Novel Cyclic Lipopeptide Antibiotics: Effects of Acyl Chain Length and Position

 , , ,
, , ,  and
and
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Optimization of Synthesis
2.1.1. Composition of the Growth Media in MIC Determinations
2.1.2. Fatty Acid Positioning Affects the Antimicrobial Activity
2.1.3. The Influence of Cationic Residues in AMPs
2.1.4. The Influence of Fatty Acid Length
2.2. Antimicrobial Activity
2.2.1. Hemolytic Activity
2.2.2. (C8)5: The Most Promising Lipopeptide Analogue of S3(B)
2.2.3. Hemolytic Activity: (C8)5 Versus S3(B)
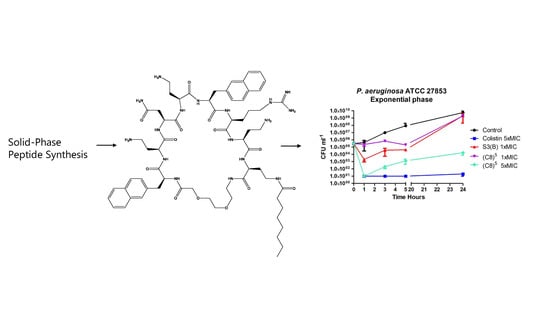
2.2.4. Time-Kill Experiment of (C8)5 Against P. aeruginosa
3. Materials and Methods
3.1. Chemistry
3.2. Peptides
3.3. Microbiology and Hemolysis
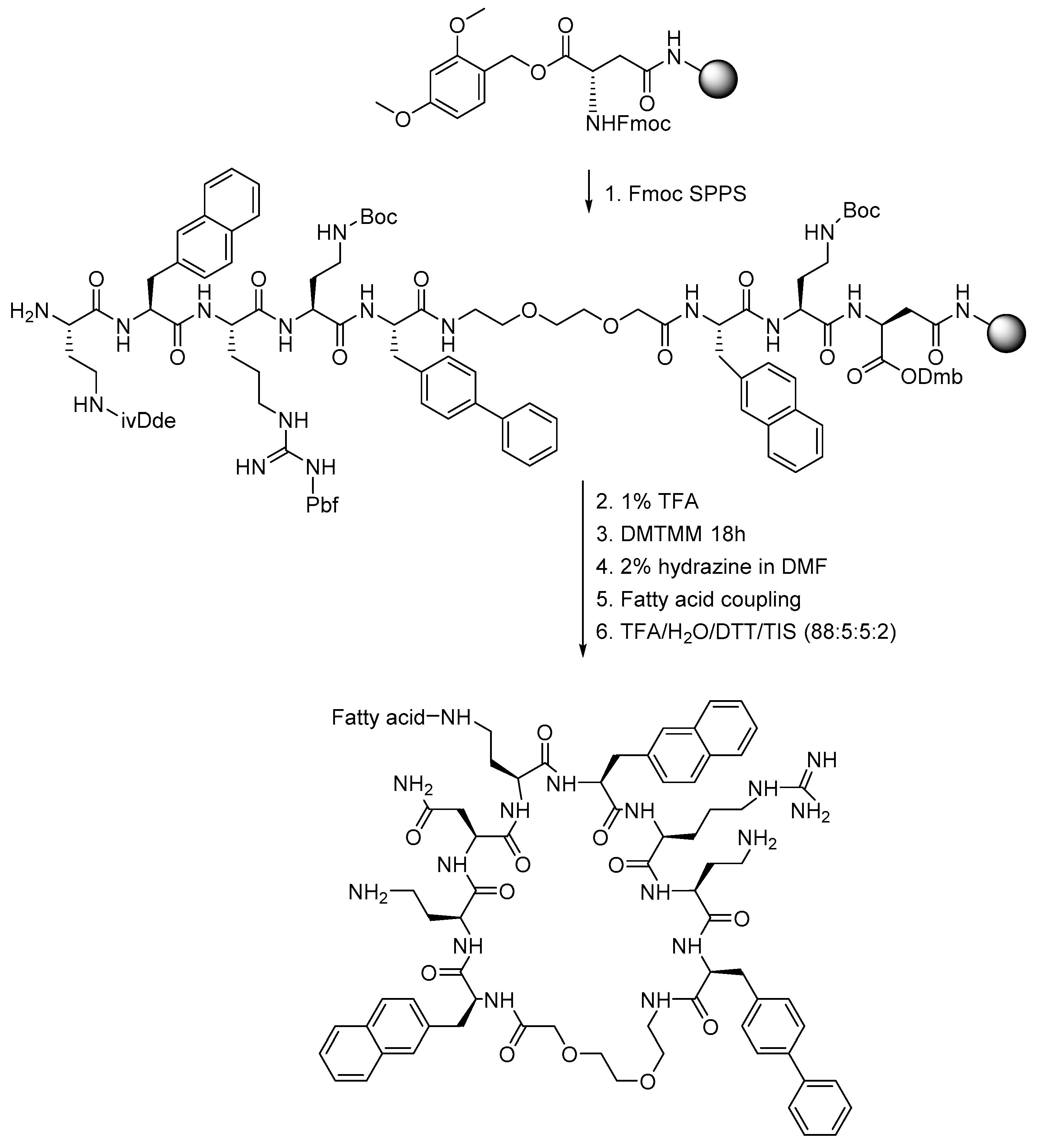
3.4. Peptide Synthesis
3.5. Fmoc Deprotection
3.6. Initiation and Preparation of Syringes
3.7. Coupling of Amino Acids
3.8. Peptide Macrocyclization
3.9. Fatty Acid Acylation
3.10. Peptide Cleavage
3.11. Minimum Inhibitory Concentration Determination
3.12. Hemolysis
3.13. Time-Kill Kinetics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ACN | Acetonitrile |
| Bip | l-biphenylalanine |
| BSA | Bovine Serum Albumin |
| CFU | Colony-forming unit |
| Dab | l-2,4-diaminobutyric acid |
| DCM | Dichloromethane |
| DIEA | Disopropylethylamine |
| Dmb | 2,4-Dimethoxybenzyl |
| DMF | Dimethylformamide |
| DMTMM.BF4 | 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium tetrafluoroborate |
| DTT | Dithiothreitol |
| EUCAST | European Committee on Antimicrobial Susceptibility Testing |
| Fmoc | 9-fluorenylmethoxycarbonyl |
| HA | Hemolytic activity |
| HATU | 1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium-3-oxid hexafluoro-phosphate, N-[(Dimethylamino)-1H-1,2,3-triazolo-[4,5-b]pyridin-1-ylmethylene]-N-methylmethan-aminium hexa-fluorophosphate N-oxide) |
| HOAt | 1-Hydroxy-7-azabenzotriazole |
| IvDde | 1-(4,4-dimethyl-2,6-dioxocyclohex-1-ylidene)-3-methylbutyl |
| LPS | Lipopolysaccharide |
| MALDI-TOF MS | Matrix-assisted linear desorption Time-Of-Flight Mass Spectrometry |
| MHB | Mueller-Hinton broth |
| MIC | Minimum Inhibitory Concentration |
| Nal | 3-(2-Naphthyl)-l-alanine |
| O2Oc | 8-amino-3,6-dioxaoctanoic acid |
| PBS | Phosphate-buffered saline |
| RAM | Rink amide |
| RBC | Red blood cell |
| RP-HPLC | Reverse Phase Analytical High Performance Liquid Chromatography |
| rpm | Rotations per minute |
| SPPS | Solid-Phase Peptide Synthesis |
| TFA | Trifluoroacetic acid |
| TIS | Triisopropylsilane |
References
- Theuretzbacher, U. Global antimicrobial resistance in Gram-negative pathogens and clinical need. Curr. Opin. Microbiol. 2017, 39, 106–112. [Google Scholar] [CrossRef]
- Mulani, M.S.; Kamble, E.E.; Kumkar, S.N.; Tawre, M.S.; Pardesi, K.R. Emerging Strategies to Combat ESKAPE Pathogens in the Era of Antimicrobial Resistance: A Review. Front. Microbiol. 2019, 10, 539. [Google Scholar] [CrossRef]
- WHO. Critically Important Antimicrobials for Human Medicine. Available online: https://www.who.int/foodsafety/publications/antimicrobials-sixth/en/ (accessed on 6 August 2020).
- Theuretzbacher, U.; Outterson, K.; Engel, A.; Karl?n, A. The global preclinical antibacterial pipeline. Nat. Rev. Microbiol. 2020, 18, 275–285. [Google Scholar] [CrossRef] [Green Version]
- Negash, K.H.; Norris, J.K.S.; Hodgkinson, J.T. Siderophore–Antibiotic Conjugate Design: New Drugs for Bad Bugs? Molecules 2019, 24, 3314. [Google Scholar] [CrossRef] [Green Version]
- Zasloff, M. Antimicrobial peptides of multicellular organisms. Nature 2002, 415, 389–395. [Google Scholar] [CrossRef]
- Mookherjee, N.; Anderson, M.A.; Haagsman, H.P.; Davidson, D.J. Antimicrobial host defence peptides: Functions and clinical potential. Nat. Rev. Drug Discov. 2020, 19, 311–332. [Google Scholar] [CrossRef]
- Haney, E.F.; Mansour, S.C.; Hancock, R.E.W. Antimicrobial Peptides: An Introduction. In Antimicrobial Peptides: Methods and Protocols; Hansen, P.R., Ed.; Springer: New York, NY, USA, 2017; pp. 3–22. [Google Scholar] [CrossRef]
- Pasupuleti, M.; Schmidtchen, A.; Malmsten, M. Antimicrobial peptides: Key components of the innate immune system. Crit. Rev. Biotechnol. 2012, 32, 143–171. [Google Scholar] [CrossRef] [Green Version]
- Epand, R.M.; Vogel, H.J. Diversity of antimicrobial peptides and their mechanisms of action. Biochim. Biophys. Acta 1999, 1462, 11–28. [Google Scholar] [CrossRef] [Green Version]
- Le, C.F.; Fang, C.M.; Sekaran, S.D. Intracellular Targeting Mechanisms by Antimicrobial Peptides. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef] [Green Version]
- Deris, Z.Z.; Akter, J.; Sivanesan, S.; Roberts, K.D.; Thompson, P.E.; Nation, R.L.; Li, J.; Velkov, T. A secondary mode of action of polymyxins against Gram-negative bacteria involves the inhibition of NADH-quinone oxidoreductase activity. J. Antibiot. (Tokyo) 2014, 67, 147–151. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.H.; Lu, T.K. Development and Challenges of Antimicrobial Peptides for Therapeutic Applications. Antibiotics 2020, 9, 24. [Google Scholar] [CrossRef] [Green Version]
- Molchanova, N.; Hansen, P.R.; Franzyk, H. Advances in Development of Antimicrobial Peptidomimetics as Potential Drugs. Molecules 2017, 22, 1430. [Google Scholar] [CrossRef] [Green Version]
- Zorzi, A.; Deyle, K.; Heinis, C. Cyclic peptide therapeutics: Past, present and future. Curr. Opin. Chem. Biol. 2017, 38, 24–29. [Google Scholar] [CrossRef] [Green Version]
- Falagas, M.E.; Kasiakou, S.K.; Saravolatz, L.D. Colistin: The Revival of Polymyxins for the Management of Multidrug-Resistant Gram-Negative Bacterial Infections. Clin. Infect. Dis. 2005, 40, 1333–1341. [Google Scholar] [CrossRef] [Green Version]
- Mishra, N.N.; Yang, S.J.; Chen, L.; Muller, C.; Saleh-Mghir, A.; Kuhn, S.; Peschel, A.; Yeaman, M.R.; Nast, C.C.; Kreiswirth, B.N.; et al. Emergence of Daptomycin Resistance in Daptomycin-Naive Rabbits with Methicillin-Resistant Staphylococcus aureus Prosthetic Joint Infection Is Associated with Resistance to Host Defense Cationic Peptides and mprF Polymorphisms. PLoS ONE 2013, 8, e71151. [Google Scholar] [CrossRef]
- Thomsen, T.T.; Mendel, H.C.; Al-Mansour, W.; Oddo, A.; Løbner-Olesen, A.; Hansen, P.R. Analogues of a cyclic antimicrobial peptide with a flexible linker show promising activity against Pseudomonas aeruginosa and Staphylococcus aureus. Antibiotics 2020, 9, 366. [Google Scholar] [CrossRef]
- Vaara, M. Polymyxins and Their Potential Next Generation as Therapeutic Antibiotics. Front. Microbiol. 2019, 10, 1689. [Google Scholar] [CrossRef]
- Velkov, T.; Roberts, K.D.; Nation, R.L.; Wang, J.; Thompson, P.E.; Li, J. Teaching ‘Old’ Polymyxins New Tricks: New-Generation Lipopeptides Targeting Gram-Negative ‘Superbugs’. ACS Chem. Biol. 2014, 9, 1172–1177. [Google Scholar] [CrossRef]
- Howard, K.T.; Chisholm, J.D. Preparation and Applications of 4-Methoxybenzyl Esters in Organic Synthesis. Org. Prep. Proced. Int. 2016, 48, 1–36. [Google Scholar] [CrossRef]
- Nielsen, S.L.; Frimodt-Moller, N.; Kragelund, B.B.; Hansen, P.R. Structure activity study of the antibacterial peptide fallaxin. Protein Sci. 2007, 16, 1969–1976. [Google Scholar] [CrossRef] [Green Version]
- Cleland, W.W. Dithiothreitol, a New Protective Reagent for SH Groups. Biochemistry 1964, 3, 480–482. [Google Scholar] [CrossRef]
- Wiegand, I.; Hilpert, K.; Hancock, R.E.W. Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat. Protoc. 2008, 3, 163–175. [Google Scholar] [CrossRef]
- Li, Y.Q.; Smith, C.; Wu, H.F.; Padhee, S.; Manoj, N.; Cardiello, J.; Qiao, Q.; Cao, C.H.; Yin, H.; Cai, J.F. Lipidated Cyclic gamma-AApeptides Display Both Antimicrobial and Anti-inflammatory Activity. ACS Chem. Biol. 2014, 9, 211–217. [Google Scholar] [CrossRef] [Green Version]
- Giangaspero, A.; Sandri, L.; Tossi, A. Amphipathic α helical antimicrobial peptides. Eur. J. Biochem. 2001, 268, 5589–5600. [Google Scholar] [CrossRef]
- Dathe, M.; Nikolenko, H.; Meyer, J.; Beyermann, M.; Bienert, M. Optimization of the antimicrobial activity of magainin peptides by modification of charge. FEBS Lett. 2001, 501, 146–150. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.X.; Guarnieri, M.T.; Vasil, A.I.; Vasil, M.L.; Mant, C.T.; Hodges, R.S. Role of peptide hydrophobicity in the mechanism of action of alpha-helical antimicrobial peptides. Antimicrob. Agents Chemother. 2007, 51, 1398–1406. [Google Scholar] [CrossRef] [Green Version]
- Chu-Kung, A.F.; Nguyen, R.; Bozzelli, K.N.; Tirrell, M. Chain length dependence of antimicrobial peptide–fatty acid conjugate activity. J. Colloid Interface Sci. 2010, 345, 160–167. [Google Scholar] [CrossRef]
- Epand, R.M. Biophysical studies of lipopeptide-membrane interactions. Biopolymers 1997, 43, 15–24. [Google Scholar] [CrossRef]
- Oddo, A.; Hansen, P.R. Hemolytic Activity of Antimicrobial Peptides. Methods Mol. Biol. 2017, 1548, 427–435. [Google Scholar] [CrossRef]
- SoftmaxPro; Molecular Devices: San Jose, CA, USA, 2020; Available online: https://www.moleculardevices.com/products/microplate-readers/acquisition-and-analysis-software/softmax-pro-software#gref (accessed on 6 August 2020).
- Oddo, A.; Thomsen, T.T.; Kjelstrup, S.; Gorey, C.; Franzyk, H.; Frimodt-Møller, N.; Løbner-Olesen, A.; Hansen, P.R. An all-D amphipathic undecapeptide shows promising activity against colistin-resistant strains of Acinetobacter baumannii and a dual mode of action. Antimicrob. Agents Chemother. 2016, 60, 592–599. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nr | ID | Peptide | EC a | SA b | AB c | PA d | KP e | %H f | %B g |
|---|---|---|---|---|---|---|---|---|---|
| S3(B) | c(Dab-Nal-Arg-Dab-Bip-O2Oc-Nal-Dab-Asn) | 16 | 8 | 16 | 8 | >64 | 77 | 71 | |
| 1 | (C10)1 | c(Dab(C10)-Nal-Arg-Dab-Bip-O2Oc-Nal-Dab-Asn) | >64 | 8 | 8 | 64 | 64 | 100 | 84 |
| 2 | (C10)2 | c(Dab-Dab(C10)-Arg-Dab-Bip-O2Oc-Nal-Dab-Asn) | 32 | 4 | 16 | 8 | >64 | 100 | 76 |
| 3 | (C12)2 | c(Dab-Dab(C12)-Arg-Dab-Bip-O2Oc-Nal-Dab-Asn) | 64 | 16 | 64 | 32 | >64 | 100 | 79 |
| 4 | (C14)2 | c(Dab-Dab(C14)-Arg-Dab-Bip-O2Oc-Nal-Dab-Asn) | >64 | 64 | >64 | >64 | >64 | 100 | 84 |
| 5 | (C10)4 | c(Dab-Nal-Arg-Dab(C10)-Bip-O2Oc-Nal-Dab-Asn) | 64 | 8 | 16 | >64 | 64 | 100 | 85 |
| 6 | (C4)5 | c(Dab-Nal-Arg-Dab-Dab(C4)-O2Oc-Nal-Dab-Asn) | 64 | >64 | >64 | 32 | >64 | 5 | 62 |
| 7 | (C6)5 | c(Dab-Nal-Arg-Dab-Dab(C6)-O2Oc-Nal-Dab-Asn) | 32 | 32 | 64 | 16 | >64 | 13 | 65 |
| 8 | (C8)5 | c(Dab-Nal-Arg-Dab-Dab(C8)-O2Oc-Nal-Dab-Asn) | 16 | 8 | 16 | 8 | >64 | 42 | 69 |
| 9 | (C10)5 | c(Dab-Nal-Arg-Dab-Dab(C10)-O2Oc-Nal-Dab-Asn) | 16 | 4 | 8 | 4 | >64 | 100 | 74 |
| 10 | (C12)5 | c(Dab-Nal-Arg-Dab-Dab(C12)-O2Oc-Nal-Dab-Asn) | 32 | 4 | 16 | 8 | >64 | 100 | 78 |
| 11 | (C14)5 | c(Dab-Nal-Arg-Dab-Dab(C14)-O2Oc-Nal-Dab-Asn) | 32 | 16 | >64 | 64 | >64 | 100 | 83 |
| 12 | (C4)7 | c(Dab-Nal-Arg-Dab-Bip-O2Oc-Dab(C4)-Dab-Asn) | 64 | 64 | 64 | 64 | >64 | 4 | 64 |
| 13 | (C6)7 | c(Dab-Nal-Arg-Dab-Bip-O2Oc-Dab(C6)-Dab-Asn) | 32 | 16 | 64 | 16 | >64 | 8 | 69 |
| 14 | (C8)7 | c(Dab-Nal-Arg-Dab-Bip-O2Oc-Dab(C8)-Dab-Asn) | 16 | 8 | 16 | 8 | >64 | 56 | 71 |
| 15 | (C10)7 | c(Dab-Nal-Arg-Dab-Bip-O2Oc-Dab(C10)-Dab-Asn) | 16 | 4 | 8 | 8 | >64 | 100 | 74 |
| 16 | (C12)7 | c(Dab-Nal-Arg-Dab-Bip-O2Oc-Dab(C12)-Dab-Asn) | 16 | 4 | 16 | 8 | >64 | 96 | 79 |
| 17 | (C14)7 | c(Dab-Nal-Arg-Dab-Bip-O2Oc-Dab(C14)-Dab-Asn) | 64 | 16 | >64 | 64 | >64 | 98 | 84 |
| 18 | (C10)8 | c(Dab-Nal-Arg-Dab-Bip-O2Oc-Nal-Dab(C10)-Asn) | 64 | 4 | 16 | >64 | >64 | 100 | 84 |
| 19 | Ref | Colistin | 0.25 | N/A | 0.25 | 0.25 | 0.5 | N/A | N/A |
| 20 | Ref | Vancomycin | N/A | 0.5 | N/A | N/A | N/A | N/A | N/A |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jensen, S.K.; Thomsen, T.T.; Oddo, A.; Franzyk, H.; Løbner-Olesen, A.; Hansen, P.R. Novel Cyclic Lipopeptide Antibiotics: Effects of Acyl Chain Length and Position. Int. J. Mol. Sci. 2020, 21, 5829. https://doi.org/10.3390/ijms21165829
Jensen SK, Thomsen TT, Oddo A, Franzyk H, Løbner-Olesen A, Hansen PR. Novel Cyclic Lipopeptide Antibiotics: Effects of Acyl Chain Length and Position. International Journal of Molecular Sciences. 2020; 21(16):5829. https://doi.org/10.3390/ijms21165829
Chicago/Turabian StyleJensen, Signe Kaustrup, Thomas T. Thomsen, Alberto Oddo, Henrik Franzyk, Anders Løbner-Olesen, and Paul R. Hansen. 2020. "Novel Cyclic Lipopeptide Antibiotics: Effects of Acyl Chain Length and Position" International Journal of Molecular Sciences 21, no. 16: 5829. https://doi.org/10.3390/ijms21165829
APA StyleJensen, S. K., Thomsen, T. T., Oddo, A., Franzyk, H., Løbner-Olesen, A., & Hansen, P. R. (2020). Novel Cyclic Lipopeptide Antibiotics: Effects of Acyl Chain Length and Position. International Journal of Molecular Sciences, 21(16), 5829. https://doi.org/10.3390/ijms21165829