Long Noncoding Competing Endogenous RNA Networks in Age-Associated Cardiovascular Diseases




Abstract
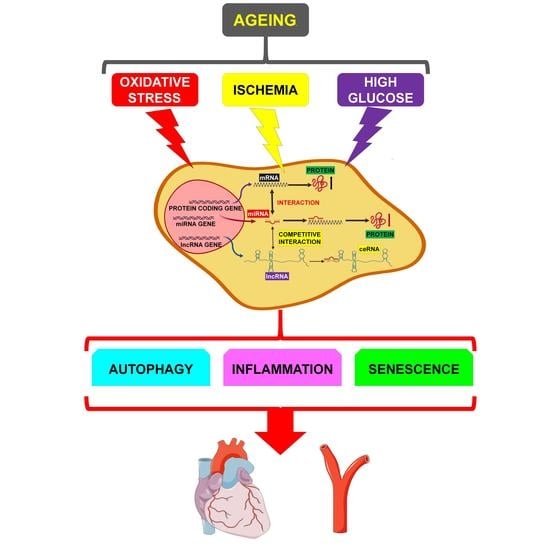
:
1. Introduction
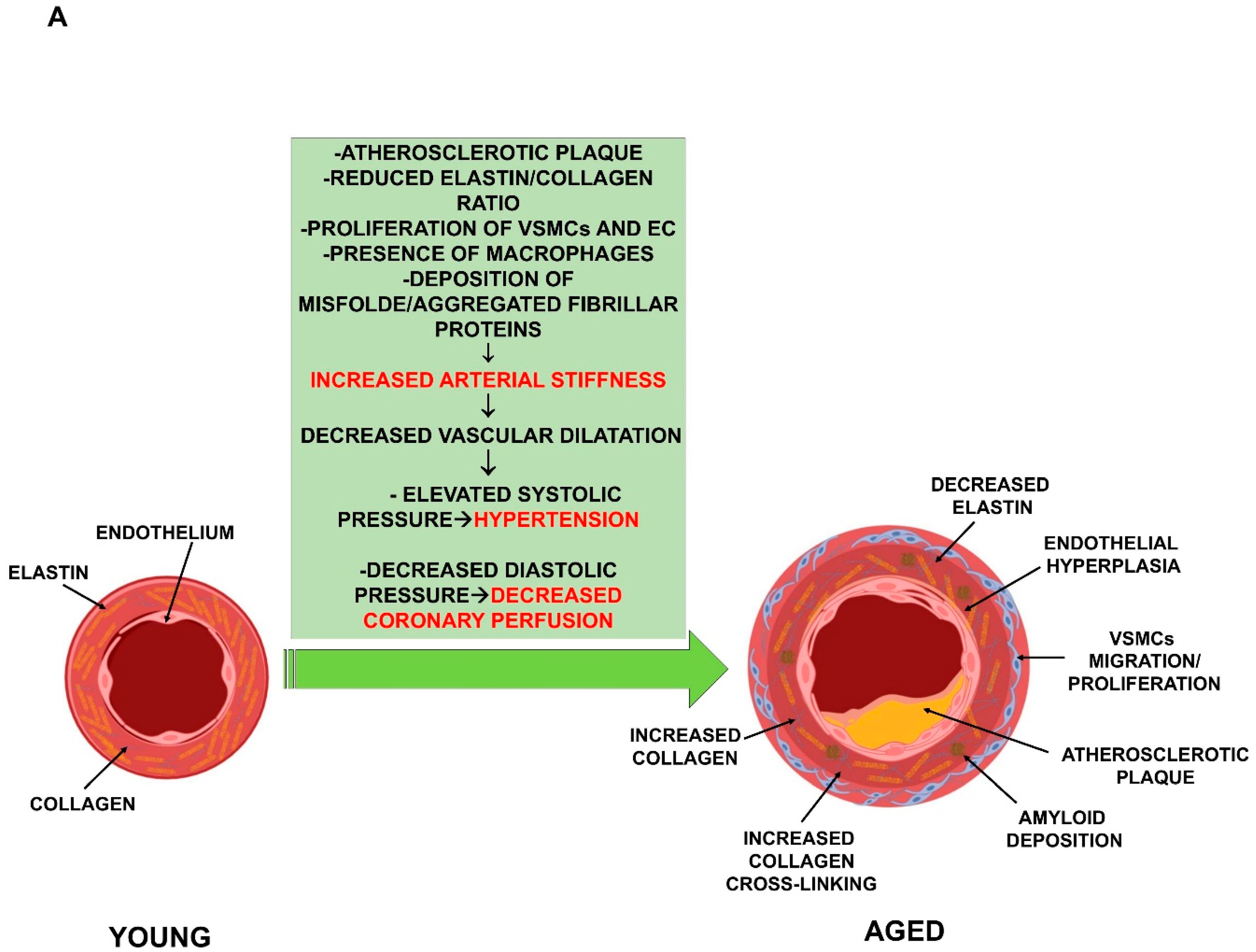
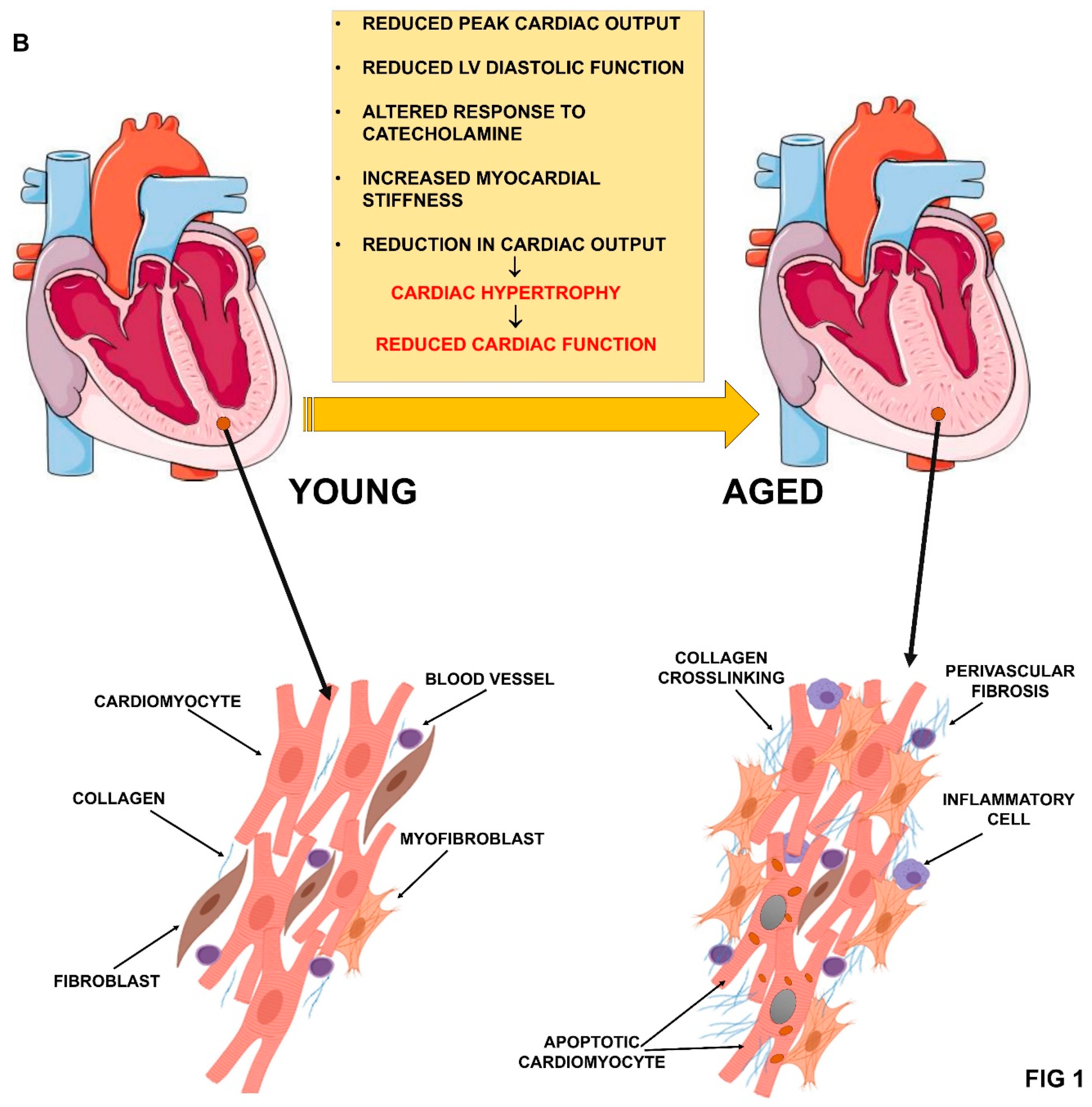
2. Aging and Cardiovascular System Deterioration
2.1. Vascular Functional Impairment
2.2. Cardiac Function Impairment
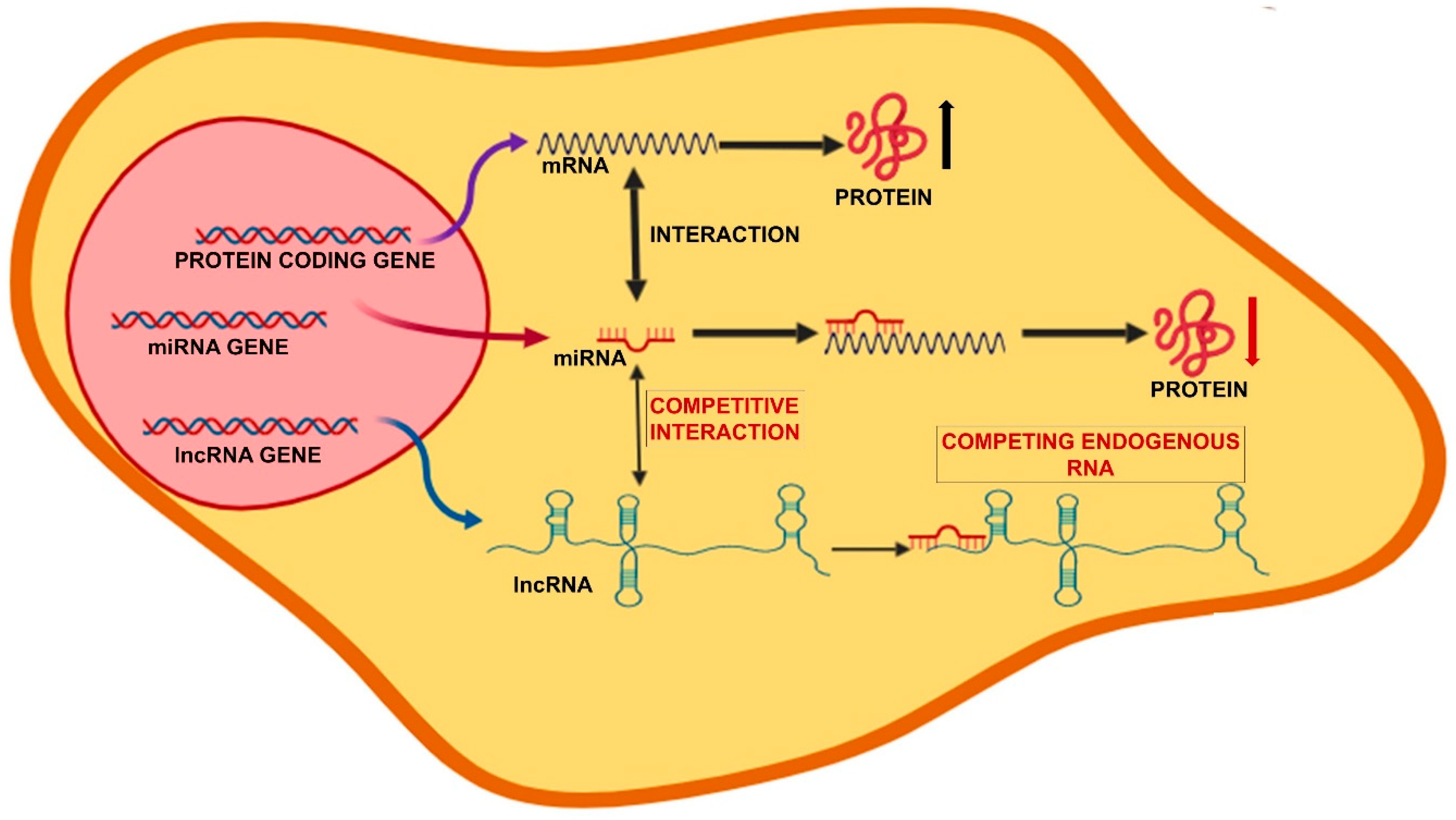
3. The Noncoding RNAs Network
3.1. microRNAs
3.2. Long Noncoding RNAs
4. miRNA/lncRNA/mRNA Network Modulation by Age-Related Mechanisms
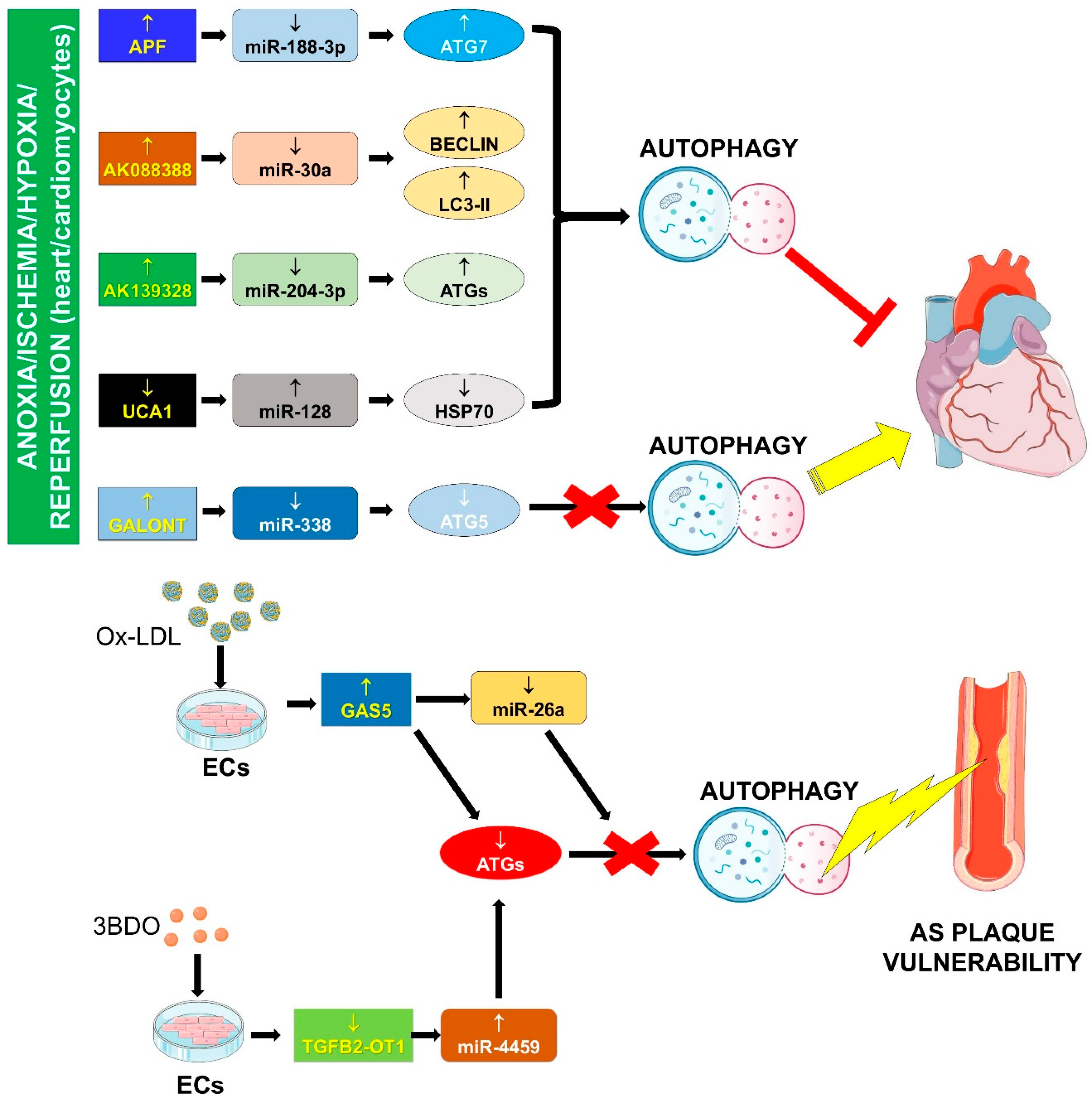
4.1. Autophagy Impairment
4.1.1. APF/miR-188-3p/ATG7
4.1.2. AK088388/miR-30a/Beclin-1 and LC3-II
4.1.3. AK139328/miR-204-3p/ATGs
4.1.4. BACE1-AS/miRNAs/BACE1
4.1.5. Galont/miR-338/ATG5
4.1.6. GAS5/miR-26a/ATGs
4.1.7. UCA1/miR-128/HSP70
4.1.8. TGFB2-OT1/miR-4459/ATG13
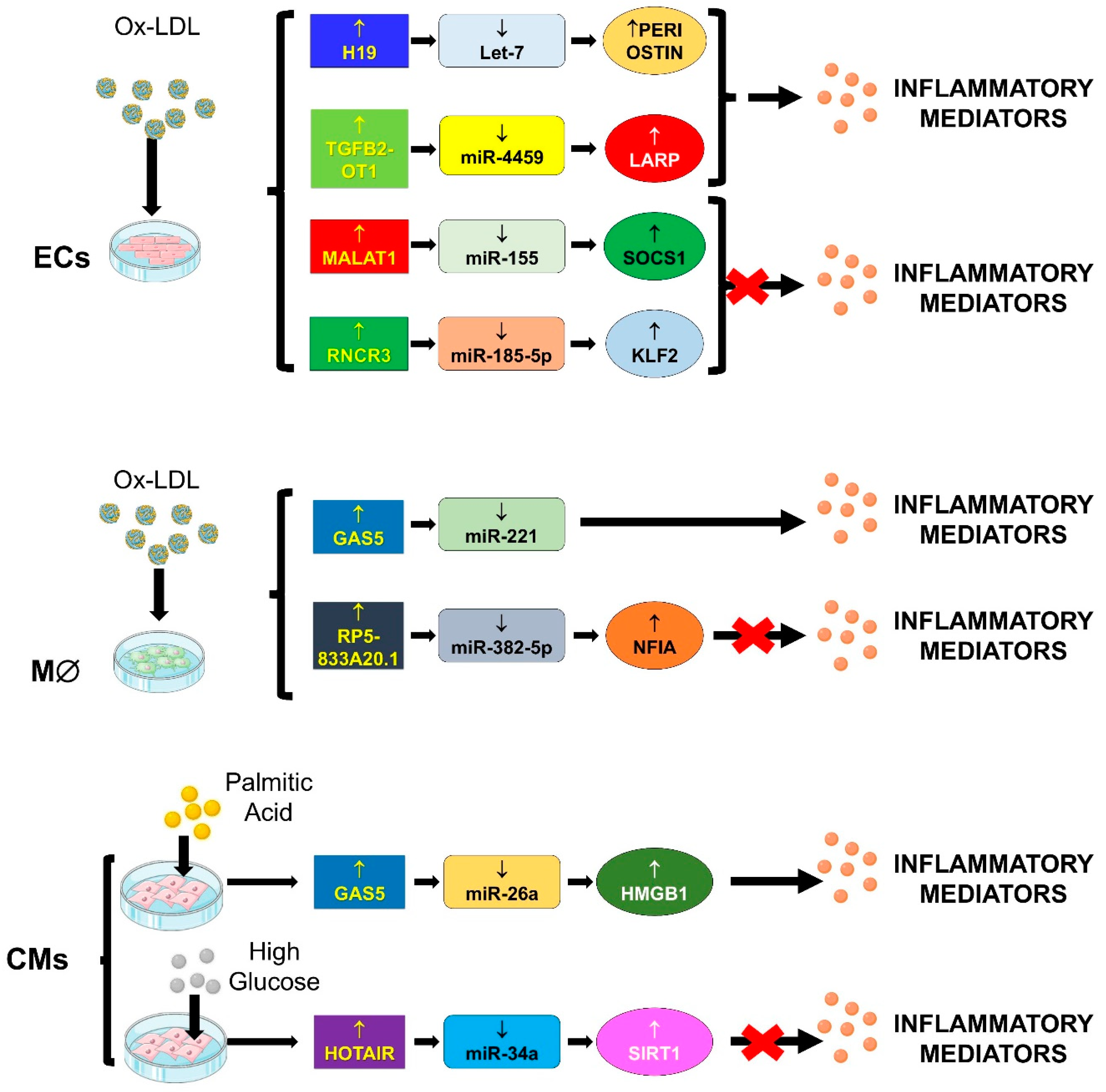
4.2. “Inflammageing”
4.2.1. TGFB2-OT1/miR-4459, miR-3960 and miR-4488/CERS1, NAT8L, ATG13 and LARP
4.2.2. GAS5/miR-26a/HMGB1
4.2.3. GAS5/miR-221/MMPs
4.2.4. H19/let-7/PERIOSTIN
4.2.5. HOTAIR/miR-34a/SIRT1
4.2.6. MALAT-1/miR-155/SOCS1
4.2.7. RNCR3/miR-185-5p/KLF-2
4.2.8. RP5-833A20.1/miR-382/NFIA
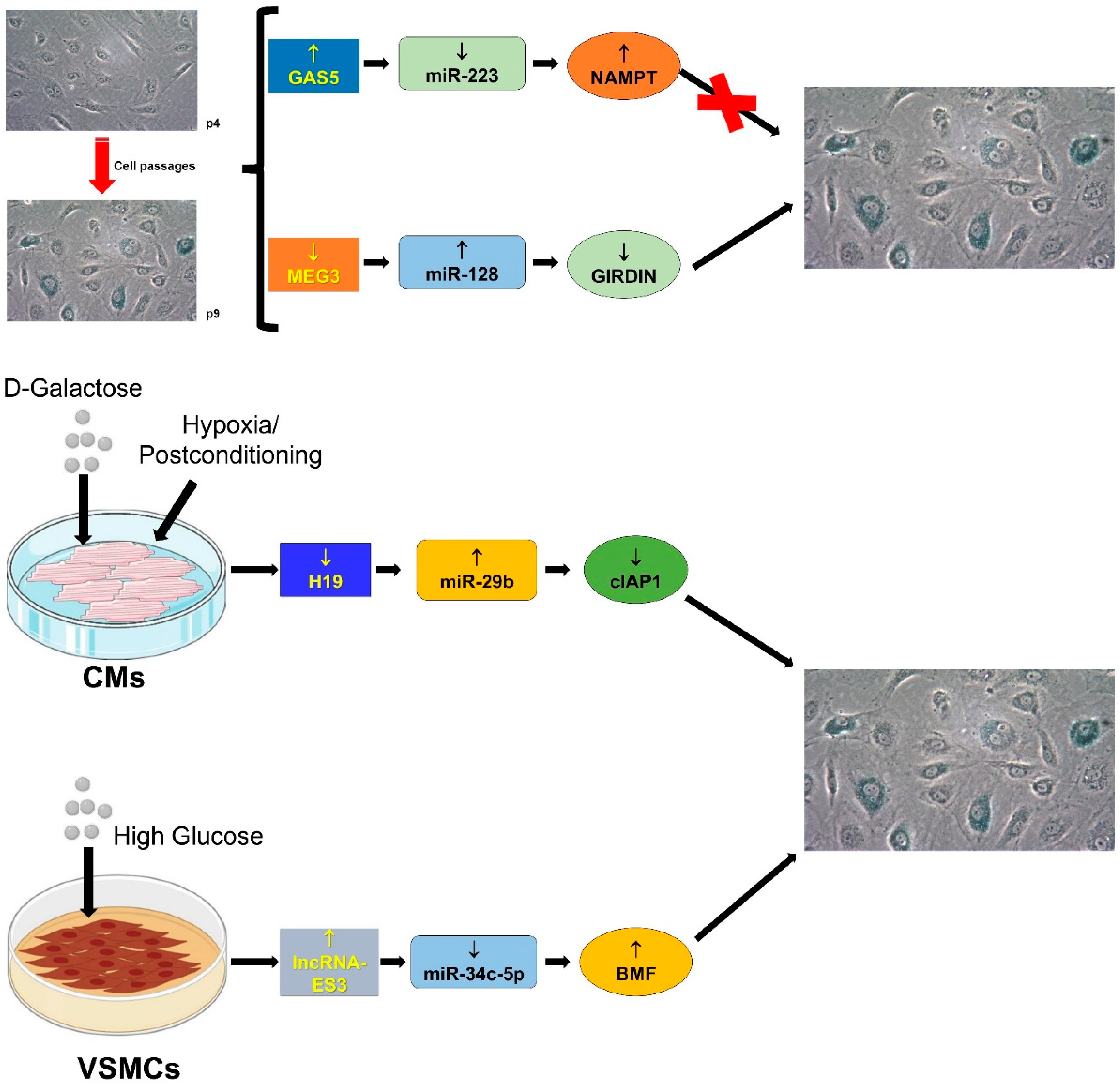
4.3. Cellular Senescence
4.3.1. GAS5/miR-223/NAMPT
4.3.2. H19/miR-29b-3p/cIAP1
4.3.3. lncRNA-ES3/miR-34c-5p/BMF
4.3.4. MEG3/miR-128/Girdin
5. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- World Health Organization (WHO). Cardiovascular Diseases (CVDs) Fact Sheet; WHO: Geneva, Switzerland, 2018. [Google Scholar]
- Paneni, F.; Diaz Canestro, C.; Libby, P.; Luscher, T.F.; Camici, G.G. The Aging Cardiovascular System: Understanding it at the Cellular and Clinical Levels. J. Am. Coll. Cardiol. 2017, 69, 1952–1967. [Google Scholar] [CrossRef] [PubMed]
- Heidenreich, P.A.; Albert, N.M.; Allen, L.A.; Bluemke, D.A.; Butler, J.; Fonarow, G.C.; Ikonomidis, J.S.; Khavjou, O.; Konstam, M.A.; Maddox, T.M.; et al. Forecasting the Impact of Heart Failure in the United States: A Policy Statement from the American Heart Association. Circ. Heart Fail. 2013, 6, 606–619. [Google Scholar] [CrossRef] [PubMed]
- Lakatta, E.G. Cardiovascular Regulatory Mechanisms in Advanced Age. Physiol. Rev. 1993, 73, 413–467. [Google Scholar] [CrossRef] [PubMed]
- Najjar, S.S.; Scuteri, A.; Lakatta, E.G. Arterial Aging: Is it an Immutable Cardiovascular Risk Factor? Hypertension 2005, 46, 454–462. [Google Scholar] [CrossRef] [PubMed]
- Christensen, K.; Doblhammer, G.; Rau, R.; Vaupel, J.W. Ageing Populations: The Challenges Ahead. Lancet 2009, 374, 1196–1208. [Google Scholar] [CrossRef]
- Lakatta, E.G. Arterial and Cardiac Aging: Major Shareholders in Cardiovascular Disease Enterprises: Part III: Cellular and Molecular Clues to Heart and Arterial Aging. Circulation 2003, 107, 490–497. [Google Scholar] [CrossRef] [PubMed]
- Mann, D.; Zipes, D.; Libby, P.; Bonow, R. Cardiovascular Disease in the Elderly. In Braunwald’s Heart Disease: A Textbook of Cardiovascular Medicine; Anonymous, Ed.; Saunders Elsevier: Philadelphia, PA, USA, 2007; pp. 1923–1953. [Google Scholar]
- Afilalo, J.; Karunananthan, S.; Eisenberg, M.J.; Alexander, K.P.; Bergman, H. Role of Frailty in Patients with Cardiovascular Disease. Am. J. Cardiol. 2009, 103, 1616–1621. [Google Scholar] [CrossRef]
- Lakatta, E.G. So! what’s Aging? is Cardiovascular Aging a Disease? J. Mol. Cell. Cardiol. 2015, 83, 1–13. [Google Scholar] [CrossRef]
- Greco, S.; Gorospe, M.; Martelli, F. Noncoding RNA in Age-Related Cardiovascular Diseases. J. Mol. Cell. Cardiol. 2015, 83, 142–155. [Google Scholar] [CrossRef]
- Fabian, M.R.; Sonenberg, N.; Filipowicz, W. Regulation of mRNA Translation and Stability by microRNAs. Annu. Rev. Biochem. 2010, 79, 351–379. [Google Scholar] [CrossRef] [Green Version]
- Greco, S.; Salgado Somoza, A.; Devaux, Y.; Martelli, F. Long Noncoding RNAs and Cardiac Disease. Antioxid. Redox Signal. 2018, 29, 880–901. [Google Scholar] [CrossRef] [PubMed]
- Zampetaki, A.; Albrecht, A.; Steinhofel, K. Long Non-Coding RNA Structure and Function: Is there a Link? Front. Physiol. 2018, 9, 1201. [Google Scholar] [CrossRef] [PubMed]
- Fuster, V.; Badimon, L.; Badimon, J.J.; Chesebro, J.H. The Pathogenesis of Coronary Artery Disease and the Acute Coronary Syndromes (2). N. Engl. J. Med. 1992, 326, 310–318. [Google Scholar] [PubMed]
- Mitchell, G.F.; Parise, H.; Benjamin, E.J.; Larson, M.G.; Keyes, M.J.; Vita, J.A.; Vasan, R.S.; Levy, D. Changes in Arterial Stiffness and Wave Reflection with Advancing Age in Healthy Men and Women: The Framingham Heart Study. Hypertension 2004, 43, 1239–1245. [Google Scholar] [CrossRef] [PubMed]
- Thorp, E.B. Mechanisms of Failed Apoptotic Cell Clearance by Phagocyte Subsets in Cardiovascular Disease. Apoptosis 2010, 15, 1124–1136. [Google Scholar] [CrossRef] [PubMed]
- Ley, K.; Miller, Y.I.; Hedrick, C.C. Monocyte and Macrophage Dynamics during Atherogenesis. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1506–1516. [Google Scholar] [CrossRef]
- Lakatta, E.G.; Levy, D. Arterial and Cardiac Aging: Major Shareholders in Cardiovascular Disease Enterprises: Part II: The Aging Heart in Health: Links to Heart Disease. Circulation 2003, 107, 346–354. [Google Scholar] [CrossRef]
- Lee, H.Y.; Oh, B.H. Aging and Arterial Stiffness. Circ. J. 2010, 74, 2257–2262. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, P.M. Hemodynamic Aging as the Consequence of Structural Changes Associated with Early Vascular Aging (EVA). Aging Dis. 2014, 5, 109–113. [Google Scholar]
- Aronson, D. Cross-Linking of Glycated Collagen in the Pathogenesis of Arterial and Myocardial Stiffening of Aging and Diabetes. J. Hypertens. 2003, 21, 3–12. [Google Scholar] [CrossRef]
- Donato, A.J.; Eskurza, I.; Silver, A.E.; Levy, A.S.; Pierce, G.L.; Gates, P.E.; Seals, D.R. Direct Evidence of Endothelial Oxidative Stress with Aging in Humans: Relation to Impaired Endothelium-Dependent Dilation and Upregulation of Nuclear Factor-kappaB. Circ. Res. 2007, 100, 1659–1666. [Google Scholar] [CrossRef] [PubMed]
- Krabbe, K.S.; Pedersen, M.; Bruunsgaard, H. Inflammatory Mediators in the Elderly. Exp. Gerontol. 2004, 39, 687–699. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Manas, L.; El-Assar, M.; Vallejo, S.; Lopez-Doriga, P.; Solis, J.; Petidier, R.; Montes, M.; Nevado, J.; Castro, M.; Gomez-Guerrero, C.; et al. Endothelial Dysfunction in Aged Humans is Related with Oxidative Stress and Vascular Inflammation. Aging Cell 2009, 8, 226–238. [Google Scholar] [CrossRef] [PubMed]
- Montezano, A.C.; Dulak-Lis, M.; Tsiropoulou, S.; Harvey, A.; Briones, A.M.; Touyz, R.M. Oxidative Stress and Human Hypertension: Vascular Mechanisms, Biomarkers, and Novel Therapies. Can. J. Cardiol. 2015, 31, 631–641. [Google Scholar] [CrossRef]
- Harvey, A.; Montezano, A.C.; Lopes, R.A.; Rios, F.; Touyz, R.M. Vascular Fibrosis in Aging and Hypertension: Molecular Mechanisms and Clinical Implications. Can. J. Cardiol. 2016, 32, 659–668. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.; Pelisek, J.; Jin, Z.G. Atherosclerosis is an Epigenetic Disease. Trends Endocrinol. Metab. 2018, 29, 739–742. [Google Scholar] [CrossRef]
- Xu, S.; Kamato, D.; Little, P.J.; Nakagawa, S.; Pelisek, J.; Jin, Z.G. Targeting Epigenetics and Non-Coding RNAs in Atherosclerosis: From Mechanisms to Therapeutics. Pharmacol. Ther. 2019, 196, 15–43. [Google Scholar] [CrossRef]
- Zhang, W.; Song, M.; Qu, J.; Liu, G.H. Epigenetic Modifications in Cardiovascular Aging and Diseases. Circ. Res. 2018, 123, 773–786. [Google Scholar] [CrossRef]
- Sessions, A.O.; Engler, A.J. Mechanical Regulation of Cardiac Aging in Model Systems. Circ. Res. 2016, 118, 1553–1562. [Google Scholar] [CrossRef] [Green Version]
- Piek, A.; de Boer, R.A.; Sillje, H.H. The Fibrosis-Cell Death Axis in Heart Failure. Heart Fail. Rev. 2016, 21, 199–211. [Google Scholar] [CrossRef]
- Lakatta, E.G.; Levy, D. Arterial and Cardiac Aging: Major Shareholders in Cardiovascular Disease Enterprises: Part I: Aging Arteries: A “Set Up” for Vascular Disease. Circulation 2003, 107, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Yeon, S.B.; Salton, C.J.; Gona, P.; Chuang, M.L.; Blease, S.J.; Han, Y.; Tsao, C.W.; Danias, P.G.; Levy, D.; O’Donnell, C.J.; et al. Impact of Age, Sex, and Indexation Method on MR Left Ventricular Reference Values in the Framingham Heart Study Offspring Cohort. J. Magn. Reson. Imaging 2015, 41, 1038–1045. [Google Scholar] [CrossRef] [PubMed]
- Hees, P.S.; Fleg, J.L.; Lakatta, E.G.; Shapiro, E.P. Left Ventricular Remodeling with Age in Normal Men Versus Women: Novel Insights using Three-Dimensional Magnetic Resonance Imaging. Am. J. Cardiol. 2002, 90, 1231–1236. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. Cardiac Fibrosis: Cell Biological Mechanisms, Molecular Pathways and Therapeutic Opportunities. Mol. Asp. Med. 2018, 65, 70–99. [Google Scholar] [CrossRef] [PubMed]
- Swynghedauw, B.; Besse, S.; Assayag, P.; Carre, F.; Chevalier, B.; Charlemagne, D.; Delcayre, C.; Hardouin, S.; Heymes, C.; Moalic, J.M. Molecular and Cellular Biology of the Senescent Hypertrophied and Failing Heart. Am. J. Cardiol. 1995, 76, 2D–7D. [Google Scholar] [CrossRef]
- Terman, A.; Brunk, U.T. Oxidative Stress, Accumulation of Biological ‘Garbage’, and Aging. Antioxid. Redox Signal. 2006, 8, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Mallat, Z.; Fornes, P.; Costagliola, R.; Esposito, B.; Belmin, J.; Lecomte, D.; Tedgui, A. Age and Gender Effects on Cardiomyocyte Apoptosis in the Normal Human Heart. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2001, 56, M719–M723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eschenhagen, T.; Bolli, R.; Braun, T.; Field, L.J.; Fleischmann, B.K.; Frisen, J.; Giacca, M.; Hare, J.M.; Houser, S.; Lee, R.T.; et al. Cardiomyocyte Regeneration: A Consensus Statement. Circulation 2017, 136, 680–686. [Google Scholar] [CrossRef] [PubMed]
- Graham, E.; Bergmann, O. Dating the Heart: Exploring Cardiomyocyte Renewal in Humans. Physiology (Bethesda) 2017, 32, 33–41. [Google Scholar] [CrossRef]
- Zacchigna, S.; Giacca, M. Extra- and Intracellular Factors Regulating Cardiomyocyte Proliferation in Postnatal Life. Cardiovasc. Res. 2014, 102, 312–320. [Google Scholar] [CrossRef]
- Gatica, D.; Chiong, M.; Lavandero, S.; Klionsky, D.J. Molecular Mechanisms of Autophagy in the Cardiovascular System. Circ. Res. 2015, 116, 456–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, J.; Zhang, Y. Targeting Autophagy in Aging and Aging-Related Cardiovascular Diseases. Trends Pharmacol. Sci. 2018, 39, 1064–1076. [Google Scholar] [CrossRef] [PubMed]
- McLean, C.A.; Cherny, R.A.; Fraser, F.W.; Fuller, S.J.; Smith, M.J.; Beyreuther, K.; Bush, A.I.; Masters, C.L. Soluble Pool of Abeta Amyloid as a Determinant of Severity of Neurodegeneration in Alzheimer’s Disease. Ann. Neurol. 1999, 46, 860–866. [Google Scholar] [CrossRef]
- McLendon, P.M.; Robbins, J. Desmin-related cardiomyopathy: An unfolding story. J. Physiol. Heart Circ. Physiol. 2011, 301, H1220–H1228. [Google Scholar] [CrossRef] [PubMed]
- Agnetti, G.; Halperin, V.L.; Kirk, J.A.; Chakir, K.; Guo, Y.; Lund, L.; Nicolini, F.; Gherli, T.; Guarnieri, C.; Caldarera, C.M.; et al. Desmin Modifications Associate with Amyloid-Like Oligomers Deposition in Heart Failure. Cardiovasc. Res. 2014, 102, 24–34. [Google Scholar] [CrossRef] [PubMed]
- McLendon, P.M.; Robbins, J. Proteotoxicity and Cardiac Dysfunction. Circ. Res. 2015, 116, 1863–1882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gianni, D.; Li, A.; Tesco, G.; McKay, K.M.; Moore, J.; Raygor, K.; Rota, M.; Gwathmey, J.K.; Dec, G.W.; Aretz, T.; et al. Protein Aggregates and Novel Presenilin Gene Variants in Idiopathic Dilated Cardiomyopathy. Circulation 2010, 121, 1216–1226. [Google Scholar] [CrossRef] [Green Version]
- Pattison, J.S.; Sanbe, A.; Maloyan, A.; Osinska, H.; Klevitsky, R.; Robbins, J. Cardiomyocyte Expression of a Polyglutamine Preamyloid Oligomer Causes Heart Failure. Circulation 2008, 117, 2743–2751. [Google Scholar] [CrossRef]
- Greco, S.; Zaccagnini, G.; Fuschi, P.; Voellenkle, C.; Carrara, M.; Sadeghi, I.; Bearzi, C.; Maimone, B.; Castelvecchio, S.; Stellos, K.; et al. Increased BACE1-AS Long Noncoding RNA and Beta-Amyloid Levels in Heart Failure. Cardiovasc. Res. 2017, 113, 453–463. [Google Scholar] [CrossRef]
- Gonzalez-Lopez, E.; Gallego-Delgado, M.; Guzzo-Merello, G.; de Haro-Del Moral, F.J.; Cobo-Marcos, M.; Robles, C.; Bornstein, B.; Salas, C.; Lara-Pezzi, E.; Alonso-Pulpon, L.; et al. Wild-Type Transthyretin Amyloidosis as a Cause of Heart Failure with Preserved Ejection Fraction. Eur. Heart J. 2015, 36, 2585–2594. [Google Scholar] [CrossRef]
- Connors, L.H.; Sam, F.; Skinner, M.; Salinaro, F.; Sun, F.; Ruberg, F.L.; Berk, J.L.; Seldin, D.C. Heart Failure Resulting from Age-Related Cardiac Amyloid Disease Associated with Wild-Type Transthyretin: A Prospective, Observational Cohort Study. Circulation 2016, 133, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Rapezzi, C.; Merlini, G.; Quarta, C.C.; Riva, L.; Longhi, S.; Leone, O.; Salvi, F.; Ciliberti, P.; Pastorelli, F.; Biagini, E.; et al. Systemic Cardiac Amyloidoses: Disease Profiles and Clinical Courses of the 3 Main Types. Circulation 2009, 120, 1203–1212. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, S.F.; Mirzoyev, S.A.; Edwards, W.D.; Dogan, A.; Grogan, D.R.; Dunlay, S.M.; Roger, V.L.; Gertz, M.A.; Dispenzieri, A.; Zeldenrust, S.R.; et al. Left Ventricular Amyloid Deposition in Patients with Heart Failure and Preserved Ejection Fraction. JACC Heart Fail. 2014, 2, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Michels da Silva, D.; Langer, H.; Graf, T. Inflammatory and Molecular Pathways in Heart Failure-Ischemia, HFpEF and Transthyretin Cardiac Amyloidosis. Int. J. Mol. Sci. 2019, 20, 2322. [Google Scholar] [CrossRef] [PubMed]
- Krol, J.; Loedige, I.; Filipowicz, W. The Widespread Regulation of microRNA Biogenesis, Function and Decay. Nat. Rev. Genet. 2010, 11, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Gorospe, M.; Abdelmohsen, K. MicroRegulators Come of Age in Senescence. Trends Genet. 2011, 27, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Amort, T.; Souliere, M.F.; Wille, A.; Jia, X.Y.; Fiegl, H.; Worle, H.; Micura, R.; Lusser, A. Long Non-Coding RNAs as Targets for Cytosine Methylation. RNA Biol. 2013, 10, 1003–1008. [Google Scholar] [CrossRef]
- Ma, L.; Bajic, V.B.; Zhang, Z. On the Classification of Long Non-Coding RNAs. RNA Biol. 2013, 10, 925–933. [Google Scholar] [CrossRef]
- Rinn, J.L.; Chang, H.Y. Genome Regulation by Long Noncoding RNAs. Annu. Rev. Biochem. 2012, 81, 145–166. [Google Scholar] [CrossRef] [Green Version]
- Clark, M.B.; Mattick, J.S. Long Noncoding RNAs in Cell Biology. Semin. Cell Dev. Biol. 2011, 22, 366–376. [Google Scholar] [CrossRef]
- Memczak, S.; Jens, M.; Elefsinioti, A.; Torti, F.; Krueger, J.; Rybak, A.; Maier, L.; Mackowiak, S.D.; Gregersen, L.H.; Munschauer, M.; et al. Circular RNAs are a Large Class of Animal RNAs with Regulatory Potency. Nature 2013, 495, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A ceRNA Hypothesis: The Rosetta Stone of a Hidden RNA Language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wee, L.M.; Flores-Jasso, C.F.; Salomon, W.E.; Zamore, P.D. Argonaute Divides its RNA Guide into Domains with Distinct Functions and RNA-Binding Properties. Cell 2012, 151, 1055–1067. [Google Scholar] [CrossRef] [PubMed]
- Devaux, Y.; Zangrando, J.; Schroen, B.; Creemers, E.E.; Pedrazzini, T.; Chang, C.P.; Dorn, G.W., 2nd; Thum, T.; Heymans, S. Cardiolinc network. Long Noncoding RNAs in Cardiac Development and Ageing. Nat. Rev. Cardiol. 2015, 12, 415–425. [Google Scholar] [PubMed]
- Kim, J.; Kim, K.M.; Noh, J.H.; Yoon, J.H.; Abdelmohsen, K.; Gorospe, M. Long Noncoding RNAs in Diseases of Aging. Biochim. Biophys. Acta 2016, 1859, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Grootaert, M.O.J.; Roth, L.; Schrijvers, D.M.; De Meyer, G.R.Y.; Martinet, W. Defective Autophagy in Atherosclerosis: To Die or to Senesce? Oxid. Med. Cell. Longev. 2018, 2018, 7687083. [Google Scholar] [CrossRef]
- Libby, P. Mechanisms of Acute Coronary Syndromes and their Implications for Therapy. N. Engl. J. Med. 2013, 368, 2004–2013. [Google Scholar] [CrossRef]
- Mei, Y.; Thompson, M.D.; Cohen, R.A.; Tong, X. Autophagy and Oxidative Stress in Cardiovascular Diseases. Biochim. Biophys. Acta 2015, 1852, 243–251. [Google Scholar] [CrossRef]
- Wang, K.; Liu, C.Y.; Zhou, L.Y.; Wang, J.X.; Wang, M.; Zhao, B.; Zhao, W.K.; Xu, S.J.; Fan, L.H.; Zhang, X.J.; et al. APF lncRNA Regulates Autophagy and Myocardial Infarction by Targeting miR-188-3p. Nat. Commun. 2015, 6, 6779. [Google Scholar] [CrossRef]
- Wang, J.J.; Bie, Z.D.; Sun, C.F. Long Noncoding RNA AK088388 Regulates Autophagy through miR-30a to Affect Cardiomyocyte Injury. J. Cell. Biochem. 2019, 120, 10155–10163. [Google Scholar] [CrossRef]
- Yu, S.Y.; Dong, B.; Fang, Z.F.; Hu, X.Q.; Tang, L.; Zhou, S.H. Knockdown of lncRNA AK139328 Alleviates Myocardial Ischaemia/Reperfusion Injury in Diabetic Mice Via Modulating miR-204-3p and Inhibiting Autophagy. J. Cell. Mol. Med. 2018, 22, 4886–4898. [Google Scholar] [CrossRef] [PubMed]
- Faghihi, M.A.; Zhang, M.; Huang, J.; Modarresi, F.; Van der Brug, M.P.; Nalls, M.A.; Cookson, M.R.; St-Laurent, G., 3rd; Wahlestedt, C. Evidence for Natural Antisense Transcript-Mediated Inhibition of microRNA Function. Genome Biol. 2010, 11, R56. [Google Scholar] [CrossRef] [PubMed]
- Zeng, T.; Ni, H.; Yu, Y.; Zhang, M.; Wu, M.; Wang, Q.; Wang, L.; Xu, S.; Xu, Z.; Xu, C.; et al. BACE1-AS Prevents BACE1 mRNA Degradation through the Sequestration of BACE1-Targeting miRNAs. J. Chem. Neuroanat. 2019, 98, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Yin, G.; Yang, X.; Li, Q.; Guo, Z. GATA1 Activated lncRNA (Galont) Promotes Anoxia/Reoxygenation-Induced Autophagy and Cell Death in Cardiomyocytes by Sponging miR-338. J. Cell. Biochem. 2018, 119, 4161–4169. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.; Fan, T.; Liu, L.; Zhang, L. Knockdown of Growth-Arrest Specific Transcript 5 Restores Oxidized Low-Density Lipoprotein-Induced Impaired Autophagy Flux Via Upregulating miR-26a in Human Endothelial Cells. Eur. J. Pharmacol. 2019, 843, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Ge, D.; Han, L.; Huang, S.; Peng, N.; Wang, P.; Jiang, Z.; Zhao, J.; Su, L.; Zhang, S.; Zhang, Y.; et al. Identification of a Novel MTOR Activator and Discovery of a Competing Endogenous RNA Regulating Autophagy in Vascular Endothelial Cells. Autophagy 2014, 10, 957–971. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Liu, R.; Niu, Q.; Wang, H.; Yang, Z.; Bao, Y. Morphine Postconditioning Alleviates Autophage in Ischemia-Reperfusion Induced Cardiac Injury through Up-Regulating lncRNA UCA1. Biomed. Pharmacother. 2018, 108, 1357–1364. [Google Scholar] [CrossRef] [PubMed]
- Yue, Q.; Zhao, C.; Wang, Y.; Zhao, L.; Zhu, Q.; Li, G.; Wu, N.; Jia, D.; Ma, C. Downregulation of Growth Arrest specific Transcript 5 Alleviates Palmitic Acid induced Myocardial Inflammatory Injury through the miR26a/HMGB1/NFkappaB Axis. Mol. Med. Rep. 2018, 18, 5742–5750. [Google Scholar]
- Ye, J.; Wang, C.; Wang, D.; Yuan, H. LncRBA GSA5, Up-Regulated by Ox-LDL, Aggravates Inflammatory Response and MMP Expression in THP-1 Macrophages by Acting Like a Sponge for miR-221. Exp. Cell Res. 2018, 369, 348–355. [Google Scholar] [CrossRef]
- Cao, L.; Zhang, Z.; Li, Y.; Zhao, P.; Chen, Y. LncRNA H19/miR-Let-7 Axis Participates in the Regulation of Ox-LDL-Induced Endothelial Cell Injury Via Targeting Periostin. Int. Immunopharmacol. 2019, 72, 496–503. [Google Scholar] [CrossRef]
- Gao, L.; Wang, X.; Guo, S.; Xiao, L.; Liang, C.; Wang, Z.; Li, Y.; Liu, Y.; Yao, R.; Liu, Y.; et al. LncRNA HOTAIR Functions as a Competing Endogenous RNA to Upregulate SIRT1 by Sponging miR-34a in Diabetic Cardiomyopathy. J. Cell. Physiol. 2019, 234, 4944–4958. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Sun, Y.; Zhong, L.; Xiao, Z.; Yang, M.; Chen, M.; Wang, C.; Xie, X.; Chen, X. The Suppression of Ox-LDL-Induced Inflammatory Cytokine Release and Apoptosis of HCAECs by Long Non-Coding RNA-MALAT1 Via Regulating microRNA-155/SOCS1 Pathway. Nutr. Metab. Cardiovasc. Dis. 2018, 28, 1175–1187. [Google Scholar] [CrossRef] [PubMed]
- Shan, K.; Jiang, Q.; Wang, X.Q.; Wang, Y.N.; Yang, H.; Yao, M.D.; Liu, C.; Li, X.M.; Yao, J.; Liu, B.; et al. Role of Long Non-Coding RNA-RNCR3 in Atherosclerosis-Related Vascular Dysfunction. Cell Death Dis. 2016, 7, e2248. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.W.; Zhao, J.Y.; Li, S.F.; Huang, J.L.; Qiu, Y.R.; Ma, X.; Wu, S.G.; Chen, Z.P.; Hu, Y.R.; Yang, J.Y.; et al. RP5-833A20.1/miR-382-5p/NFIA-Dependent Signal Transduction Pathway Contributes to the Regulation of Cholesterol Homeostasis and Inflammatory Reaction. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 87–101. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Lu, W.; Ge, D.; Meng, N.; Li, Y.; Su, L.; Zhang, S.; Zhang, Y.; Zhao, B.; Miao, J. A New microRNA Signal Pathway Regulated by Long Noncoding RNA TGFB2-OT1 in Autophagy and Inflammation of Vascular Endothelial Cells. Autophagy 2015, 11, 2172–2183. [Google Scholar] [CrossRef]
- Yao, J.; Shi, Z.; Ma, X.; Xu, D.; Ming, G. lncRNA GAS5/miR-223/NAMPT Axis Modulates the Cell Proliferation and Senescence of Endothelial Progenitor Cells through PI3K/AKT Signaling. J. Cell. Biochem. 2019, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Cheng, L.; Xu, L.; Zhang, Y.; Yang, Y.; Fu, Q.; Mi, W.; Li, H. The Lncrna, H19 Mediates the Protective Effect of Hypoxia Postconditioning Against Hypoxia-Reoxygenation Injury to Senescent Cardiomyocytes by Targeting MicroRNA-29b-3p. Shock 2018. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Zhan, J.K.; Zhong, J.Y.; Wang, Y.J.; Wang, Y.; Li, S.; He, J.Y.; Tan, P.; Chen, Y.Y.; Liu, X.B.; et al. lncRNA-ES3/miR-34c-5p/BMF Axis is Involved in Regulating High-Glucose-Induced Calcification/Senescence of VSMCs. Aging (Albany NY) 2019, 11, 523–535. [Google Scholar] [CrossRef]
- Lin, C.; Ear, J.; Midde, K.; Lopez-Sanchez, I.; Aznar, N.; Garcia-Marcos, M.; Kufareva, I.; Abagyan, R.; Ghosh, P. Structural Basis for Activation of Trimeric Gi Proteins by Multiple Growth Factor Receptors Via GIV/Girdin. Mol. Biol. Cell 2014, 25, 3654–3671. [Google Scholar] [CrossRef]
- De Duve, C.; Wattiaux, R. Functions of Lysosomes. Annu. Rev. Physiol. 1966, 28, 435–492. [Google Scholar] [CrossRef]
- Wohlgemuth, S.E.; Calvani, R.; Marzetti, E. The Interplay between Autophagy and Mitochondrial Dysfunction in Oxidative Stress-Induced Cardiac Aging and Pathology. J. Mol. Cell. Cardiol. 2014, 71, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Rifki, O.F.; Hill, J.A. Cardiac Autophagy: Good with the Bad. J. Cardiovasc. Pharmacol. 2012, 60, 248–252. [Google Scholar] [CrossRef] [PubMed]
- Hamacher-Brady, A.; Brady, N.R.; Gottlieb, R.A. The Interplay between Pro-Death and Pro-Survival Signaling Pathways in Myocardial Ischemia/Reperfusion Injury: Apoptosis Meets Autophagy. Cardiovasc. Drugs Ther. 2006, 20, 445–462. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Wang, Y.; Chen, Y.; Cao, F. The Role of the Autophagy in Myocardial Ischemia/Reperfusion Injury. Biochim. Biophys. Acta 2015, 1852, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Essick, E.E.; Sam, F. Oxidative Stress and Autophagy in Cardiac Disease, Neurological Disorders, Aging and Cancer. Oxid. Med. Cell. Longev. 2010, 3, 168–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Meyer, G.R.; De Keulenaer, G.W.; Martinet, W. Role of Autophagy in Heart Failure Associated with Aging. Heart Fail. Rev. 2010, 15, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Yang, W.; Guo, Y.; Chen, W.; Zheng, P.; Zeng, J.; Tong, W. Exosomal lncRNA GAS5 Regulates the Apoptosis of Macrophages and Vascular Endothelial Cells in Atherosclerosis. PLoS ONE 2017, 12, e0185406. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Meng, N.; Zhao, B.; Zhao, J.; Zhang, Y.; Zhang, S.; Miao, J. Protective Effects of a Synthesized Butyrolactone Derivative Against Chloroquine-Induced Autophagic Vesicle Accumulation and the Disturbance of Mitochondrial Membrane Potential and Na+,K+-ATPase Activity in Vascular Endothelial Cells. Chem. Res. Toxicol. 2009, 22, 471–475. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Bonafe, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflamm-Aging. an Evolutionary Perspective on Immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Ferrucci, L.; Fabbri, E. Inflammageing: Chronic Inflammation in Ageing, Cardiovascular Disease, and Frailty. Nat. Rev. Cardiol. 2018, 15, 505–522. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Candales, A.; Hernandez Burgos, P.M.; Hernandez-Suarez, D.F.; Harris, D. Linking Chronic Inflammation with Cardiovascular Disease: From Normal Aging to the Metabolic Syndrome. J. Nat. Sci. 2017, 3, e341. [Google Scholar] [PubMed]
- Zuliani, G.; Morieri, M.L.; Volpato, S.; Maggio, M.; Cherubini, A.; Francesconi, D.; Bandinelli, S.; Paolisso, G.; Guralnik, J.M.; Ferrucci, L. Insulin Resistance and Systemic Inflammation, but Not Metabolic Syndrome Phenotype, Predict 9 Years Mortality in Older Adults. Atherosclerosis 2014, 235, 538–545. [Google Scholar] [CrossRef] [PubMed]
- Geovanini, G.R.; Libby, P. Atherosclerosis and Inflammation: Overview and Updates. Clin. Sci. (Lond.) 2018, 132, 1243–1252. [Google Scholar] [CrossRef]
- Lucas, A.R.; Korol, R.; Pepine, C.J. Inflammation in Atherosclerosis: Some Thoughts about Acute Coronary Syndromes. Circulation 2006, 113, e728–e732. [Google Scholar] [CrossRef] [PubMed]
- Boldin, M.P.; Baltimore, D. MicroRNAs, New Effectors and Regulators of NF-kappaB. Immunol. Rev. 2012, 246, 205–220. [Google Scholar] [CrossRef]
- Chew, C.L.; Conos, S.A.; Unal, B.; Tergaonkar, V. Noncoding RNAs: Master Regulators of Inflammatory Signaling. Trends Mol. Med. 2018, 24, 66–84. [Google Scholar] [CrossRef] [PubMed]
- Magagula, L.; Gagliardi, M.; Naidoo, J.; Mhlanga, M. Lnc-Ing Inflammation to Disease. Biochem. Soc. Trans. 2017, 45, 953–962. [Google Scholar] [CrossRef]
- Guzik, T.J.; Touyz, R.M. Oxidative Stress, Inflammation, and Vascular Aging in Hypertension. Hypertension 2017, 70, 660–667. [Google Scholar] [CrossRef]
- Zhang, J.; Rane, G.; Dai, X.; Shanmugam, M.K.; Arfuso, F.; Samy, R.P.; Lai, M.K.; Kappei, D.; Kumar, A.P.; Sethi, G. Ageing and the Telomere Connection: An Intimate Relationship with Inflammation. Ageing Res. Rev. 2016, 25, 55–69. [Google Scholar] [CrossRef]
- Razani, B.; Feng, C.; Coleman, T.; Emanuel, R.; Wen, H.; Hwang, S.; Ting, J.P.; Virgin, H.W.; Kastan, M.B.; Semenkovich, C.F. Autophagy Links Inflammasomes to Atherosclerotic Progression. Cell. Metab. 2012, 15, 534–544. [Google Scholar] [CrossRef] [Green Version]
- Deretic, V.; Saitoh, T.; Akira, S. Autophagy in Infection, Inflammation and Immunity. Nat. Rev. Immunol. 2013, 13, 722–737. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Mizushima, N.; Virgin, H.W. Autophagy in Immunity and Inflammation. Nature 2011, 469, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Guardia, D.; Palomer, X.; Coll, T.; Serrano, L.; Rodriguez-Calvo, R.; Davidson, M.M.; Merlos, M.; El Kochairi, I.; Michalik, L.; Wahli, W.; et al. PPARbeta/Delta Activation Blocks Lipid-Induced Inflammatory Pathways in Mouse Heart and Human Cardiac Cells. Biochim. Biophys. Acta 2011, 1811, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Ying, Y.; Zhu, H.; Liang, Z.; Ma, X.; Li, S. GLP1 Protects Cardiomyocytes from Palmitate-Induced Apoptosis Via Akt/GSK3b/B-Catenin Pathway. J. Mol. Endocrinol. 2015, 55, 245–262. [Google Scholar] [CrossRef]
- Rinn, J.L.; Kertesz, M.; Wang, J.K.; Squazzo, S.L.; Xu, X.; Brugmann, S.A.; Goodnough, L.H.; Helms, J.A.; Farnham, P.J.; Segal, E.; et al. Functional Demarcation of Active and Silent Chromatin Domains in Human HOX Loci by Noncoding RNAs. Cell 2007, 129, 1311–1323. [Google Scholar] [CrossRef] [Green Version]
- Gao, L.; Liu, Y.; Guo, S.; Yao, R.; Wu, L.; Xiao, L.; Wang, Z.; Liu, Y.; Zhang, Y. Circulating Long Noncoding RNA HOTAIR is an Essential Mediator of Acute Myocardial Infarction. Cell. Physiol. Biochem. 2017, 44, 1497–1508. [Google Scholar]
- Greco, S.; Zaccagnini, G.; Perfetti, A.; Fuschi, P.; Valaperta, R.; Voellenkle, C.; Castelvecchio, S.; Gaetano, C.; Finato, N.; Beltrami, A.P.; et al. Long Noncoding RNA Dysregulation in Ischemic Heart Failure. J. Transl. Med. 2016, 14, 183. [Google Scholar] [CrossRef]
- Lai, Y.; He, S.; Ma, L.; Lin, H.; Ren, B.; Ma, J.; Zhu, X.; Zhuang, S. HOTAIR Functions as a Competing Endogenous RNA to Regulate PTEN Expression by Inhibiting miR-19 in Cardiac Hypertrophy. Mol. Cell. Biochem. 2017, 432, 179–187. [Google Scholar] [CrossRef]
- Zhang, W.; Huang, Q.; Zeng, Z.; Wu, J.; Zhang, Y.; Chen, Z. Sirt1 Inhibits Oxidative Stress in Vascular Endothelial Cells. Oxid. Med. Cell. Longev. 2017, 2017, 7543973. [Google Scholar] [CrossRef]
- Kitada, M.; Ogura, Y.; Koya, D. The Protective Role of Sirt1 in Vascular Tissue: Its Relationship to Vascular Aging and Atherosclerosis. Aging (Albany NY) 2016, 8, 2290–2307. [Google Scholar] [CrossRef]
- Michalik, K.M.; You, X.; Manavski, Y.; Doddaballapur, A.; Zornig, M.; Braun, T.; John, D.; Ponomareva, Y.; Chen, W.; Uchida, S.; et al. Long Noncoding RNA MALAT1 Regulates Endothelial Cell Function and Vessel Growth. Circ. Res. 2014, 114, 1389–1397. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Jin, X.; Xiang, Y.; Chen, Y.; Shen, C.X.; Zhang, Y.C.; Li, Y.G. The lncRNA MALAT1 Protects the Endothelium Against Ox-LDL-Induced Dysfunction Via Upregulating the Expression of the miR-22-3p Target Genes CXCR2 and AKT. FEBS Lett. 2015, 589, 3189–3196. [Google Scholar] [CrossRef] [PubMed]
- Puthanveetil, P.; Chen, S.; Feng, B.; Gautam, A.; Chakrabarti, S. Long Non-Coding RNA MALAT1 Regulates Hyperglycaemia Induced Inflammatory Process in the Endothelial Cells. J. Cell. Mol. Med. 2015, 19, 1418–1425. [Google Scholar] [CrossRef]
- Tili, E.; Michaille, J.J.; Cimino, A.; Costinean, S.; Dumitru, C.D.; Adair, B.; Fabbri, M.; Alder, H.; Liu, C.G.; Calin, G.A.; et al. Modulation of miR-155 and miR-125b Levels Following Lipopolysaccharide/TNF-Alpha Stimulation and their Possible Roles in Regulating the Response to Endotoxin Shock. J. Immunol. 2007, 179, 5082–5089. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Yang, L.; Liang, X.; Zhu, G. MicroRNA-155 Promotes Atherosclerosis Inflammation Via Targeting SOCS1. Cell. Physiol. Biochem. 2015, 36, 1371–1381. [Google Scholar] [CrossRef] [PubMed]
- Dekker, R.J.; Boon, R.A.; Rondaij, M.G.; Kragt, A.; Volger, O.L.; Elderkamp, Y.W.; Meijers, J.C.; Voorberg, J.; Pannekoek, H.; Horrevoets, A.J. KLF2 Provokes a Gene Expression Pattern that Establishes Functional Quiescent Differentiation of the Endothelium. Blood 2006, 107, 4354–4363. [Google Scholar] [CrossRef] [PubMed]
- Hayflick, L.; Moorhead, P.S. The Serial Cultivation of Human Diploid Cell Strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Ben-Porath, I.; Weinberg, R.A. The Signals and Pathways Activating Cellular Senescence. Int. J. Biochem. Cell Biol. 2005, 37, 961–976. [Google Scholar] [CrossRef] [PubMed]
- Sack, M.N.; Fyhrquist, F.Y.; Saijonmaa, O.J.; Fuster, V.; Kovacic, J.C. Basic Biology of Oxidative Stress and the Cardiovascular System: Part 1 of a 3-Part Series. J. Am. Coll. Cardiol. 2017, 70, 196–211. [Google Scholar] [CrossRef] [PubMed]
- de Lange, T. Shelterin: The Protein Complex that Shapes and Safeguards Human Telomeres. Genes Dev. 2005, 19, 2100–2110. [Google Scholar] [CrossRef] [PubMed]
- von Zglinicki, T. Oxidative Stress Shortens Telomeres. Trends Biochem. Sci. 2002, 27, 339–344. [Google Scholar] [CrossRef]
- Carnevali, S.; Petruzzelli, S.; Longoni, B.; Vanacore, R.; Barale, R.; Cipollini, M.; Scatena, F.; Paggiaro, P.; Celi, A.; Giuntini, C. Cigarette Smoke Extract Induces Oxidative Stress and Apoptosis in Human Lung Fibroblasts. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003, 284, L955–L963. [Google Scholar] [CrossRef] [PubMed]
- van Deursen, J.M. The Role of Senescent Cells in Ageing. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J.; d’Adda di Fagagna, F. Cellular Senescence: When Bad Things Happen to Good Cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Ji, S. Cellular Senescence: Molecular Mechanisms and Pathogenicity. J. Cell. Physiol. 2018, 233, 9121–9135. [Google Scholar] [CrossRef]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; LeBrasseur, N.K.; Childs, B.G.; van de Sluis, B.; Kirkland, J.L.; van Deursen, J.M. Clearance of p16Ink4a-Positive Senescent Cells Delays Ageing-Associated Disorders. Nature 2011, 479, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Panda, A.C.; Abdelmohsen, K.; Gorospe, M. SASP Regulation by Noncoding RNA. Mech. Ageing Dev. 2017, 168, 37–43. [Google Scholar] [CrossRef]
- Grammatikakis, I.; Panda, A.C.; Abdelmohsen, K.; Gorospe, M. Long Noncoding RNAs(lncRNAs) and the Molecular Hallmarks of Aging. Aging (Albany NY) 2014, 6, 992–1009. [Google Scholar] [CrossRef]
- Song, X.; Bao, M.; Li, D.; Li, Y.M. Advanced Glycation in D-Galactose Induced Mouse Aging Model. Mech. Ageing Dev. 1999, 108, 239–251. [Google Scholar] [CrossRef]
- Donato, M.; Evelson, P.; Gelpi, R.J. Protecting the Heart from Ischemia/Reperfusion Injury: An Update on Remote Ischemic Preconditioning and Postconditioning. Curr. Opin. Cardiol. 2017, 32, 784–790. [Google Scholar] [CrossRef]
- Lefer, D.J.; Marban, E. Is Cardioprotection Dead? Circulation 2017, 136, 98–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rennenberg, R.J.; Kessels, A.G.; Schurgers, L.J.; van Engelshoven, J.M.; de Leeuw, P.W.; Kroon, A.A. Vascular Calcifications as a Marker of Increased Cardiovascular Risk: A Meta-Analysis. Vasc. Health Risk Manag. 2009, 5, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Burton, D.G.; Matsubara, H.; Ikeda, K. Pathophysiology of Vascular Calcification: Pivotal Role of Cellular Senescence in Vascular Smooth Muscle Cells. Exp. Gerontol. 2010, 45, 819–824. [Google Scholar] [CrossRef] [PubMed]
- Qiu, G.Z.; Tian, W.; Fu, H.T.; Li, C.P.; Liu, B. Long Noncoding RNA-MEG3 is Involved in Diabetes Mellitus-Related Microvascular Dysfunction. Biochem. Biophys. Res. Commun. 2016, 471, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Boon, R.A.; Hofmann, P.; Michalik, K.M.; Lozano-Vidal, N.; Berghauser, D.; Fischer, A.; Knau, A.; Jae, N.; Schurmann, C.; Dimmeler, S. Long Noncoding RNA Meg3 Controls Endothelial Cell Aging and Function: Implications for Regenerative Angiogenesis. J. Am. Coll. Cardiol. 2016, 68, 2589–2591. [Google Scholar] [CrossRef] [PubMed]
- Simion, V.; Haemmig, S.; Feinberg, M.W. LncRNAs in Vascular Biology and Disease. Vasc. Pharmacol. 2018, 114, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Lan, Y.; Li, Y.J.; Li, D.J.; Li, P.; Wang, J.Y.; Diao, Y.P.; Ye, G.D.; Li, Y.F. Long Non-Coding RNA MEG3 Prevents Vascular Endothelial Cell Senescence by Impairing miR-128-Dependent Girdin Down-Regulation. Am. J. Physiol. Cell. Physiol. 2018, 316, C830–C843. [Google Scholar] [CrossRef] [PubMed]
- Noren Hooten, N.; Abdelmohsen, K.; Gorospe, M.; Ejiogu, N.; Zonderman, A.B.; Evans, M.K. microRNA Expression Patterns Reveal Differential Expression of Target Genes with Age. PLoS ONE 2010, 5, e10724. [Google Scholar] [CrossRef] [PubMed]
- Dykes, I.M.; Emanueli, C. Transcriptional and Post-Transcriptional Gene Regulation by Long Non-Coding RNA. Genom. Proteom. Bioinform. 2017, 15, 177–186. [Google Scholar] [CrossRef]
- Voellenkle, C.; Garcia-Manteiga, J.M.; Pedrotti, S.; Perfetti, A.; De Toma, I.; Da Silva, D.; Maimone, B.; Greco, S.; Fasanaro, P.; Creo, P.; et al. Implication of Long Noncoding RNAs in the Endothelial Cell Response to Hypoxia Revealed by RNA-Sequencing. Sci. Rep. 2016, 6, 24141. [Google Scholar] [CrossRef]
- Merryman, W.D.; Clark, C.R. Lnc-Ing NOTCH1 to Idiopathic Calcific Aortic Valve Disease. Circulation 2016, 134, 1863–1865. [Google Scholar] [CrossRef] [PubMed]
- Keniry, A.; Oxley, D.; Monnier, P.; Kyba, M.; Dandolo, L.; Smits, G.; Reik, W. The H19 lincRNA is a Developmental Reservoir of miR-675 that Suppresses Growth and Igf1r. Nat. Cell Biol. 2012, 14, 659–665. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, H.; Yao, B.; Xu, W.; Chen, J.; Zhou, X. lncRNA H19/miR-675 Axis Regulates Cardiomyocyte Apoptosis by Targeting VDAC1 in Diabetic Cardiomyopathy. Sci. Rep. 2016, 6, 36340. [Google Scholar] [CrossRef] [PubMed]
- Cai, B.; Ma, W.; Bi, C.; Yang, F.; Zhang, L.; Han, Z.; Huang, Q.; Ding, F.; Li, Y.; Yan, G.; et al. Long Noncoding RNA H19 Mediates Melatonin Inhibition of Premature Senescence of C-Kit+ Cardiac Progenitor Cells by Promoting miR-675. J. Pineal Res. 2016, 61, 82–95. [Google Scholar] [CrossRef] [PubMed]
- Carrara, M.; Fuschi, P.; Ivan, C.; Martelli, F. Circular RNAs: Methodological Challenges and Perspectives in Cardiovascular Diseases. J. Cell. Mol. Med. 2018, 22, 5176–5187. [Google Scholar] [CrossRef] [PubMed]
- Li, H.M.; Ma, X.L.; Li, H.G. Intriguing Circles: Conflicts and Controversies in Circular RNA Research. Wiley Interdiscip. Rev. RNA 2019, e1538. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.B.; Jensen, T.I.; Clausen, B.H.; Bramsen, J.B.; Finsen, B.; Damgaard, C.K.; Kjems, J. Natural RNA Circles Function as Efficient microRNA Sponges. Nature 2013, 495, 384–388. [Google Scholar] [CrossRef]
- Gandhi, S.; Ruehle, F.; Stoll, M. Evolutionary Patterns of Non-Coding RNA in Cardiovascular Biology. Non-Coding RNA 2019, 5, 15. [Google Scholar] [CrossRef]
- Bassett, A.R.; Akhtar, A.; Barlow, D.P.; Bird, A.P.; Brockdorff, N.; Duboule, D.; Ephrussi, A.; Ferguson-Smith, A.C.; Gingeras, T.R.; Haerty, W.; et al. Considerations when Investigating lncRNA Function in Vivo. Elife 2014, 3, e03058. [Google Scholar] [CrossRef]
- Boon, R.A.; Jae, N.; Holdt, L.; Dimmeler, S. Long Noncoding RNAs: From Clinical Genetics to Therapeutic Targets? J. Am. Coll. Cardiol. 2016, 67, 1214–1226. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Process | lncRNA | miRNA | Prediction Tool | miRNA Target (Direct/Indirect) | Pathology/Condition | Ref. |
|---|---|---|---|---|---|---|
| Autophagy | APF | miR-188-3p | RNAhybrid | ATG7 (direct) | -A/R in mouse CMs -I/R (mouse) | [71] |
| AK088388 | miR-30a | miRANDA TargetScan | Beclin-1 (direct) LC3-II (indirect) | -H/R in mouse CMs | [72] | |
| AK139328 | miR-204-3p | nd | ATG proteins (indirect) | -Diabetic mouse -I/R (mouse) | [73] | |
| BACE1-AS | miR-29/miR-107/miR-124/miR-485/miR-761 | miRANDA | BACE1 (direct) | -human DCM -HAOECs -mouse cardiomyocytes | [51,74,75] | |
| Galont | miR-338 | RNAhybrid | ATG5 (direct) | A/R cardiomyocytes (mouse) | [76] | |
| GAS5 | miR-26a | RNAhybrid | ATG proteins (indirect) | -ox-LDL stimulation of human ECs -plasma of AS patients | [77] | |
| TGFB2-OT1 | miR-4459 | Mirbase MicroInspector | ATG13 (direct) | -3BDO stimulation of human ECs | [78] | |
| UCA1 | miR-128 | nd | HSP70 (direct) | -I/R (mouse) -H/R in mouse CMs | [79] | |
| Inflammation | GAS5 | miR-26a | Starbase v2.0 LncBase Predicted v.2 tool | HMGB1 | -PA treated mouse CMs | [80] |
| GAS5 | miR-221 | nd | IL-1β, TNF-α, MMP-2, MMP-9 (indirect) | -Ox/LDL stimulation of THP-1 human cells -Human AS plaques | [81] | |
| H19 | let-7 | nd | Periostin | -Ox/LDL stimulation of human HUVEC | [82] | |
| HOTAIR | miR-34a | miRANDA | SIRT1 (direct) | -STZ mice -high glucose treated rat CMs | [83] | |
| MALAT-1 | miR-155 | TargetScan | SOCS1 (direct) | - ox-LDL stimulation of human HAOECs | [84] | |
| RNCR3 | miR-185-5p | TargetScan | KLF-2 (direct) | -Human aortic lesions -Aorta from apoE−/− mice - ox-LDL stimulation of human EC and VSMCs | [85] | |
| RP5-833A20.1 | miR-382-5p | miRBase, PicTar, TargetScan, RNAhybrid | NFIA (direct) | -Ox/Ac-LDL stimulation of THP-1 human cells -Aorta from apoE−/− mice | [86] | |
| TGFB2-OT1 | miR-4459 | MicroInspector | LARP/CERS1/ NAT8L/ ATG13 (direct) | -LPS/ox-LDL stimulation of human ECs | [87] | |
| Senescence | GAS5 | miR-223 | miRWalk miRANDA TargetScan microT-CDS | NAMPT (direct) | -Human EPCs (late passages) | [88] |
| H19 | miR-29b-3p | nd | cIAP1 (direct) | -D-galactose/H-Post treatment of CMs -I/Post (mouse) | [89] | |
| LncRNA-ES3 | miR-34c-5p | nd | BMF | -high glucose stimulated human aorta VSMCs | [90] | |
| MEG3 | miR-128 | nd | GIRDIN (direct) | -Coronary artery aged mice -HUVECs (late passages) | [91] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Greco, S.; Gaetano, C.; Martelli, F. Long Noncoding Competing Endogenous RNA Networks in Age-Associated Cardiovascular Diseases. Int. J. Mol. Sci. 2019, 20, 3079. https://doi.org/10.3390/ijms20123079
Greco S, Gaetano C, Martelli F. Long Noncoding Competing Endogenous RNA Networks in Age-Associated Cardiovascular Diseases. International Journal of Molecular Sciences. 2019; 20(12):3079. https://doi.org/10.3390/ijms20123079
Chicago/Turabian StyleGreco, Simona, Carlo Gaetano, and Fabio Martelli. 2019. "Long Noncoding Competing Endogenous RNA Networks in Age-Associated Cardiovascular Diseases" International Journal of Molecular Sciences 20, no. 12: 3079. https://doi.org/10.3390/ijms20123079
APA StyleGreco, S., Gaetano, C., & Martelli, F. (2019). Long Noncoding Competing Endogenous RNA Networks in Age-Associated Cardiovascular Diseases. International Journal of Molecular Sciences, 20(12), 3079. https://doi.org/10.3390/ijms20123079