Recombinant Bacillus caldovelox Arginase Mutant (BCA-M) Induces Apoptosis, Autophagy, Cell Cycle Arrest and Growth Inhibition in Human Cervical Cancer Cells

 and
and
Abstract
:1. Introduction
2. Results
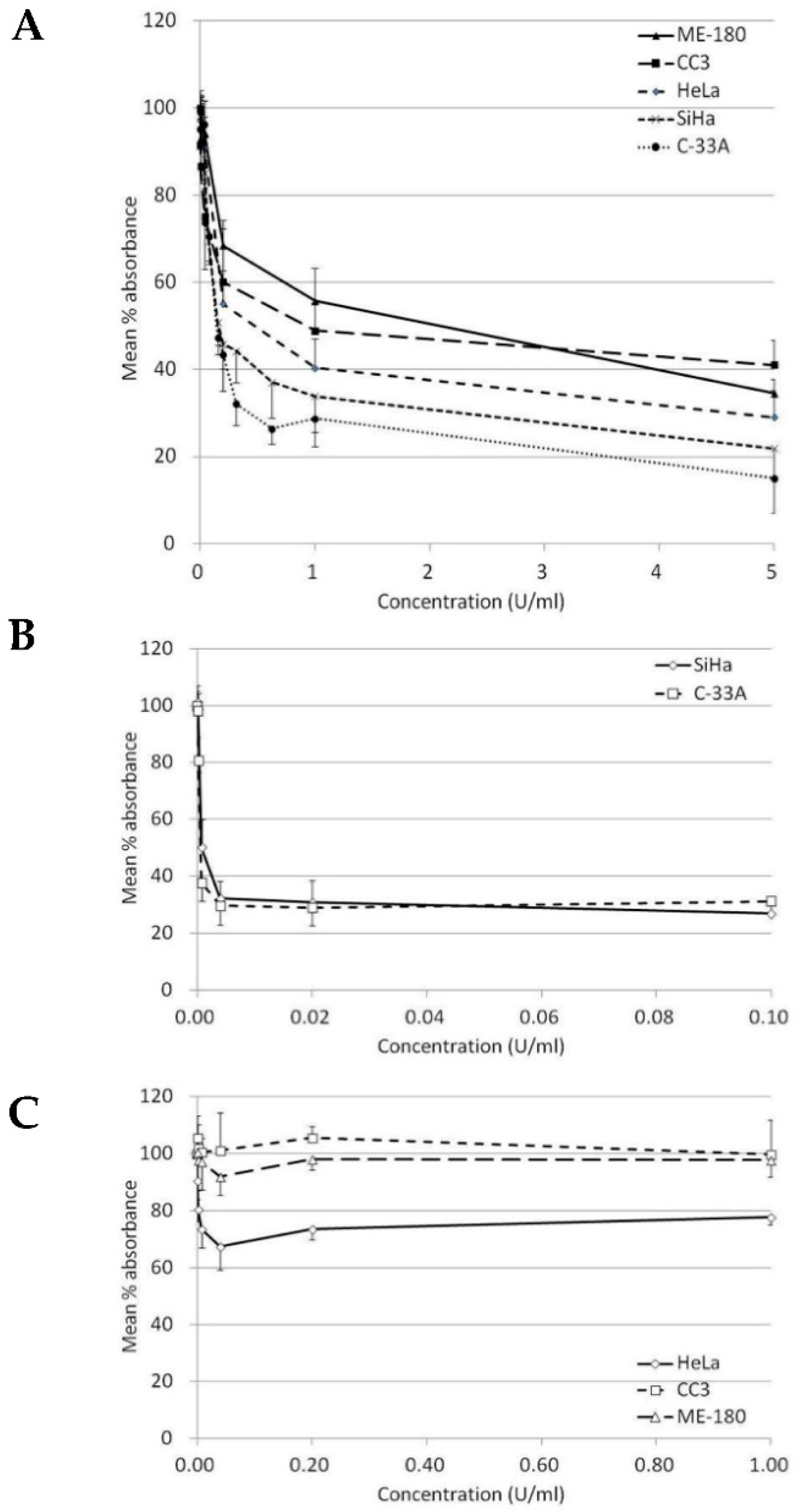
2.1. Recombinant Bacillus Arginase Mutant (BCA-M) Suppressed the Growth of Human Cervical Cancer Cells
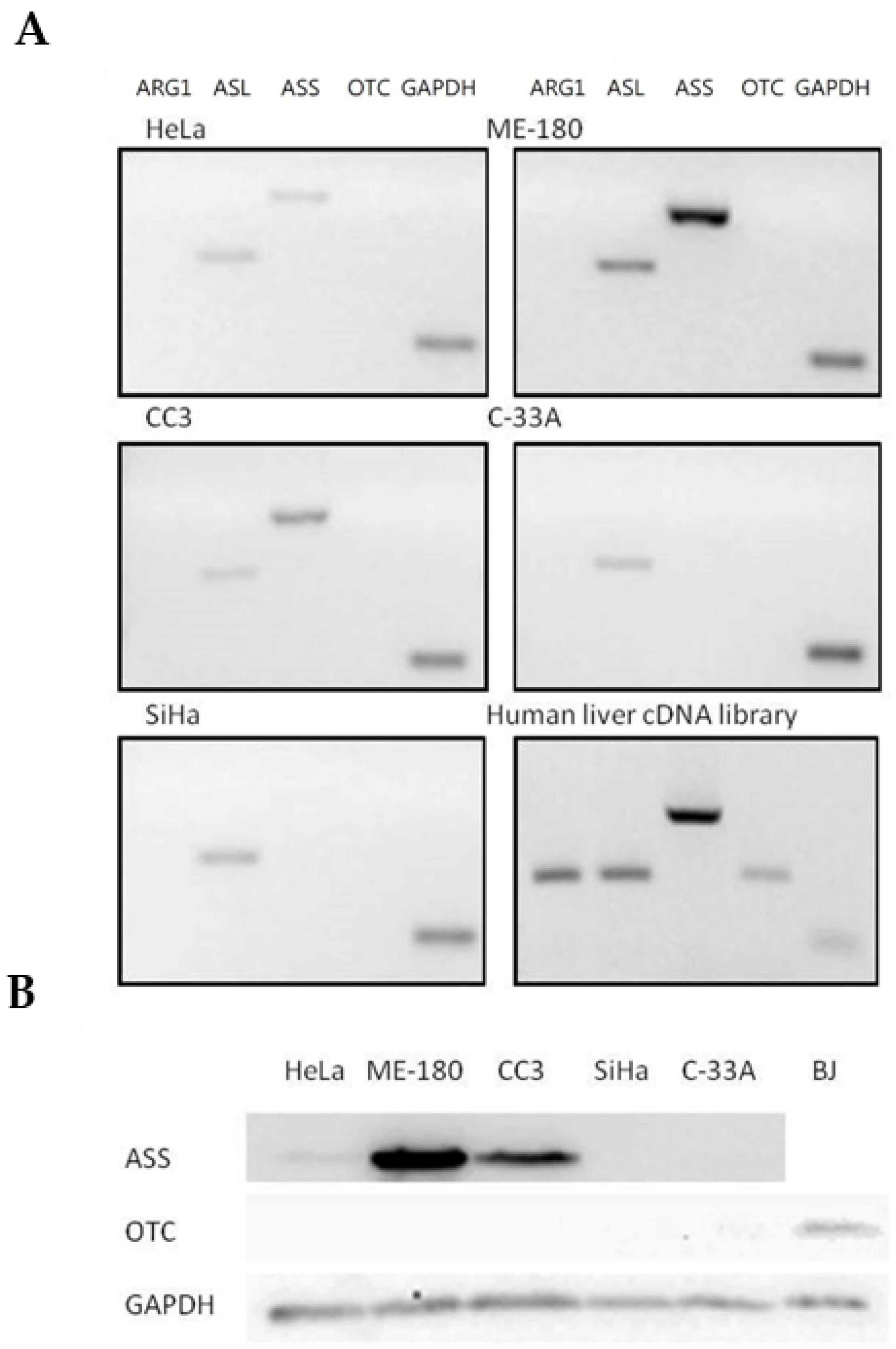
2.2. Expression Profiles of Major Urea Cycle Genes in Human Cervical Cancer Cells
2.3. Over-Expression of OTC Conferred Significant Resistance Towards the Growth-Suppressive Effect of BCA-M on HeLa Cells
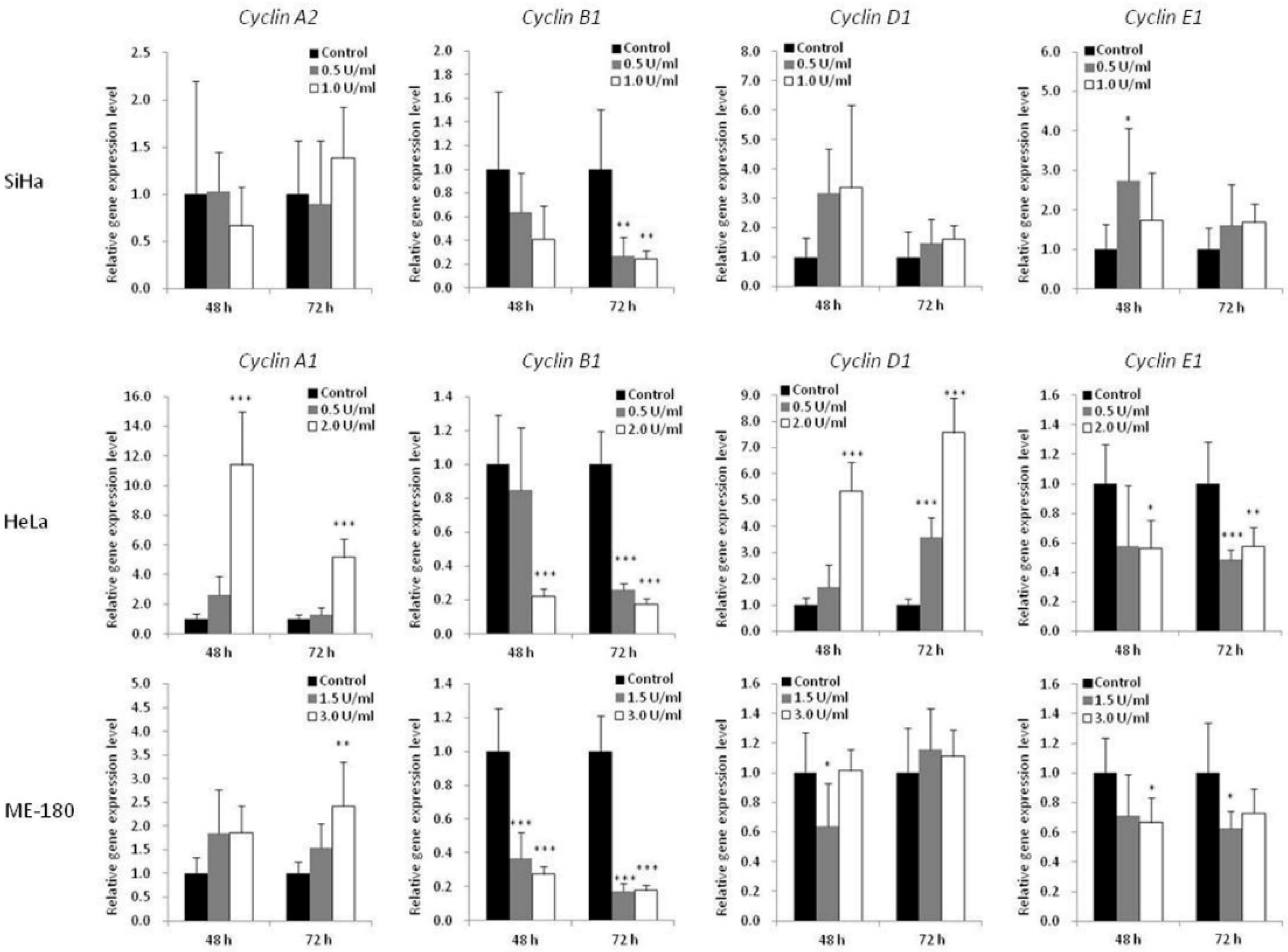
2.4. BCA-M-Induced Cell Cycle Arrest in Human Cervical Cancer Cells
2.5. BCA-M Induces Apoptosis in Human Cervical Cancer Cells
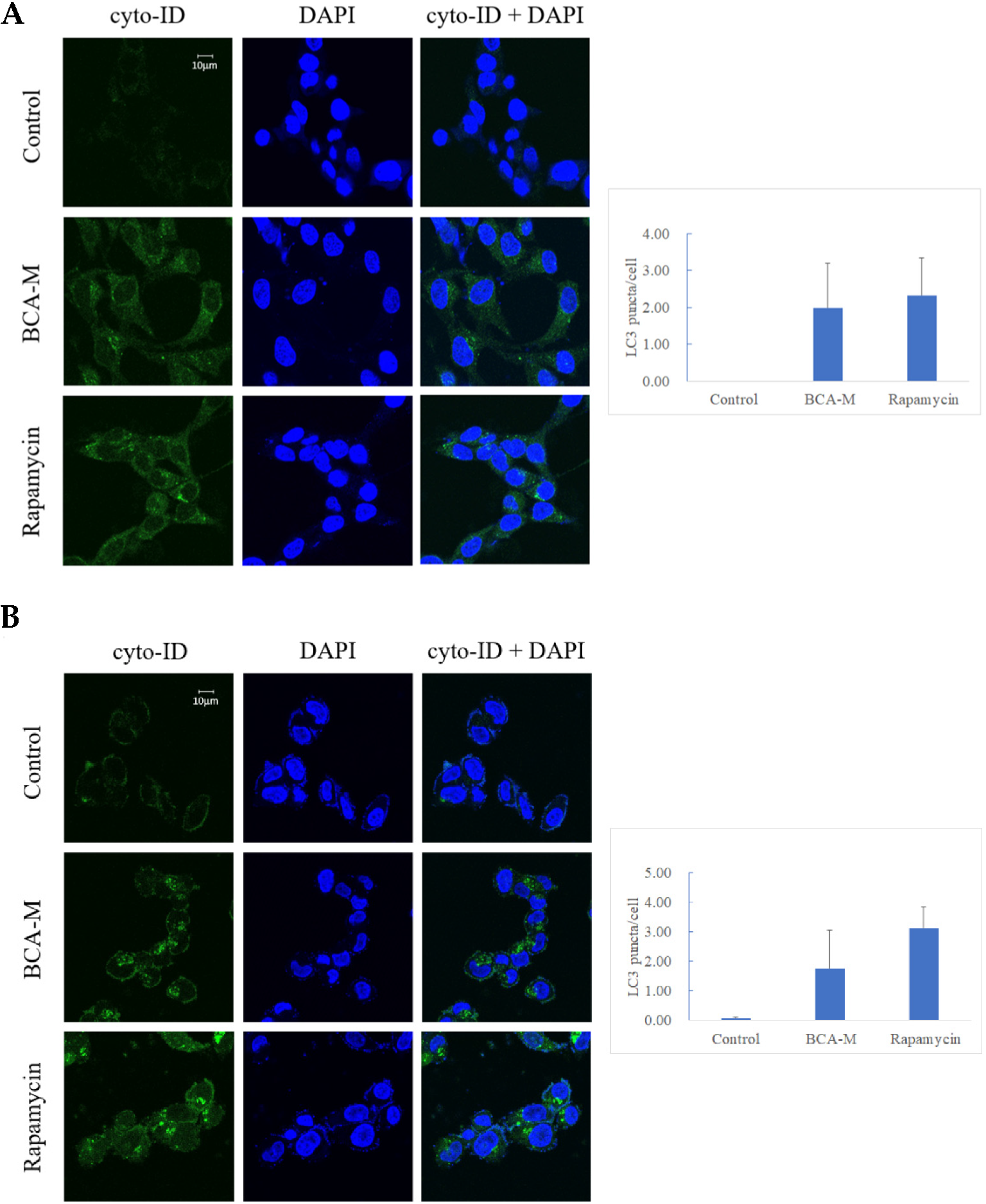
2.6. BCA-M-Induced Autophagy in HeLa and SiHa Cells and the Growth Inhibitory Effects Was Enhanced Synergistically by Its Combination to CQ
3. Discussion
4. Materials and Methods
4.1. Reagents and Materials
4.2. Preparation of Drugs
4.3. Cell Culture
4.4. Cell Proliferation Assay
4.5. Reverse Transcription-Polymerase Chain Reaction (RT-PCR) Analysis
4.6. Western Blotting Analysis
4.7. Cell Cycle Analysis
4.8. Quantitative Real-Time RT-PCR Analysis
4.9. Apoptosis Induction Analysis
4.10. Autophagy Detection
4.11. Generation of Recombinant Adenoviruses for OTC Rescue
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ferenczy, A.; Franco, E. Persistent human papillomavirus infection and cervical neoplasia. Lancet Oncol. 2002, 3, 11–16. [Google Scholar] [CrossRef]
- Ferlay, J.; Soerjomataram, I.; Ervik, M.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. GLOBOCAN 2012 v1.0, Cancer Incidence and Mortality Worldwide: IARC CancerBase No. 11. Available online: http://globocan.iarc.fr (accessed on 17 February 2020).
- Cheng, P.N.; Lam, T.L.; Lam, W.M.; Tsui, S.M.; Cheng, A.W.; Lo, W.H.; Leung, Y.C. Pegylated recombinant human arginase (rhArg-peg5000mw) inhibits the in vitro and in vivo proliferation of human hepatocellular carcinoma through arginine depletion. Cancer Res. 2007, 67, 309–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, T.L.; Wong, G.K.; Chong, H.C.; Cheng, P.N.; Choi, S.C.; Chow, T.L.; Kwok, S.Y.; Poon, R.T.; Wheatley, D.N.; Lo, W.H.; et al. Recombinant human arginase inhibits proliferation of human hepatocellular carcinoma by inducing cell cycle arrest. Cancer Lett. 2009, 277, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Lam, T.L.; Wong, G.K.; Chow, H.Y.; Chong, H.C.; Chow, T.L.; Kwok, S.Y.; Cheng, P.N.; Wheatley, D.N.; Lo, W.H.; Leung, Y.C. Recombinant human arginase inhibits the in vitro and in vivo proliferation of human melanoma by inducing cell cycle arrest and apoptosis. Pigment Cell Melanoma Res. 2011, 24, 366–376. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.F.; Kim, C.F.; Tam, S.Y.; Choi, M.C.; So, P.K.; Wong, K.Y.; Leung, Y.C.; Lo, W.H. A bioengineered arginine-depleting enzyme as a long-lasting therapeutic agent against cancer. Appl. Microbiol. Biotechnol. 2020, 104, 3921–3934. [Google Scholar] [CrossRef] [PubMed]
- Alexandrou, C.; Al-Aqbi, S.S.; Higgins, J.A.; Boyle, W.; Karmokar, A.; Andreadi, C.; Luo, J.L.; Moore, D.A.; Viskaduraki, M.; Blades, M.; et al. Sensitivity of colorectal cancer to arginine deprivation therapy is shaped by differential expression of urea cycle enzymes. Sci. Rep. 2018, 8, 12096. [Google Scholar] [CrossRef] [PubMed]
- De Santo, C.; Cheng, P.; Beggs, A.; Egan, S.; Bessudo, A.; Mussai, F. Metabolic therapy with PEG-arginase induces a sustained complete remission in immunotherapy-resistant melanoma. J. Hematol. Oncol. 2018, 11, 68. [Google Scholar] [CrossRef]
- Xu, S.; Lam, S.K.; Cheng, P.N.M.; Ho, J.C.M. Recombinant human arginase induces apoptosis through oxidative stress and cell cycle arrest in small cell lung cancer. Cancer Sci. 2018, 109, 3471–3482. [Google Scholar] [CrossRef] [Green Version]
- Chung, S.-F.; Kim, C.-F.; Kwok, S.-Y.; Tam, S.-Y.; Chen, Y.W.; Chong, H.-C.; Leung, S.-L.; So, P.-K.; Wong, K.-Y.; Leung, Y.-C.; et al. Mono-PEGylation of a thermostable arginine-depleting enzyme for the treatment of lung cancer. Int. J. Mol. Sci. 2020, 21, 4234. [Google Scholar] [CrossRef]
- Morris, S.M. Recent advances in arginine metabolism. Curr. Opin. Clin. Nutr. Metab. Care 2004, 7, 45–51. [Google Scholar] [CrossRef]
- Wu, G.; Morris, S.M. Arginine metabolism: Nitric oxide and beyond. Biochem. J. 1998, 336, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Morris, S.M. Arginine metabolism: Boundaries of our knowledge. J. Nutr. 2007, 137, 1602S–1609S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, S.M. Regulation of enzymes of the urea cycle and arginine metabolism. Annu. Rev. Nutr. 2002, 22, 87–105. [Google Scholar] [CrossRef] [PubMed]
- Sugimura, K.; Ohno, T.; Kusuyama, T.; Azuma, I. High sensitivity of human melanoma cell lines to the growth inhibitory activity of mycoplasmal arginine deiminase in vitro. Melanoma Res. 1992, 2, 191–196. [Google Scholar] [CrossRef]
- Scott, L.; Lamb, J.; Smith, S.; Wheatley, D.N. Single amino acid (arginine) deprivation: Rapid and selective death of cultured transformed and malignant cells. Br. J. Cancer 2000, 83, 800–810. [Google Scholar] [CrossRef] [Green Version]
- Wheatley, D.N. Controlling cancer by restricting arginine availability—Arginine-catabolizing enzymes as anticancer agents. Anticancer Drugs 2004, 15, 825–833. [Google Scholar] [CrossRef]
- Shen, W.T.; Zhang, X.Y.; Fu, X.; Fan, J.J.; Luan, J.Y.; Cao, Z.L.; Yang, P.; Xu, Z.Y.; Ju, D.W. A novel and promising therapeutic approach for NSCLC: Recombinant human arginase alone or combined with autophagy inhibitor. Cell Death Dis. 2017, 8, e2720. [Google Scholar] [CrossRef]
- Wang, Z.; Shi, X.; Li, Y.; Fan, J.; Zeng, X.; Xian, Z.; Wang, Z.; Sun, Y.; Wang, S.; Song, P.; et al. Blocking autophagy enhanced cytotoxicity induced by recombinant human arginase in triple-negative breast cancer cells. Cell Death Dis. 2014, 5, e1563. [Google Scholar] [CrossRef]
- Lamb, J.; Wheatley, D.N. Single amino acid (arginine) deprivation induces G1 arrest associated with inhibition of cdk4 expression in cultured human diploid fibroblasts. Exp. Cell Res. 2000, 255, 238–249. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Qin, S.; Ryoo, B.Y.; Lu, S.N.; Yen, C.J.; Feng, Y.H.; Lim, H.Y.; Izzo, F.; Colombo, M.; Sarker, D.; et al. Phase III randomized study of second line ADI-PEG 20 plus best supportive care versus placebo plus best supportive care in patients with advanced hepatocellular carcinoma. Ann. Oncol. 2018, 29, 1402–1408. [Google Scholar] [CrossRef]
- Al-Koussa, H.; Al-Haddad, M.; Abi-Habib, R.; El-Sibai, M. Human recombinant arginase I [HuArgI (Co)-PEG5000]-induced arginine depletion inhibits colorectal cancer cell migration and invasion. Int. J. Mol. Sci. 2019, 20, 6018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mortimore, G.E.; Schworer, C.M. Induction of autophagy by amino-acid deprivation in perfused rat liver. Nature 1977, 270, 174–176. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.H.; Coates, J.M.; Bowles, T.L.; McNerney, G.P.; Sutcliffe, J.; Jung, J.U.; Gandour-Edwards, R.; Chuang, F.Y.; Bold, R.J.; Kung, H.J. Arginine deiminase as a novel therapy for prostate cancer induces autophagy and caspase-independent apoptosis. Cancer Res. 2009, 69, 700–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savaraj, N.; You, M.; Wu, C.; Wangpaichitr, M.; Kuo, M.T.; Feun, L.G. Arginine deprivation, autophagy, apoptosis (AAA) for the treatment of melanoma. Curr. Mol. Med. 2010, 10, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Shintani, T.; Klionsky, D.J. Autophagy in health and disease: A double-edged sword. Science 2004, 306, 990–995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, T.C.; Talalay, P. Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Adv. Enzym. Regul. 1984, 22, 27–55. [Google Scholar] [CrossRef]
- Chou, T.C. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev. 2006, 58, 621–681. [Google Scholar] [CrossRef]
- Husson, A.; Brasse-Lagnel, C.; Fairand, A.; Renouf, S.; Lavoinne, A. Argininosuccinate synthetase from the urea cycle to the citrulline-NO cycle. Eur. J. Biochem. 2003, 270, 1887–1899. [Google Scholar] [CrossRef]
- Wheatley, D.N.; Kilfeather, R.; Stitt, A.; Campbell, E. Integrity and stability of the citrulline-arginine pathway in normal and tumour cell lines. Cancer Lett. 2005, 227, 141–152. [Google Scholar] [CrossRef]
- Bowles, T.L.; Kim, R.; Galante, J.; Parsons, C.M.; Virudachalam, S.; Kung, H.J.; Bold, R.J. Pancreatic cancer cell lines deficient in argininosuccinate synthetase are sensitive to arginine deprivation by arginine deiminase. Int. J. Cancer 2008, 123, 1950–1955. [Google Scholar] [CrossRef] [Green Version]
- Feun, L.; You, M.; Wu, C.J.; Kuo, M.T.; Wangpaichitr, M.; Spector, S.; Savaraj, N. Arginine deprivation as a targeted therapy for cancer. Curr. Pharm. Des. 2008, 14, 1049–1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manca, A.; Sini, M.C.; Izzo, F.; Ascierto, P.A.; Tatangelo, F.; Botti, G.; Gentilcore, G.; Capone, M.; Mozzillo, N.; Rozzo, C.; et al. Induction of arginosuccinate synthetase (ASS) expression affects the antiproliferative activity of arginine deiminase (ADI) in melanoma cells. Oncol. Rep. 2011, 25, 1495–1502. [Google Scholar] [PubMed] [Green Version]
- Meissner, J.D. Nucleotide sequences and further characterization of human papillomavirus DNA present in the CaSki, SiHa and HeLa cervical carcinoma cell lines. J. Gen. Virol. 1999, 80, 1725–1733. [Google Scholar] [CrossRef]
- Yaginuma, Y.; Westphal, H. Analysis of the p53 gene in human uterine carcinoma cell-lines. Cancer Res. 1991, 51, 6506–6509. [Google Scholar]
- Bourgo, R.J.; Braden, W.A.; Wells, S.I.; Knudsen, E.S. Activation of the retinoblastoma tumor suppressor mediates cell cycle inhibition and cell death in specific cervical cancer cell lines. Mol. Carcinogen. 2009, 48, 45–55. [Google Scholar] [CrossRef] [Green Version]
- Pines, J. Cyclins: Wheels within wheels. Cell Growth Differ. 1991, 2, 305–310. [Google Scholar] [PubMed]
- Santamaria, D.; Ortega, S. Cyclins and CDKS in development and cancer: Lessons from genetically modified mice. Front. Biosci. 2006, 11, 1164–1188. [Google Scholar] [CrossRef] [Green Version]
- Tyner, A.L.; Gartel, A.L. Roles of cyclin kinase inhibitors in G1 phase progression. In G1 Phase Progression; Boonstra, J., Ed.; Landes Biosciences: Georgetown, TX, USA, 2003; pp. 58–76. [Google Scholar]
- Hulleman, E.; Boonstra, J. Regulation of G1 phase progression by growth factors and the extracellular matrix. Cell Mol. Life Sci. 2001, 58, 80–93. [Google Scholar] [CrossRef]
- Petersen, B.O.; Lukas, J.; Sorensen, C.S.; Bartek, J.; Helin, K. Phosphorylation of mammalian CDC6 by cyclin A/CDK2 regulates its subcellular localization. EMBO J. 1999, 18, 396–410. [Google Scholar] [CrossRef]
- Coverley, D.; Pelizon, C.; Trewick, S.; Laskey, R.A. Chromatin-bound Cdc6 persists in S and G2 phases in human cells, while soluble Cdc6 is destroyed in a cyclin A-cdk2 dependent process. J. Cell Sci. 2000, 113, 1929–1938. [Google Scholar]
- Graña, X.; Reddy, E.P. Cell cycle control in mammalian cells: Role of cyclins, cyclin dependent kinases (CDKs), growth suppressor genes and cyclin-dependent kinase inhibitors (CKIs). Oncogene 1995, 11, 211–219. [Google Scholar] [PubMed]
- Kitkumthorn, N.; Yanatatsanajit, P.; Kiatpongsan, S.; Phokaew, C.; Triratanachat, S.; Trivijitsilp, P.; Termrungruanglert, W.; Tresukosol, D.; Niruthisard, S.; Mutirangura, A. Cyclin A1 promoter hypermethylation in human papillomavirus-associated cervical cancer. BMC Cancer 2006, 6, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maiuri, M.C.; Zalckvar, E.; Kimchi, A.; Kroemer, G. Self-eating and self-killing: Crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2007, 8, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.B.; Zeng, X.; Wang, S.F.; Fan, J.J.; Wang, Z.Y.; Song, P.; Mei, X.B.; Ju, D.W. Blocking autophagy enhanced leukemia cell death induced by recombinant human arginase. Tumor Biol. 2016, 37, 6627–6635. [Google Scholar] [CrossRef]
- Nasreddine, G.; El-Sibai, M.; Abi-Habib, R.J. Cytotoxicity of [HuArgI (co)-PEG5000]-induced arginine deprivation to ovarian Cancer cells is autophagy dependent. Investig. New Drugs 2020, 38, 10–19. [Google Scholar] [CrossRef]
- Leung, Y.C.; Lo, W.H. Site-Directed Pegylation of Arginases and The Use Thereof as Anti-Cancer and Anti-Viral Agents. U.S. Patent 8,507,245, 13 August 2013. [Google Scholar]
- Noh, E.J.; Kang, S.W.; Shin, Y.J.; Kim, D.C.; Park, I.S.; Kim, M.Y.; Chun, B.G.; Min, B.H. Characterization of mycoplasma arginine deiminase expressed in E. coli and its inhibitory regulation of nitric oxide synthesis. Mol. Cells 2002, 13, 137–143. [Google Scholar]
- Boyde, T.R.; Rahmatullah, M. Optimization of conditions for the colorimetric determination of citrulline, using diacetyl monoxime. Anal. Biochem. 1980, 107, 424–431. [Google Scholar] [CrossRef]
- Wybenga, D.R.; Di Giorgio, J.; Pileggi, V.J. Manual and automated methods for urea nitrogen measurement in whole serum. Clin. Chem. 1971, 17, 891–895. [Google Scholar] [CrossRef]
- Chou, T.C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [Green Version]
- Krishnan, A.; Hariharan, R.; Nair, S.A.; Pillai, M.R. Fluoxetine mediates G0/G1 arrest by inducing functional inhibition of cyclin dependent kinase subunit (CKS) 1. Biochem. Pharmacol. 2008, 75, 1924–1934. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | BCA-M | ADI | Relative Gene Expression b | ||||
|---|---|---|---|---|---|---|---|
| IC50 Value a, U/mL (Max. %) | IC50 Value a, Milli-Units/mL (Max. %) | GAPDH | Arg I | OTC | ASL | ASS | |
| C-33A | 0.19 ± 0.06 (85.0%) | 1.47 ± 0.7 (71.0%) | 1.00 | UD | UD | 0.22 | UD |
| SiHa | 0.31 ± 0.12 (78.1%) | 2.99 ± 2.12 (73.0%) | 1.00 | UD | UD | 0.50 | UD |
| HeLa | 0.53 ± 0.13 (70.9%) | N/A (32.6%) | 1.00 | UD | UD | 0.35 | 0.19 |
| CC3 | 1.36 ± 0.89 (58.9%) | N/A (0.4%) | 1.00 | UD | UD | 0.18 | 0.66 |
| ME-180 | 1.42 ± 0.33 (65.4%) | N/A (8.1%) | 1.00 | UD | UD | 0.80 | 2.31 |
| Cell | Treatment | Drug | IC50 Value a | C.I. |
|---|---|---|---|---|
| SiHa | BCA-M alone | BCA-M | 0.31 ± 0.12 U/mL | |
| CQ alone | CQ | 25.7 ± 4.7 μM | ||
| BCA-M + CQ | BCA-M | 0.17 ± 0.05 U/mL | 0.78 ± 0.06 | |
| CQ | 3.37 ± 0.95 μM | |||
| HeLa | BCA-M alone | BCA-M | 0.53 ± 0.13 U/mL | |
| CQ alone | CQ | 27.1 ± 6.2 μM | ||
| BCA-M + CQ | BCA-M | 0.30 ± 0.09 U/mL | 0.70 ± 0.21 | |
| CQ | 1.49 ± 0.46 μM |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chung, S.-F.; Kim, C.-F.; Chow, H.-Y.; Chong, H.-C.; Tam, S.-Y.; Leung, Y.-C.; Lo, W.-H. Recombinant Bacillus caldovelox Arginase Mutant (BCA-M) Induces Apoptosis, Autophagy, Cell Cycle Arrest and Growth Inhibition in Human Cervical Cancer Cells. Int. J. Mol. Sci. 2020, 21, 7445. https://doi.org/10.3390/ijms21207445
Chung S-F, Kim C-F, Chow H-Y, Chong H-C, Tam S-Y, Leung Y-C, Lo W-H. Recombinant Bacillus caldovelox Arginase Mutant (BCA-M) Induces Apoptosis, Autophagy, Cell Cycle Arrest and Growth Inhibition in Human Cervical Cancer Cells. International Journal of Molecular Sciences. 2020; 21(20):7445. https://doi.org/10.3390/ijms21207445
Chicago/Turabian StyleChung, Sai-Fung, Chi-Fai Kim, Ho-Yin Chow, Hiu-Chi Chong, Suet-Ying Tam, Yun-Chung Leung, and Wai-Hung Lo. 2020. "Recombinant Bacillus caldovelox Arginase Mutant (BCA-M) Induces Apoptosis, Autophagy, Cell Cycle Arrest and Growth Inhibition in Human Cervical Cancer Cells" International Journal of Molecular Sciences 21, no. 20: 7445. https://doi.org/10.3390/ijms21207445
APA StyleChung, S. -F., Kim, C. -F., Chow, H. -Y., Chong, H. -C., Tam, S. -Y., Leung, Y. -C., & Lo, W. -H. (2020). Recombinant Bacillus caldovelox Arginase Mutant (BCA-M) Induces Apoptosis, Autophagy, Cell Cycle Arrest and Growth Inhibition in Human Cervical Cancer Cells. International Journal of Molecular Sciences, 21(20), 7445. https://doi.org/10.3390/ijms21207445