009. [Google Scholar] [CrossRef] [PubMed]
- Sando, S.; Narita, A.; Hayami, M.; Aoyama, Y. Transcription monitoring using fused RNA with a dye-binding light-up aptamer as a tag: A blue fluorescent RNA. Chem. Commun. 2008, 44, 3858–3860. [Google Scholar] [CrossRef] [PubMed]
- Paige, J.S.; Wu, K.Y.; Jaffrey, S.R. RNA mimics of green fluorescent protein. Science 2011, 333, 642–646. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Strack, R.L.; Svensen, N.; Jaffrey, S.R. Plug-and-play fluorophores extend the spectral properties of Spinach. J. Am. Chem. Soc. 2014, 136, 1198–1201. [Google Scholar] [CrossRef] [PubMed]
- Filonov, G.S.; Moon, J.D.; Svensen, N.; Jaffrey, S.R. Broccoli: Rapid selection of an RNA mimic of green fluorescent protein by fluorescence-based selection and directed evolution. J. Am. Chem. Soc. 2014, 136, 16299–16308. [Google Scholar] [CrossRef] [PubMed]
- Arora, A.; Sunbul, M.; Jaschke, A. Dual-colour imaging of RNAs using quencher- and fluorophore-binding aptamers. Nucleic Acids Res. 2015, 43, e144. [Google Scholar] [CrossRef] [PubMed]
- Dolgosheina, E.V.; Jeng, S.C.; Panchapakesan, S.S.; Cojocaru, R.; Chen, P.S.; Wilson, P.D.; Hawkins, N.; Wiggins, P.A.; Unrau, P.J. RNA Mango aptamer-fluorophore: A bright, high-affinity complex for RNA labeling and tracking. ACS Chem. Biol. 2014, 9, 2412–2420. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Filonov, G.S.; Kim, H.; Hirsch, M.; Li, X.; Moon, J.D.; Jaffrey, S.R. Imaging RNA polymerase III transcription using a photostable RNA-fluorophore complex. Nat. Chem. Biol. 2017, 13, 1187–1194. [Google Scholar] [CrossRef] [PubMed]
- Murata, A.; Sato, S.; Kawazoe, Y.; Uesugi, M. Small-molecule fluorescent probes for specific RNA targets. Chem. Commun. 2011, 47, 4712–4714. [Google Scholar] [CrossRef] [PubMed]
- Constantin, T.P.; Silva, G.L.; Robertson, K.L.; Hamilton, T.P.; Fague, K.; Waggoner, A.S.; Armitage, B.A. Synthesis of new fluorogenic cyanine dyes and incorporation into RNA fluoromodules. Org. Lett. 2008, 10, 1561–1564. [Google Scholar] [CrossRef] [PubMed]
- Dolgosheina, E.V.; Unrau, P.J. Fluorophore-binding RNA aptamers and their applications. Wiley Interdiscip. Rev. RNA 2016, 7, 843–851. [Google Scholar] [CrossRef] [PubMed]
- Klymchenko, A.S. Solvatochromic and fluorogenic dyes as environment-sensitive probes: Design and biological applications. Acc. Chem. Res. 2017, 50, 366–375. [Google Scholar] [CrossRef] [PubMed]
- Lo̊ber, G. The fluorescence of dye-nucleic acid complexes. J. Lumin. 1981, 22, 221–265. [Google Scholar] [CrossRef]
- Olmsted, J., 3rd; Kearns, D.R. Mechanism of ethidium bromide fluorescence enhancement on binding to nucleic acids. Biochemistry 1977, 16, 3647–3654. [Google Scholar] [CrossRef] [PubMed]
- Jin, R.; Breslauer, K.J. Characterization of the minor groove environment in a drug-DNA complex: Bisbenzimide bound to the poly[d(AT)].Poly[d(AT)]duplex. Proc. Natl. Acad. Sci. USA 1988, 85, 8939–8942. [Google Scholar] [CrossRef] [PubMed]
- Sando, S.; Narita, A.; Aoyama, Y. Light-up hoechst-DNA aptamer pair: Generation of an aptamer-selective fluorophore from a conventional DNA-staining dye. Chembiochem 2007, 8, 1795–1803. [Google Scholar] [CrossRef] [PubMed]
- Kraus, G.A.; Jeon, I.; Nilsen-Hamilton, M.; Awad, A.M.; Banerjee, J.; Parvin, B. Fluorinated analogs of malachite green: Synthesis and toxicity. Molecules 2008, 13, 986–994. [Google Scholar] [CrossRef] [PubMed]
- Lux, J.; Pena, E.J.; Bolze, F.; Heinlein, M.; Nicoud, J.F. Malachite green derivatives for two-photon RNA detection. Chembiochem 2012, 13, 1206–1213. [Google Scholar] [CrossRef] [PubMed]
- Ilgu, M.; Ray, J.; Bendickson, L.; Wang, T.; Geraskin, I.M.; Kraus, G.A.; Nilsen-Hamilton, M. Light-up and FRET aptamer reporters; evaluating their applications for imaging transcription in eukaryotic cells. Methods 2016, 98, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Saurabh, S.; Perez, A.M.; Comerci, C.J.; Shapiro, L.; Moerner, W.E. Super-resolution imaging of live bacteria cells using a genetically directed, highly photostable fluoromodule. J. Am. Chem. Soc. 2016, 138, 10398–10401. [Google Scholar] [CrossRef] [PubMed]
- Silva, G.L.; Ediz, V.; Yaron, D.; Armitage, B.A. Experimental and computational investigation of unsymmetrical cyanine dyes: Understanding torsionally responsive fluorogenic dyes. J. Am. Chem. Soc. 2007, 129, 5710–5718. [Google Scholar] [CrossRef] [PubMed]
- Armitage, B.A. Cyanine dye–nucleic acid interactions. In Heterocyclic Polymethine Dyes: Synthesis, Properties and Applications; Strekowski, L., Ed.; Springer: Berlin/Heidelberg, Germany, 2008; pp. 11–29. [Google Scholar]
- Lee, L.G.; Chen, C.H.; Chiu, L.A. Thiazole orange: A new dye for reticulocyte analysis. Cytometry 1986, 7, 508–517. [Google Scholar] [CrossRef] [PubMed]
- Nygren, J.; Svanvik, N.; Kubista, M. The interactions between the fluorescent dye thiazole orange and DNA. Biopolymers 1998, 46, 39–51. [Google Scholar] [CrossRef]
- Walker, C.L.; Lukyanov, K.A.; Yampolsky, I.V.; Mishin, A.S.; Bommarius, A.S.; Duraj-Thatte, A.M.; Azizi, B.; Tolbert, L.M.; Solntsev, K.M. Fluorescence imaging using synthetic GFP chromophores. Curr. Opin. Chem. Biol. 2015, 27, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Feng, G.; Luo, C.; Yi, H.; Yuan, L.; Lin, B.; Luo, X.; Hu, X.; Wang, H.; Lei, C.; Nie, Z.; et al. DNA mimics of red fluorescent proteins (RFP) based on G-quadruplex-confined synthetic RFP chromophores. Nucleic Acids Res. 2017, 45, 10380–10392. [Google Scholar] [CrossRef] [PubMed]
- Sparano, B.A.; Koide, K. A strategy for the development of small-molecule-based sensors that strongly fluoresce when bound to a specific RNA. J. Am. Chem. Soc. 2005, 127, 14954–14955. [Google Scholar] [CrossRef] [PubMed]
- Sparano, B.A.; Koide, K. Fluorescent sensors for specific RNA: A general paradigm using chemistry and combinatorial biology. J. Am. Chem. Soc. 2007, 129, 4785–4794. [Google Scholar] [CrossRef] [PubMed]
- Sunbul, M.; Jaschke, A. Contact-mediated quenching for RNA imaging in bacteria with a fluorophore-binding aptamer. Angew. Chem. 2013, 52, 13401–13404. [Google Scholar] [CrossRef] [PubMed]
- Ellington, A.D.; Szostak, J.W. In vitro selection of RNA molecules that bind specific ligands. Nature 1990, 346, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Tuerk, C.; Gold, L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 1990, 249, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Stoltenburg, R.; Reinemann, C.; Strehlitz, B. Selex—A (r)evolutionary method to generate high-affinity nucleic acid ligands. Biomol. Eng. 2007, 24, 381–403. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.H.; Szostak, J.W. Isolation of high-affinity GTP aptamers from partially structured RNA libraries. Proc. Natl. Acad. Sci. USA 2002, 99, 11616–11621. [Google Scholar] [CrossRef] [PubMed]
- Trachman, R.J., 3rd; Demeshkina, N.A.; Lau, M.W.L.; Panchapakesan, S.S.S.; Jeng, S.C.Y.; Unrau, P.J.; Ferre-D’Amare, A.R. Structural basis for high-affinity fluorophore binding and activation by RNA Mango. Nat. Chem. Biol. 2017, 13, 807–813. [Google Scholar] [CrossRef] [PubMed]
- Strack, R.L.; Disney, M.D.; Jaffrey, S.R. A superfolding Spinach2 reveals the dynamic nature of trinucleotide repeat-containing RNA. Nat. Methods 2013, 10, 1219–1224. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Lee, K.H.; Jeon, J.; Dragulescu-Andrasi, A.; Xiao, F.; Rao, J. Combining SELEX screening and rational design to develop light-up fluorophore-RNA aptamer pairs for RNA tagging. ACS Chem. Biol. 2010, 5, 1065–1074. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.; Huang, X.; Wu, L.; Chen, G.; Dong, J.; Cui, X.; Tang, Z. Selection of intracellularly functional RNA mimics of green fluorescent protein using fluorescence-activated cell sorting. J. Mol. Evol. 2015, 81, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Ketterer, S.; Fuchs, D.; Weber, W.; Meier, M. Systematic reconstruction of binding and stability landscapes of the fluorogenic aptamer Spinach. Nucleic Acids Res. 2015, 43, 9564–9572. [Google Scholar] [CrossRef] [PubMed]
- Autour, A.; Westhof, E.; Ryckelynck, M. iSpinach: A fluorogenic RNA aptamer optimized for in vitro applications. Nucleic Acids Res. 2016, 44, 2491–2500. [Google Scholar] [CrossRef] [PubMed]
- Autour, A.; Ryckelynck, M. Ultrahigh-throughput improvement and discovery of enzymes using droplet-based microfluidic screening. Micromachines 2017, 8, 128. [Google Scholar] [CrossRef]
- Ryckelynck, M.; Baudrey, S.; Rick, C.; Marin, A.; Coldren, F.; Westhof, E.; Griffiths, A.D. Using droplet-based microfluidics to improve the catalytic properties of RNA under multiple-turnover conditions. RNA 2015, 21, 458–469. [Google Scholar] [CrossRef] [PubMed]
- Trachman, R.J., 3rd; Truong, L.; Ferre-D’Amare, A.R. Structural principles of fluorescent RNA aptamers. Trends Pharmacol.Sci. 2017, 38, 928–939. [Google Scholar] [CrossRef] [PubMed]
- Baugh, C.; Grate, D.; Wilson, C. 2.8 a crystal structure of the malachite green aptamer. J. Mol. Biol. 2000, 301, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Suslov, N.B.; Li, N.S.; Shelke, S.A.; Evans, M.E.; Koldobskaya, Y.; Rice, P.A.; Piccirilli, J.A. A G-quadruplex-containing RNA activates fluorescence in a GFP-like fluorophore. Nat. Chem. Biol. 2014, 10, 686–691. [Google Scholar] [CrossRef] [PubMed]
- Warner, K.D.; Chen, M.C.; Song, W.; Strack, R.L.; Thorn, A.; Jaffrey, S.R.; Ferre-D’Amare, A.R. Structural basis for activity of highly efficient RNA mimics of green fluorescent protein. Nat. Struct. Mol. Biol. 2014, 21, 658–663. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Millan, P.; Autour, A.; Ennifar, E.; Westhof, E.; Ryckelynck, M. Crystal structure and fluorescence properties of the iSpinach aptamer in complex with dfhbi. RNA 2017, 23, 1788–1795. [Google Scholar] [CrossRef] [PubMed]
- Warner, K.D.; Sjekloca, L.; Song, W.; Filonov, G.S.; Jaffrey, S.R.; Ferre-D’Amare, A.R. A homodimer interface without base pairs in an RNA mimic of red fluorescent protein. Nat. Chem. Biol. 2017, 13, 1195–1201. [Google Scholar] [CrossRef] [PubMed]
- Jeng, S.C.; Chan, H.H.; Booy, E.P.; McKenna, S.A.; Unrau, P.J. Fluorophore ligand binding and complex stabilization of the RNA Mango and RNA Spinach aptamers. RNA 2016, 22, 1884–1892. [Google Scholar] [CrossRef] [PubMed]
- Han, K.Y.; Leslie, B.J.; Fei, J.; Zhang, J.; Ha, T. Understanding the photophysics of the Spinach-DFHBI RNA aptamer-fluorogen complex to improve live-cell RNA imaging. J. Am. Chem. Soc. 2013, 135, 19033–19038. [Google Scholar] [CrossRef] [PubMed]
- Ageely, E.A.; Kartje, Z.J.; Rohilla, K.J.; Barkau, C.L.; Gagnon, K.T. Quadruplex-flanking stem structures modulate the stability and metal ion preferences of RNA mimics of GFP. ACS Chem. Biol. 2016, 11, 2398–2406. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Watanabe, M.; Katsuda, Y.; Murata, A.; Wang, D.O.; Uesugi, M. Live-cell imaging of endogenous mRNAs with a small molecule. Angew. Chem. 2015, 54, 1855–1858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ong, W.Q.; Citron, Y.R.; Sekine, S.; Huang, B. Live cell imaging of endogenous mRNA using RNA-based fluorescence “turn-on” probe. ACS Chem. Biol. 2017, 12, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Bhadra, S.; Ellington, A.D. A Spinach molecular beacon triggered by strand displacement. RNA 2014, 20, 1183–1194. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Doyle, F.; Wurz, Z.E.; Tenenbaum, S.A.; Hammond, R.K.; Caplan, J.L.; Meyers, B.C. Fastmir: An RNA-based sensor for in vitro quantification and live-cell localization of small RNAs. Nucleic Acids Res. 2017, 45, e130. [Google Scholar] [CrossRef] [PubMed]
- Ying, Z.M.; Wu, Z.; Tu, B.; Tan, W.; Jiang, J.H. Genetically encoded fluorescent RNA sensor for ratiometric imaging of microRNA in living tumor cells. J. Am. Chem. Soc. 2017, 139, 9779–9782. [Google Scholar] [CrossRef] [PubMed]
- Kolpashchikov, D.M. Binary malachite green aptamer for fluorescent detection of nucleic acids. J. Am. Chem. Soc. 2005, 127, 12442–12443. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, N.; Kolpashchikov, D.M. A universal split Spinach aptamer (USSA) for nucleic acid analysis and DNA computation. Chem. Commun. 2017, 53, 4977–4980. [Google Scholar] [CrossRef] [PubMed]
- Stojanovic, M.N.; Kolpashchikov, D.M. Modular aptameric sensors. J. Am. Chem. Soc. 2004, 126, 9266–9270. [Google Scholar] [CrossRef] [PubMed]
- Kellenberger, C.A.; Wilson, S.C.; Sales-Lee, J.; Hammond, M.C. RNA-based fluorescent biosensors for live cell imaging of second messengers cyclic di-GMP and cyclic AMP-GMP. J. Am. Chem. Soc. 2013, 135, 4906–4909. [Google Scholar] [CrossRef] [PubMed]
- Ponchon, L.; Dardel, F. Recombinant RNA technology: The tRNA scaffold. Nat. Methods 2007, 4, 571–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filonov, G.S.; Kam, C.W.; Song, W.; Jaffrey, S.R. In-gel imaging of RNA processing using Broccoli reveals optimal aptamer expression strategies. Chem. Biol. 2015, 22, 649–660. [Google Scholar] [CrossRef] [PubMed]
- Masuda, I.; Igarashi, T.; Sakaguchi, R.; Nitharwal, R.G.; Takase, R.; Han, K.Y.; Leslie, B.J.; Liu, C.; Gamper, H.; Ha, T.; et al. A genetically encoded fluorescent tRNA is active in live-cell protein synthesis. Nucleic Acids Res. 2017, 45, 4081–4093. [Google Scholar] [CrossRef] [PubMed]
- Okuda, M.; Fourmy, D.; Yoshizawa, S. Use of baby Spinach and Broccoli for imaging of structured cellular RNAs. Nucleic Acids Res. 2017, 45, 1404–1415. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Filonov, G.S.; Noto, J.J.; Schmidt, C.A.; Hatkevich, T.L.; Wen, Y.; Jaffrey, S.R.; Matera, A.G. Metazoan tRNA introns generate stable circular RNAs in vivo. RNA 2015, 21, 1554–1565. [Google Scholar] [CrossRef] [PubMed]
- Pothoulakis, G.; Ceroni, F.; Reeve, B.; Ellis, T. The Spinach RNA aptamer as a characterization tool for synthetic biology. ACS Synth. Biol. 2013, 3, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Ellefson, J.W.; Meyer, A.J.; Hughes, R.A.; Cannon, J.R.; Brodbelt, J.S.; Ellington, A.D. Directed evolution of genetic parts and circuits by compartmentalized partnered replication. Nat. Biotechnol. 2014, 32, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Fei, J.; Leslie, B.J.; Han, K.Y.; Kuhlman, T.E.; Ha, T. Tandem Spinach array for mRNA imaging in living bacterial cells. Sci. Rep. 2015, 5, 17295. [Google Scholar] [CrossRef] [PubMed]
- Guet, D.; Burns, L.T.; Maji, S.; Boulanger, J.; Hersen, P.; Wente, S.R.; Salamero, J.; Dargemont, C. Combining Spinach-tagged RNA and gene localization to image gene expression in live yeast. Nat. Commun. 2015, 6, 8882. [Google Scholar] [CrossRef] [PubMed]
- Tsvetkova, I.B.; Yi, G.; Yi, Y.; Kao, C.C.; Dragnea, B.G. Segmented GFP-like aptamer probes for functional imaging of viral genome trafficking. Virus Res. 2015, 210, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Burch, B.D.; Garrido, C.; Margolis, D.M. Detection of human immunodeficiency virus RNAs in living cells using Spinach RNA aptamers. Virus Res. 2017, 228, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Nilaratanakul, V.; Hauer, D.A.; Griffin, D.E. Development and characterization of sindbis virus with encoded fluorescent RNA aptamer Spinach2 for imaging of replication and immune-mediated changes in intracellular viral RNA. J. Gen. Virol. 2017, 98, 992–1003. [Google Scholar] [CrossRef] [PubMed]
- Guzman-Zapata, D.; Dominguez-Anaya, Y.; Macedo-Osorio, K.S.; Tovar-Aguilar, A.; Castrejon-Flores, J.L.; Duran-Figueroa, N.V.; Badillo-Corona, J.A. mRNA imaging in the chloroplast of chlamydomonas reinhardtii using the light-up aptamer Spinach. J. Biotechnol. 2017, 251, 186–188. [Google Scholar] [CrossRef] [PubMed]
- Aw, S.S.; Tang, M.X.; Teo, Y.N.; Cohen, S.M. A conformation-induced fluorescence method for microRNA detection. Nucleic Acids Res. 2016, 44, e92. [Google Scholar] [CrossRef] [PubMed]
- Alam, K.K.; Tawiah, K.D.; Lichte, M.F.; Porciani, D.; Burke, D.H. A fluorescent split aptamer for visualizing RNA-RNA assembly in vivo. ACS Synth. Biol. 2017, 6, 1710–1721. [Google Scholar] [CrossRef] [PubMed]
- Breaker, R.R. Engineered allosteric ribozymes as biosensor components. Curr. Opin. Biotechnol. 2002, 13, 31–39. [Google Scholar] [CrossRef]
- Nakayama, S.; Luo, Y.; Zhou, J.; Dayie, T.K.; Sintim, H.O. Nanomolar fluorescent detection of c-di-GMP using a modular aptamer strategy. Chem. Commun. 2012, 48, 9059–9061. [Google Scholar] [CrossRef] [PubMed]
- Inuzuka, S.; Matsumura, S.; Ikawa, Y. Optimization of RNA-based c-di-GMP fluorescent sensors through tuning their structural modules. J. Biosci. Bioeng. 2016, 122, 183–187. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.C.; Wilson, S.C.; Hammond, M.C. Next-generation RNA-based fluorescent biosensors enable anaerobic detection of cyclic di-GMP. Nucleic Acids Res. 2016, 44, e139. [Google Scholar] [CrossRef] [PubMed]
- Kellenberger, C.A.; Chen, C.; Whiteley, A.T.; Portnoy, D.A.; Hammond, M.C. RNA-based fluorescent biosensors for live cell imaging of second messenger cyclic di-AMP. J. Am. Chem. Soc. 2015, 137, 6432–6435. [Google Scholar] [CrossRef] [PubMed]
- Kellenberger, C.A.; Wilson, S.C.; Hickey, S.F.; Gonzalez, T.L.; Su, Y.; Hallberg, Z.F.; Brewer, T.F.; Iavarone, A.T.; Carlson, H.K.; Hsieh, Y.F.; et al. Gemm-I riboswitches from geobacter sense the bacterial second messenger cyclic AMP-GMP. Proc. Natl. Acad. Sci. USA 2015, 112, 5383–5388. [Google Scholar] [CrossRef] [PubMed]
- Hallberg, Z.F.; Wang, X.C.; Wright, T.A.; Nan, B.; Ad, O.; Yeo, J.; Hammond, M.C. Hybrid promiscuous (hypr) ggdef enzymes produce cyclic AMP-GMP (3′, 3′-cgamp). Proc. Natl. Acad. Sci. USA 2016, 113, 1790–1795. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Zaveri, A.; Visweswariah, S.S.; Krishnan, Y. A fluorescent nucleic acid nanodevice quantitatively images elevated cyclic adenosine monophosphate in membrane-bound compartments. Small 2014, 10, 4276–4280. [Google Scholar] [CrossRef] [PubMed]
- Paige, J.S.; Nguyen-Duc, T.; Song, W.; Jaffrey, S.R. Fluorescence imaging of cellular metabolites with RNA. Science 2012, 335, 1194. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Hickey, S.F.; Keyser, S.G.; Hammond, M.C. In vitro and in vivo enzyme activity screening via RNA-based fluorescent biosensors for S-adenosyl-l-homocysteine (SAH). J. Am. Chem. Soc. 2016, 138, 7040–7047. [Google Scholar] [CrossRef] [PubMed]
- You, M.; Litke, J.L.; Jaffrey, S.R. Imaging metabolite dynamics in living cells using a Spinach-based riboswitch. Proc. Natl. Acad. Sci. USA 2015, 112, E2756–E2765. [Google Scholar] [CrossRef] [PubMed]
- Porter, E.B.; Polaski, J.T.; Morck, M.M.; Batey, R.T. Recurrent RNA motifs as scaffolds for genetically encodable small-molecule biosensors. Nat. Chem. Biol. 2017, 13, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Strack, R.L.; Jaffrey, S.R. Imaging bacterial protein expression using genetically encoded RNA sensors. Nat. Methods 2013, 10, 873–875. [Google Scholar] [CrossRef] [PubMed]
- Ketterer, S.; Gladis, L.; Kozica, A.; Meier, M. Engineering and characterization of fluorogenic glycine riboswitches. Nucleic Acids Res. 2016, 44, 5983–5992. [Google Scholar] [CrossRef] [PubMed]
- Hofer, K.; Langejurgen, L.V.; Jaschke, A. Universal aptamer-based real-time monitoring of enzymatic RNA synthesis. J. Am. Chem. Soc. 2013, 135, 13692–13694. [Google Scholar] [CrossRef] [PubMed]
- Van Nies, P.; Nourian, Z.; Kok, M.; van Wijk, R.; Moeskops, J.; Westerlaken, I.; Poolman, J.M.; Eelkema, R.; van Esch, J.H.; Kuruma, Y.; et al. Unbiased tracking of the progression of mRNA and protein synthesis in bulk and in liposome-confined reactions. Chembiochem 2013, 14, 1963–1966. [Google Scholar] [CrossRef] [PubMed]
- Chizzolini, F.; Forlin, M.; Cecchi, D.; Mansy, S.S. Gene position more strongly influences cell-free protein expression from operons than T7 transcriptional promoter strength. ACS Synth. Biol. 2014, 3, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Chizzolini, F.; Forlin, M.; Yeh Martin, N.; Berloffa, G.; Cecchi, D.; Mansy, S.S. Cell-free translation is more variable than transcription. ACS Synth. Biol. 2017, 6, 638–647. [Google Scholar] [CrossRef] [PubMed]
- Svensen, N.; Jaffrey, S.R. Fluorescent RNA aptamers as a tool to study RNA-modifying enzymes. Cell Chem. Biol. 2016, 23, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Bose, D.; Su, Y.; Marcus, A.; Raulet, D.H.; Hammond, M.C. An RNA-based fluorescent biosensor for high-throughput analysis of the cGAS-cGAMP-sting pathway. Cell Chem. Biol. 2016, 23, 1539–1549. [Google Scholar] [CrossRef] [PubMed]
- Abatemarco, J.; Sarhan, M.F.; Wagner, J.M.; Lin, J.L.; Liu, L.; Hassouneh, W.; Yuan, S.F.; Alper, H.S.; Abate, A.R. RNA-aptamers-in-droplets (RAPID) high-throughput screening for secretory phenotypes. Nat. Commun. 2017, 8, 332. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Ellington, A.D.; Chen, X. Rational, modular adaptation of enzyme-free DNA circuits to multiple detection methods. Nucleic Acids Res. 2011, 39, e110. [Google Scholar] [CrossRef] [PubMed]
- Bhadra, S.; Ellington, A.D. Design and application of cotranscriptional non-enzymatic RNA circuits and signal transducers. Nucleic Acids Res. 2014, 42, e58. [Google Scholar] [CrossRef] [PubMed]
- Rogers, T.A.; Andrews, G.E.; Jaeger, L.; Grabow, W.W. Fluorescent monitoring of RNA assembly and processing using the split-Spinach aptamer. ACS Synth. Biol. 2015, 4, 162–166. [Google Scholar] [CrossRef] [PubMed]
- Akter, F.; Yokobayashi, Y. RNA signal amplifier circuit with integrated fluorescence output. ACS Synth. Biol. 2015, 4, 655–658. [Google Scholar] [CrossRef] [PubMed]
- Auslander, S.; Fuchs, D.; Hurlemann, S.; Auslander, D.; Fussenegger, M. Engineering a ribozyme cleavage-induced split fluorescent aptamer complementation assay. Nucleic Acids Res. 2016, 44, e94. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, A.; Tanaka, T.; Furuta, H.; Matsumura, S.; Ikawa, Y. Use of a fluorescent aptamer RNA as an exonic sequence to analyze self-splicing ability of agroup I intron from structured RNAs. Biology 2016, 5, 43. [Google Scholar] [CrossRef] [PubMed]
- Afonin, K.A.; Danilov, E.O.; Novikova, I.V.; Leontis, N.B. TokenRNA: A new type of sequence-specific, label-free fluorescent biosensor for folded RNA molecules. Chembiochem 2008, 9, 1902–1905. [Google Scholar] [CrossRef] [PubMed]
- Shu, D.; Shu, Y.; Haque, F.; Abdelmawla, S.; Guo, P. Thermodynamically stable RNA three-way junction for constructing multifunctional nanoparticles for delivery of therapeutics. Nat. Nanotechnol. 2011, 6, 658–667. [Google Scholar] [CrossRef] [PubMed]
- Reif, R.; Haque, F.; Guo, P. Fluorogenic RNA nanoparticles for monitoring RNA folding and degradation in real time in living cells. Nucleic Acid Ther. 2012, 22, 428–437. [Google Scholar] [PubMed]
- Shu, D.; Khisamutdinov, E.F.; Zhang, L.; Guo, P. Programmable folding of fusion RNA in vivo and in vitro driven by pRNA 3WJ motif of phi29 DNA packaging motor. Nucleic Acids Res. 2014, 42, e10. [Google Scholar] [CrossRef] [PubMed]
- Panchapakesan, S.S.S.; Ferguson, M.L.; Hayden, E.J.; Chen, X.; Hoskins, A.A.; Unrau, P.J. Ribonucleoprotein purification and characterization using RNA Mango. RNA 2017, 23, 1592–1599. [Google Scholar] [CrossRef] [PubMed]
- You, M.; Jaffrey, S.R. Structure and mechanism of RNA mimics of green fluorescent protein. Annu. Rev. Biophys. 2015, 44, 187–206. [Google Scholar] [CrossRef] [PubMed]
- DasGupta, S.; Shelke, S.A.; Li, N.S.; Piccirilli, J.A. Spinach RNA aptamer detects lead(II) with high selectivity. Chem. Commun. 2015, 51, 9034–9037. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fluorogen | Light-Up Aptamer | KD (nM) | Ex./Em. (nm) | ε 1 (M−1/cm) | Φcomplex 2 | Brightness 3 | Relative Brightness 4 | Ref. |
|---|---|---|---|---|---|---|---|---|
| GFP | / | / | 395/508 | 21,000 | 0.770 | 16.20 | 0.60 | [26] |
| eGFP | / | / | 490/508 | 39,200 | 0.680 | 26.60 | 1.00 | [26] |
| OTB | DiR2s-Apt | 662 | 380/421 | 73,000 | 0.510 | 37.23 | 1.40 | [27] |
| Hoescht | Apt II-mini3-4 c | 35 | 345/470 | n.a. | 0.260 | n.a. | n.a. | [28] |
| DFHBI | Spinach | 540 | 469/501 | 24,300 | 0.720 | 17.50 | 0.65 | [29] |
| DFHBI-1T | Spinach2 | 560 | 482/505 | 31,000 | 0.940 | 29.10 | 1.10 | [30] |
| DFHBI-1T | Broccoli | 360 | 472/507 | 29,600 | 0.940 | 27.80 | 1.04 | [31] |
| DFHBI-2T | Spinach2 | 1300 | 500/523 | 29,000 | 0.120 | 3.48 | 0.10 | [30] |
| RG-DN | DNB | 4480 | 507/534 | 37,350 | 0.320 | 11.90 | 0.44 | [32] |
| TO-1 | Mango | 3 | 510/535 | 77,500 | 0.140 | 10.85 | 0.40 | [33] |
| DFHO | Corn | 70 | 505/545 | 29,000 | 0.250 | 7.25 | 0.27 | [34] |
| CY3-BHQ1 | BHQ apt (A1) | n.a. | 520/565 | n.a. | n.a. | n.a. | n.a. | [35] |
| DFHO | Red-Broccoli | 206 | 518/582 | 35,000 | 0.340 | 11.90 | 0.44 | [34] |
| TMR-DN | DNB | 350 | 555/582 | 47,150 | 0.900 | 42.43 | 1.60 | [32] |
| SR-DN | DNB | 800 | 572/591 | 50,250 | 0.980 | 49.24 | 1.80 | [32] |
| DIR | DIR apt | 86 | 600/646 | 134,000 | 0.260 | 34.80 | 1.30 | [36] |
| Mal. Green | MG aptamer | 117 | 630/650 | 150,000 | 0.187 | 28.00 | 1.05 | [19] |
| DIR-pro | DIR2s-Apt | 252 | 600/658 | 164,000 | 0.330 | 54.12 | 2.00 | [27] |
| TO-3 | Mango | 6–8 | 637/658 | 9300 | n.a. | n.a. | n.a. | [33] |
| Patent Blue | SRB apt | 23 | n.a./665 | n.a. | 0.034 | n.a. | n.a. | [19] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bouhedda, F.; Autour, A.; Ryckelynck, M. Light-Up RNA Aptamers and Their Cognate Fluorogens: From Their Development to Their Applications. Int. J. Mol. Sci. 2018, 19, 44. https://doi.org/10.3390/ijms19010044
Bouhedda F, Autour A, Ryckelynck M. Light-Up RNA Aptamers and Their Cognate Fluorogens: From Their Development to Their Applications. International Journal of Molecular Sciences. 2018; 19(1):44. https://doi.org/10.3390/ijms19010044
Chicago/Turabian StyleBouhedda, Farah, Alexis Autour, and Michael Ryckelynck. 2018. "Light-Up RNA Aptamers and Their Cognate Fluorogens: From Their Development to Their Applications" International Journal of Molecular Sciences 19, no. 1: 44. https://doi.org/10.3390/ijms19010044
APA StyleBouhedda, F., Autour, A., & Ryckelynck, M. (2018). Light-Up RNA Aptamers and Their Cognate Fluorogens: From Their Development to Their Applications. International Journal of Molecular Sciences, 19(1), 44. https://doi.org/10.3390/ijms19010044