Proton Pump Inhibitor Use, Hypergastrinemia, and Gastric Carcinoids—What Is the Relationship?

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Observations in Humans
3. Evidence of Causation
4. Summary
Conflicts of Interest
Appendix A. Gastric Carcinoids Associated with the Use of PPI: Summary of Features of Reported Cases
References
- Merchant, S.H.; VanderJagt, T.; Lathorp, S.; Amin, M.B. Sporadic duodenal bulb gastrin-cell tumors: Association with Helicobacter pylori gastritis and Long-term use of proton pump inhibitors. Am. J. Surg. Pathol. 2006, 30, 1581–1587. [Google Scholar] [CrossRef] [PubMed]
- Sunderasan, S.; Meininger, C.A.; Kang, A.J.; Photenhauer, A.L.; Hayes, M.M.; Sahoo, N.; Grembecka, J.; Cierpicki, T.; Ding, T.; Giordano, T.J.; et al. Gastrin induces nuclear export and proteasome degradation of Menin in enteric glial cells. Gastroenterology 2017, 153, 1555–1567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearse, A.G. The cytochemistry and ultrastructure of polypeptide hormone-producing cells of the APUD series and the embryologic, physiologic and pathologic implications of the concept. J. Histochem. Cytochem. 1969, 17, 303–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dasari, A.; Shen, C.; Halperin, D.; Zhao, B.; Zhou, S.; Xu, Y.; Shih, T.; Yao, J.C. Trends in the incidence, prevalence and survival outcomes in patients with Neuroendocrine tumors in the United States. JAMA Oncol. 2017, 3, 1335–1342. [Google Scholar] [CrossRef]
- Ellis, L.; Shale, M.J.; Coleman, M.P. Carcinoid tumors in the gastrointestinal tract: Trends in incidence in England since 1971. Am. J. Gastroenterol. 2010, 105, 2563–2569. [Google Scholar] [CrossRef]
- Modlin, I.M.; Kidd, M.; Latich, I.; Zikusoka, M.N.; Shapiro, M.D. Current status of gastrointestinak carcinoids. Gastroenterology 2005, 128, 1717–1751. [Google Scholar] [CrossRef]
- Scherubl, H.; Cadiot, G.; Jensen, R.T.; Rosch, T.; Stölzel, U.; Klöppel, G. Neuroendocrine tumors of the stomach (gastric carcinoids) are on the rise: Small tumors, small problems? Endoscopy 2010, 42, 664–671. [Google Scholar] [CrossRef]
- Jensen, R.T.; Norton, J.A.; Oberg, K. Neuroendocrine Tumors. In Sleisenger and Fordtran’s Textbook of Gastroenterology and Liver Disease, 10th ed.; Feldman, L.S., Brandt, L.J., Eds.; Elsevier: Philladelphia, PA, USA, 2016; pp. 501–541. [Google Scholar]
- Rimdi, G.; Arnold, R.; Bosman, F.T.; Capella, C.; Bosman, F.T. WHO Classification of Tumours of the Digestive System; IARC Press: Lyon, France, 2010; pp. 13–14. [Google Scholar]
- Tang, L.H.; Untch, B.R.; Reidy, D.L.; O’Reilly, E. Well differentiated neuroendocrine tumors with a morphologically high grade component: A pathway distinct from poorly differentiated neuroendocrine carcinoma. Clin. Cancer Res. 2016, 22, 1011–1017. [Google Scholar] [CrossRef] [Green Version]
- Lee, L.; Ramos-Alvarez, I.; Ito, T.; Jensen, R.T. Insights into effects/risks of chronic hypergastrinemia and lifelong PPI treatment in man based on studies of patients with Zollinger-Ellison Syndrome. Int. J. Mol. Sci. 2019, 20, 5128. [Google Scholar] [CrossRef] [Green Version]
- Nandy, N.; Hanson, J.A.; Strickland, R.S.; McCarthy, D.M. Solitary gastric carcinoid associated with longterm use of omeprazole: A case report and review of the literature. Dig. Dis. Sci. 2016, 61, 708–712. [Google Scholar] [CrossRef]
- Kidd, M.; Gustafsson, B.; Modlin, I.M. Gastric Carcinoids (Neuroendocrine Neoplasms). Gastroenterol. Clin. N. Am. 2013, 42, 381–397. [Google Scholar] [CrossRef] [PubMed]
- Fossmark, R.; Sordal, O.; Jianu, C.S.; Qvigstad, G. Treatment of gastric carcinoid type-1with the gastrin receptor antagonist netazeoide (YF476) results in regression of tumors and normalization of serum chromogranin A. Aliment. Pharmacol. Ther. 2012, 36, 1067–1075. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, D.M. Commentary: A gastrin antagonist against carcinoids - implications for PPI-induced hypergastrinemia. Aliment. Pharmacol. Ther. 2013, 37, 276–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crystal, D.; Kamilaris, C.; Startakis, C.A. Multiple Endocrine Neoplasia Type 1 (MEN-1): An update and the significance of early genetic and clinical diagnosis. Front. Endocrinol. 2019, 10, 339–367. [Google Scholar] [CrossRef]
- Veniaminova, N.A.; Hayes, M.M.; Varrney, J.M.; Merchant, J.L. Conditional deletion of Menin results in antral G-cell hyperplasia and hypergastrinemia. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 303, G752–G764. [Google Scholar] [CrossRef] [Green Version]
- Anlauf, M.; Perren, A.; Henopp, T.; Rudolph, T. Allelic deletion of the MEN 1 gene in duodenal gastrin and somatostatin cell neoplasms and their precursor lesions. Gut 2016, 56, 637–644. [Google Scholar] [CrossRef] [Green Version]
- Cives, M.; Strosberg, J.R. Gastroenteropancreatic Neuroendocrine Tumors. CA A Cancer J. Clin. 2018, 68, 471–487. [Google Scholar] [CrossRef]
- Richards, M.L.; Gauger, P.; Thompson, N.W.; Giordano, T.J. Regression of type11 gastric carcinoids in Multiple Endocrine Neoplasia Type1patients with Zollinger-Ellison Syndrome after surgical excision of all gastrinomas. World J. Surg. 2004, 28, 652–658. [Google Scholar] [CrossRef]
- Kinoshita, Y.; Ishihara, S.; Kadowaki, Y.; Fukui, H. Reg Protein is a unique growth factor of gastric mucosal cells. J. Gastronterol. 2004, 39, 507–513. [Google Scholar] [CrossRef]
- Waldum, H.L.; Sordal, O.F.; Mjones, P.G. The enterochromaffin-like [ECL] cell—Central in Gastric physiology and pathology. Int. J. Mol. Sci. 2019, 20, 2444. [Google Scholar] [CrossRef] [Green Version]
- Fossmark, R.; Martinsen, T.C.; Waldum, H.L. Adverse effects of proton pump inhibitors—Evidence and plausibility. Int. J. Mol. Sci. 2019, 20, 5203. [Google Scholar] [CrossRef] [Green Version]
- Calvete, O.; Reyes, J.; Zuniga, S.; Paumard-Hernandez, B. Exome sequencing identifies ATP4A gene as responsible for an atypical familial type 1 gastric neuroendocrine tumor. Hum. Mol. Genet. 2015, 24, 2914–2922. [Google Scholar] [CrossRef] [PubMed]
- Fossmark, R.; Calvete, O.; Mjones, P.; Benitez, J.; Waldum, H.L. ECL-cell carcinoids and carcinoma in patients homozygous for an inactivating mutation in the gastric H(+) K(+) ATPASE alpha subunit. APMIS 2016, 124, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Ooji, A.; Ota, M.; Katsuda, S.; Nakanishi, I. An unusual case of multiple gastric carcinoids associated with diffuse endocrine cell hyperplasia and parietal hypertrophy. Endocr. Pathol. 1995, 6, 229–237. [Google Scholar]
- Shiotani, A.; Katsumata, R.; Gouda, K.; Fukushima, S. Hypergastrinemia in long-term use of proton pump inhibitors. Digestion 2018, 97, 154–162. [Google Scholar] [CrossRef] [PubMed]
- Helgadottir, H.; Metz, D.C.; Yang, Y.X.; Rhim, A.D.; Bjornsson, E.S. The effects of long-term therapy with proton pump inhibitors on meal stimulated gastrin. Dig. Liver Dis. 2014, 46, 125–130. [Google Scholar] [CrossRef]
- Bakke, I.; Qvigstad, G.; Brenna, E.; Sandvik, A.; Waldum, H.L. Gastrin has a specific proliferative effect on the rat entrochromaffin-like cell, but not on the parietal cell: A study by elutriation centrifugation. Acta Physiol. Scand. 2000, 169, 29–37. [Google Scholar] [CrossRef]
- Brenna, E.; Waldum, H.L. Trophic Effect of gastrin on the enterochromaffin like cells of the rat stomach: Establishment of a dose response relationship. Gut 1992, 33, 1303–1306. [Google Scholar] [CrossRef] [Green Version]
- Lundell, L.; Vieth, M.; Gibson, F.; Nagy, P.; Kahrilas, P.J. Systematic Review: The effects of long-term proton pump inhibitor use on serum gastrin levels and gastric histology. Aliment. Pharmacol. Ther. 2015, 42, 649–663. [Google Scholar] [CrossRef]
- Gilligan, C.; Lawton, G.; Tang, L.; West, A. Gastric carcinoid tumors: The biology and therapy of an enigmatic and controversial lesion. Am. J. Gastroenterol. 1995, 90, 330–352. [Google Scholar]
- Kidd, M.; Bodei, L.; Modlin, I.M. Chromogranin A: Any relevance in neuroendocrine tumors? Curr. Opin. Endocrinol. Diabetes Obes. 2016, 23, 28–37. [Google Scholar] [CrossRef] [PubMed]
- AL-Risi, E.S.; AL-Essry, F.S.; Mula-Abed, W.S. Chromogranin A as a biomedical marker for neuroendocrine tumors: A single center experience at Royal Hospital, Oman. Oman Med. J. 2017, 32, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Haga, Y.; Nakatsura, T.; Shibata, Y.; Samashima, H. Human Gastric carcinoid detected during long-term anti-ulcer therapy of H2 receptor antagonist and proton pump inhibitor. Dig Dis Sci. 1998, 43, 253. [Google Scholar] [CrossRef]
- Dawson, R.; Manson, J.M. Omeprazole in esophageal reflux disease. Lancet 2000, 356, 1770–1771. [Google Scholar] [CrossRef]
- Attila, T.; Santharam, R.J.; Blom, D.; Komorowski, R. Multifocal gastric carcinoid tumor in patient with pernicious anemia receiving Lansoprazole. Dig. Dis. Sci. 2005, 50, 509–513. [Google Scholar] [CrossRef]
- Jianu, C.S.; Lange, O.J.; Viset, T.; Qvigstad, G. Gastric neuroendocrine carcinoma after long-term use of proton pump inhibitor. Scand. J. Gastroenterol. 2012, 47, 64–67. [Google Scholar] [CrossRef]
- Jianu, C.S.; Fossmark, R.; Viset, T.; Qvigstad, G. Gastric carcinoids after long-term use of a proton pump inhibitor. Aliment. Pharmacol. Ther. 2012, 36, 644–649. [Google Scholar] [CrossRef]
- Lahner, E.; Pilozzi, E.; Esposito, G.; Galli, G. Gastric carcinoid in the absence of atrophic body gastritis and with low Ki-67 index: A clinical challenge. Scand. J. Gastroenterol. 2014, 49, 506–510. [Google Scholar] [CrossRef]
- Cavalcoli, F.; Zilli, A.; Ciafardini, F.; Massironi, S. Gastric neuroendocrine neoplasms and proton pump inhibitors: Fact orco-incidence? Scand. J. Gastroerol. 2014, 50, 1397–1403. [Google Scholar]
- Sanduleanu, S.; Jonkers, D.; de Bruine, A.; Hameeteman, W. Changes in Gastric mucosa and luminal environment during acid-suppressive therapy: A review in depth. Dig. Liver Dis. 2001, 33, 707–718. [Google Scholar] [CrossRef]
- Kuipers, E.J. Proton Pump Inhibitors and gastric neoplasia. Gut 2006, 55, 1217–1221. [Google Scholar] [CrossRef] [Green Version]
- Ma, C.; Shaheen, A.A.; Congley, S.E.; Andrews, C.N. Interpreting reported risks associated with use of proton pump inhibitors: Residual confounding in a 10-year analysis of national ambulatory data. Gastroenterology 2019. [Google Scholar] [CrossRef] [PubMed]
- Food and Drug Administration. Hypergastrinemia, ECL-cell Hyperplasia and Atrophic Gastritis. FDA Background. Omeprazole magnesium (Prilosec TM) Astra Zeneca with Proctor and Gamble. In Proceedings of the Non-Prescription Drugs Advisory Committee and the Gastrointestinal Drugs Advisory Committee, Gaithersburg, MD, USA, 20 October 2000; Available online: https://wayback.archive-it.org/7993/20170403184623/https://www.fda.gov/ohrms/dockets/ac/00mtbc.htm (accessed on 19 January 2020).
- Hodgson, N.; Koniaris, L.G.; Livingstone, A.S.; Franceschi, D. Gastric Carcinoids. Surg. Endosc. 2005, 19, 1610–1612. [Google Scholar] [CrossRef] [PubMed]
- Maggard, M.A.; O’Connell, J.B.; Ko, C.Y. Updated population-based review of carcinoid tumors. Ann. Surg. 2004, 240, 117–122. [Google Scholar] [CrossRef] [PubMed]
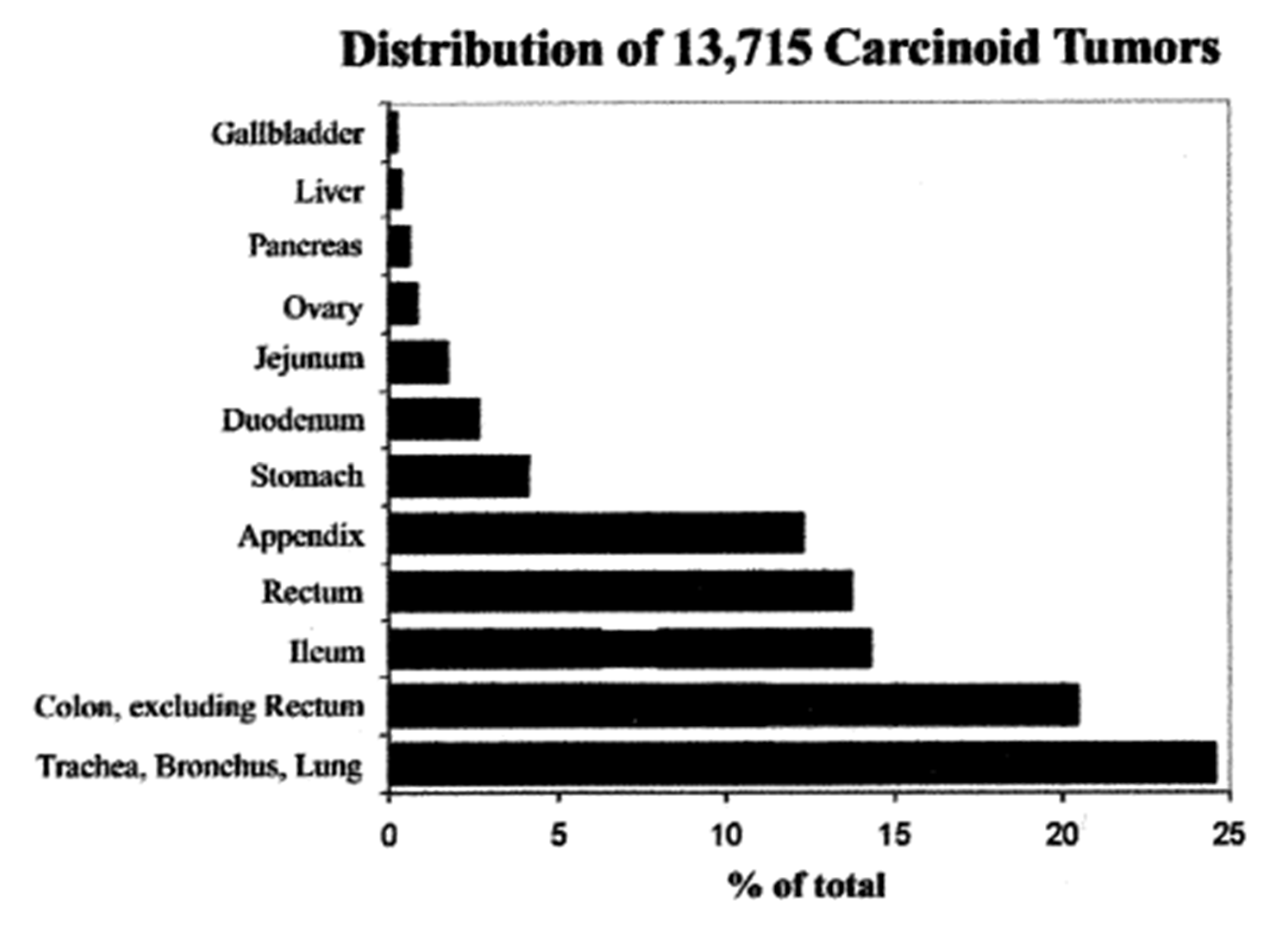
- Modlin, I.M.; Lye, K.D.; Kidd, M. A 5-decade analysis of 13,715 carcinoid tumors. Cancer 2003, 97, 934–950. [Google Scholar] [CrossRef] [PubMed]
- Waldum, H.L.; Oberg, K.; Sandvik, A.K.; Gustafsson, B.I. Not only stem cells, but also mature cells, particularly neuroendocrine cells, may develop into tumors: Time for a paradigm shift. Ther. Adv. Gastroenterol. 2018. [Google Scholar] [CrossRef]
- Cheung, K.S.; Leung, W.K. long-term use of proton pump inhibitors and risk of gastric cancer: A review of the current evidence. Ther. Adv. Gastroenterol. 2019, 2, 1–11. [Google Scholar] [CrossRef]
- Waldum, H.L.; Rehfeld, J.F. Gastric cancer and gastrin: On the interaction of Helicobacter gastritis and acid-inhibitory induced hypergastrinemia. Scand. J. Gastroenterol. 2019, 54, 1118–1123. [Google Scholar] [CrossRef]
- Tran-Duy, A.; Spaetgens, B.; Hoes, A.W.; de Wit, N.J.; Stehouwer, C.D. Use of proton pump inhibitors and risk of fundic gland polyps and gastric cancer: Systematic review and meta-analysis. Clin. Gastroenterol. Hepatol. 2016, 14, 1706–1719. [Google Scholar] [CrossRef] [Green Version]
- Brusselaers, N.; Wahlin, K.; Engstrand, L.; Lagergren, J. Maintenance therapy with proton pump inhibitors and risk of gastric cancer: A nationwide population-based cohort study in Sweden. BMJ Open 2017, e017739. [Google Scholar] [CrossRef] [Green Version]


© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
McCarthy, D.M. Proton Pump Inhibitor Use, Hypergastrinemia, and Gastric Carcinoids—What Is the Relationship? Int. J. Mol. Sci. 2020, 21, 662. https://doi.org/10.3390/ijms21020662
McCarthy DM. Proton Pump Inhibitor Use, Hypergastrinemia, and Gastric Carcinoids—What Is the Relationship? International Journal of Molecular Sciences. 2020; 21(2):662. https://doi.org/10.3390/ijms21020662
Chicago/Turabian StyleMcCarthy, Denis M. 2020. "Proton Pump Inhibitor Use, Hypergastrinemia, and Gastric Carcinoids—What Is the Relationship?" International Journal of Molecular Sciences 21, no. 2: 662. https://doi.org/10.3390/ijms21020662
APA StyleMcCarthy, D. M. (2020). Proton Pump Inhibitor Use, Hypergastrinemia, and Gastric Carcinoids—What Is the Relationship? International Journal of Molecular Sciences, 21(2), 662. https://doi.org/10.3390/ijms21020662