The Role of Chondrocyte Hypertrophy and Senescence in Osteoarthritis Initiation and Progression


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Nature of Articular Cartilage
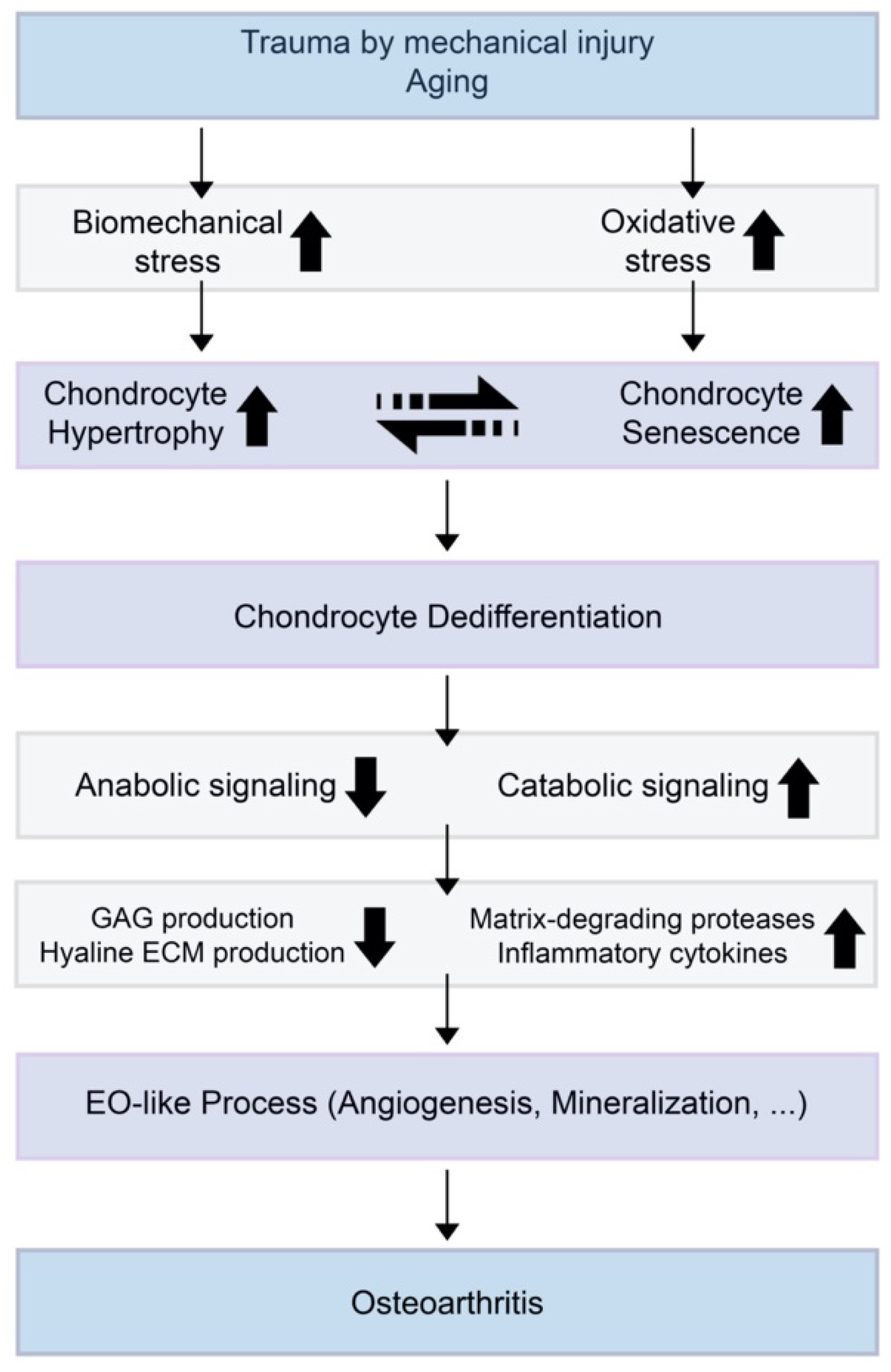
3. OA and Chondrocyte Hypertrophy
4. OA and Chondrocyte Senescence
5. Hypertrophy and Senescence-Related Markers in OA
6. Cartilage Treatment and Regeneration Strategies Targeting Hypertrophy or Senescence
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ECM | Extracellular matrix |
| OA | Osteoarthritis |
| ADAMTS | A disintegrin and metalloprotease with thrombospondin motifs |
| MMP | Matrix metallopeptidase |
| EO | Endochondral ossification |
| SOX9 | SRY-box 9 |
| COL2A1 | Collagen type II, alpha 1 |
| ACAN | Aggrecan |
| RUNX2 | Runt-related transcription factor 2 |
| SA-βgal | Senescence-associated beta-galactosidase |
| SASP | Senescence-associated secretory phenotype |
| ROS | Reactive oxygen specie |
| SIPS | Stress-induced premature senescence |
| TNF-α | Tumor necrosis factor-α |
| VEGF IHH | Vascular endothelial growth factor Indian hedgehog |
| COL10A1 | Collagen type X, alpha 1 |
| ALK5 | Activin-like kinase 5 |
| DMM | Destabilization of the medial meniscus |
| PTHrP BMP | Parathyroid hormone-related protein Bone morphogenetic protein |
| H2O2 GAG | Hydrogen peroxide Glycosaminoglycan |
| NO JNK AGE ALP | Nitric oxide c-Jun N-terminal kinases Advanced glycation end product Alkaline phosphatase |
| ACLT | Anterior cruciate ligament transection |
References
- Nam, Y.; Rim, Y.A.; Jung, S.M.; Ju, J.H. Cord blood cell-derived iPSCs as a new candidate for chondrogenic differentiation and cartilage regeneration. Stem Cell Res. Ther. 2017, 8, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rim, Y.A.; Nam, Y.; Ju, J.H. Application of Cord Blood and Cord Blood-Derived Induced Pluripotent Stem Cells for Cartilage Regeneration. Cell Transpl. 2019, 28, 529–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeon, O.H.; David, N.; Campisi, J.; Elisseeff, J.H. Senescent cells and osteoarthritis: A painful connection. J Clin. Invest. 2018, 128, 1229–1237. [Google Scholar] [CrossRef] [PubMed]
- Dreier, R. Hypertrophic differentiation of chondrocytes in osteoarthritis: The developmental aspect of degenerative joint disorders. Arthritis Res. Ther. 2010, 12, 216. [Google Scholar] [CrossRef] [Green Version]
- Riedl, M.; Witzmann, C.; Koch, M.; Lang, S.; Kerschbaum, M.; Baumann, F.; Krutsch, W.; Docheva, D.; Alt, V.; Pfeifer, C. Attenuation of Hypertrophy in Human MSCs via Treatment with a Retinoic Acid Receptor Inverse Agonist. Int. J. Mol. Sci. 2020, 21, 1444. [Google Scholar] [CrossRef] [Green Version]
- Abula, K.; Muneta, T.; Miyatake, K.; Yamada, J.; Matsukura, Y.; Inoue, M.; Sekiya, I.; Graf, D.; Economides, A.N.; Rosen, V.; et al. Elimination of BMP7 from the developing limb mesenchyme leads to articular cartilage degeneration and synovial inflammation with increased age. FEBS Lett. 2015, 589, 1240–1248. [Google Scholar] [CrossRef] [Green Version]
- Goldring, M.B. Chondrogenesis, chondrocyte differentiation, and articular cartilage metabolism in health and osteoarthritis. Ther. Adv. Musculoskelet Dis. 2012, 4, 269–285. [Google Scholar] [CrossRef]
- Archer, C.W.; Francis-West, P. The chondrocyte. Int. J. Biochem. Cell Biol. 2003, 35, 401–404. [Google Scholar] [CrossRef]
- Akkiraju, H.; Nohe, A. Role of Chondrocytes in Cartilage Formation, Progression of Osteoarthritis and Cartilage Regeneration. J. Dev. Biol. 2015, 3, 177–192. [Google Scholar] [CrossRef] [Green Version]
- Poole, C.A. Articular cartilage chondrons: Form, function and failure. J. Anat. 1997, 191 (Pt 1), 1–13. [Google Scholar] [CrossRef]
- Oh, C.D.; Lu, Y.; Liang, S.; Mori-Akiyama, Y.; Chen, D.; de Crombrugghe, B.; Yasuda, H. SOX9 regulates multiple genes in chondrocytes, including genes encoding ECM proteins, ECM modification enzymes, receptors, and transporters. PLoS ONE 2014, 9, e107577. [Google Scholar] [CrossRef] [PubMed]
- van der Kraan, P.M.; van den Berg, W.B. Chondrocyte hypertrophy and osteoarthritis: Role in initiation and progression of cartilage degeneration? Osteoarthr. Cartil. 2012, 20, 223–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackie, E.J.; Ahmed, Y.A.; Tatarczuch, L.; Chen, K.S.; Mirams, M. Endochondral ossification: How cartilage is converted into bone in the developing skeleton. Int. J. Biochem. Cell Biol. 2008, 40, 46–62. [Google Scholar] [CrossRef] [PubMed]
- van Donkelaar, C.C.; Wilson, W. Mechanics of chondrocyte hypertrophy. Biomech. Model. Mechanobiol. 2012, 11, 655–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lian, C.; Wang, X.; Qiu, X.; Wu, Z.; Gao, B.; Liu, L.; Liang, G.; Zhou, H.; Yang, X.; Peng, Y.; et al. Collagen type II suppresses articular chondrocyte hypertrophy and osteoarthritis progression by promoting integrin beta1-SMAD1 interaction. Bone Res. 2019, 7, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirsch, T.; Swoboda, B.; Nah, H. Activation of annexin II and V expression, terminal differentiation, mineralization and apoptosis in human osteoarthritic cartilage. Osteoarthr. Cartil. 2000, 8, 294–302. [Google Scholar] [CrossRef] [Green Version]
- Tchetina, E.V.; Kobayashi, M.; Yasuda, T.; Meijers, T.; Pidoux, I.; Poole, A.R. Chondrocyte hypertrophy can be induced by a cryptic sequence of type II collagen and is accompanied by the induction of MMP-13 and collagenase activity: Implications for development and arthritis. Matrix Biol. 2007, 26, 247–258. [Google Scholar] [CrossRef]
- Findlay, D.M.; Atkins, G.J. Osteoblast-chondrocyte interactions in osteoarthritis. Curr. Osteoporos Rep. 2014, 12, 127–134. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Dong, S. The Signaling Pathways Involved in Chondrocyte Differentiation and Hypertrophic Differentiation. Stem Cells Int. 2016, 2016, 2470351. [Google Scholar] [CrossRef] [Green Version]
- Sharif, M.; Whitehouse, A.; Sharman, P.; Perry, M.; Adams, M. Increased apoptosis in human osteoarthritic cartilage corresponds to reduced cell density and expression of caspase-3. Arthritis Rheum. 2004, 50, 507–515. [Google Scholar] [CrossRef]
- Charlier, E.; Relic, B.; Deroyer, C.; Malaise, O.; Neuville, S.; Collee, J.; Malaise, M.G.; De Seny, D. Insights on Molecular Mechanisms of Chondrocytes Death in Osteoarthritis. Int. J. Mol. Sci. 2016, 17, 2146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lane, L.B.; Bullough, P.G. Age-related changes in the thickness of the calcified zone and the number of tidemarks in adult human articular cartilage. J. Bone Joint Surg. Br. 1980, 62, 372–375. [Google Scholar] [CrossRef] [PubMed]
- McCulloch, K.; Litherland, G.J.; Rai, T.S. Cellular senescence in osteoarthritis pathology. Aging Cell 2017, 16, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Vinatier, C.; Dominguez, E.; Guicheux, J.; Carames, B. Role of the Inflammation-Autophagy-Senescence Integrative Network in Osteoarthritis. Front. Physiol. 2018, 9, 706. [Google Scholar] [CrossRef] [PubMed]
- Kuilman, T.; Michaloglou, C.; Mooi, W.J.; Peeper, D.S. The essence of senescence. Genes Dev. 2010, 24, 2463–2479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greene, M.A.; Loeser, R.F. Aging-related inflammation in osteoarthritis. Osteoarthr. Cartil. 2015, 23, 1966–1971. [Google Scholar] [CrossRef] [Green Version]
- Jeon, O.H.; Kim, C.; Laberge, R.M.; Demaria, M.; Rathod, S.; Vasserot, A.P.; Chung, J.W.; Kim, D.H.; Poon, Y.; David, N.; et al. Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat. Med. 2017, 23, 775–781. [Google Scholar] [CrossRef]
- Stokes, D.G.; Liu, G.; Dharmavaram, R.; Hawkins, D.; Piera-Velazquez, S.; Jimenez, S.A. Regulation of type-II collagen gene expression during human chondrocyte de-differentiation and recovery of chondrocyte-specific phenotype in culture involves Sry-type high-mobility-group box (SOX) transcription factors. Biochem. J. 2001, 360, 461–470. [Google Scholar] [CrossRef]
- Benya, P.D.; Padilla, S.R.; Nimni, M.E. Independent regulation of collagen types by chondrocytes during the loss of differentiated function in culture. Cell 1978, 15, 1313–1321. [Google Scholar] [CrossRef]
- Ashraf, S.; Cha, B.H.; Kim, J.S.; Ahn, J.; Han, I.; Park, H.; Lee, S.H. Regulation of senescence associated signaling mechanisms in chondrocytes for cartilage tissue regeneration. Osteoarthr. Cartil. 2016, 24, 196–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.; Huang, P.; Li, G.; Feng, Y.; Zhendong, L.; Zhou, C.; Hu, G.; Xu, Q. Overexpression of Pitx1 attenuates the senescence of chondrocytes from osteoarthritis degeneration cartilage-A self-controlled model for studying the etiology and treatment of osteoarthritis. Bone 2020, 131, 115177. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.A.; Buckwalter, J.A. Telomere erosion and senescence in human articular cartilage chondrocytes. J. Gerontol. A Biol. Sci. Med. Sci. 2001, 56, B172–B179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, J.S.; Waters, J.G.; Darrah, C.; Pennington, C.; Edwards, D.R.; Donell, S.T.; Clark, I.M. The role of chondrocyte senescence in osteoarthritis. Aging Cell 2002, 1, 57–65. [Google Scholar] [CrossRef]
- Muller, M. Cellular senescence: Molecular mechanisms, in vivo significance, and redox considerations. Antioxid. Redox Signal. 2009, 11, 59–98. [Google Scholar] [CrossRef]
- Erusalimsky, J.D.; Kurz, D.J. Cellular senescence in vivo: Its relevance in ageing and cardiovascular disease. Exp. Gerontol. 2005, 40, 634–642. [Google Scholar] [CrossRef]
- Xu, M.; Bradley, E.W.; Weivoda, M.M.; Hwang, S.M.; Pirtskhalava, T.; Decklever, T.; Curran, G.L.; Ogrodnik, M.; Jurk, D.; Johnson, K.O.; et al. Transplanted Senescent Cells Induce an Osteoarthritis-Like Condition in Mice. J. Gerontol. A Biol. Sci. Med. Sci. 2017, 72, 780–785. [Google Scholar] [CrossRef] [Green Version]
- Acosta, J.C.; Banito, A.; Wuestefeld, T.; Georgilis, A.; Janich, P.; Morton, J.P.; Athineos, D.; Kang, T.W.; Lasitschka, F.; Andrulis, M.; et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell. Biol. 2013, 15, 978–990. [Google Scholar] [CrossRef]
- Del Rey, M.J.; Valin, A.; Usategui, A.; Ergueta, S.; Martin, E.; Municio, C.; Canete, J.D.; Blanco, F.J.; Criado, G.; Pablos, J.L. Senescent synovial fibroblasts accumulate prematurely in rheumatoid arthritis tissues and display an enhanced inflammatory phenotype. Immun. Ageing 2019, 16, 29. [Google Scholar] [CrossRef] [Green Version]
- Johnson, K.A.; van Etten, D.; Nanda, N.; Graham, R.M.; Terkeltaub, R.A. Distinct transglutaminase 2-independent and transglutaminase 2-dependent pathways mediate articular chondrocyte hypertrophy. J. Biol. Chem. 2003, 278, 18824–18832. [Google Scholar] [CrossRef] [Green Version]
- Hennig, T.; Lorenz, H.; Thiel, A.; Goetzke, K.; Dickhut, A.; Geiger, F.; Richter, W. Reduced chondrogenic potential of adipose tissue derived stromal cells correlates with an altered TGFbeta receptor and BMP profile and is overcome by BMP-6. J. Cell Physiol. 2007, 211, 682–691. [Google Scholar] [CrossRef] [PubMed]
- Pullig, O.; Weseloh, G.; Ronneberger, D.; Kakonen, S.; Swoboda, B. Chondrocyte differentiation in human osteoarthritis: Expression of osteocalcin in normal and osteoarthritic cartilage and bone. Calcif. Tissue Int. 2000, 67, 230–240. [Google Scholar] [CrossRef] [PubMed]
- Aigner, T.; Fundel, K.; Saas, J.; Gebhard, P.M.; Haag, J.; Weiss, T.; Zien, A.; Obermayr, F.; Zimmer, R.; Bartnik, E. Large-scale gene expression profiling reveals major pathogenetic pathways of cartilage degeneration in osteoarthritis. Arthritis Rheum. 2006, 54, 3533–3544. [Google Scholar] [CrossRef] [PubMed]
- Fukui, N.; Ikeda, Y.; Ohnuki, T.; Tanaka, N.; Hikita, A.; Mitomi, H.; Mori, T.; Juji, T.; Katsuragawa, Y.; Yamamoto, S.; et al. Regional differences in chondrocyte metabolism in osteoarthritis: A detailed analysis by laser capture microdissection. Arthritis Rheum. 2008, 58, 154–163. [Google Scholar] [CrossRef]
- Cha, B.H.; Lee, J.S.; Kim, S.W.; Cha, H.J.; Lee, S.H. The modulation of the oxidative stress response in chondrocytes by Wip1 and its effect on senescence and dedifferentiation during in vitro expansion. Biomaterials 2013, 34, 2380–2388. [Google Scholar] [CrossRef]
- Philipot, D.; Guerit, D.; Platano, D.; Chuchana, P.; Olivotto, E.; Espinoza, F.; Dorandeu, A.; Pers, Y.M.; Piette, J.; Borzi, R.M.; et al. p16INK4a and its regulator miR-24 link senescence and chondrocyte terminal differentiation-associated matrix remodeling in osteoarthritis. Arthritis Res. Ther. 2014, 16, R58. [Google Scholar] [CrossRef] [Green Version]
- Shlopov, B.V.; Gumanovskaya, M.L.; Hasty, K.A. Autocrine regulation of collagenase 3 (matrix metalloproteinase 13) during osteoarthritis. Arthritis Rheum. 2000, 43, 195–205. [Google Scholar] [CrossRef]
- Wang, X.; Manner, P.A.; Horner, A.; Shum, L.; Tuan, R.S.; Nuckolls, G.H. Regulation of MMP-13 expression by RUNX2 and FGF2 in osteoarthritic cartilage. Osteoarthr. Cartil. 2004, 12, 963–973. [Google Scholar] [CrossRef] [Green Version]
- Kamekura, S.; Hoshi, K.; Shimoaka, T.; Chung, U.; Chikuda, H.; Yamada, T.; Uchida, M.; Ogata, N.; Seichi, A.; Nakamura, K.; et al. Osteoarthritis development in novel experimental mouse models induced by knee joint instability. Osteoarthr. Cartil. 2005, 13, 632–641. [Google Scholar] [CrossRef] [Green Version]
- Neuhold, L.A.; Killar, L.; Zhao, W.; Sung, M.L.; Warner, L.; Kulik, J.; Turner, J.; Wu, W.; Billinghurst, C.; Meijers, T.; et al. Postnatal expression in hyaline cartilage of constitutively active human collagenase-3 (MMP-13) induces osteoarthritis in mice. J. Clin. Invest. 2001, 107, 35–44. [Google Scholar] [CrossRef] [Green Version]
- Little, C.B.; Barai, A.; Burkhardt, D.; Smith, S.M.; Fosang, A.J.; Werb, Z.; Shah, M.; Thompson, E.W. Matrix metalloproteinase 13-deficient mice are resistant to osteoarthritic cartilage erosion but not chondrocyte hypertrophy or osteophyte development. Arthritis Rheum. 2009, 60, 3723–3733. [Google Scholar] [CrossRef] [PubMed]
- Blaney Davidson, E.N.; Remst, D.F.; Vitters, E.L.; van Beuningen, H.M.; Blom, A.B.; Goumans, M.J.; van den Berg, W.B.; van der Kraan, P.M. Increase in ALK1/ALK5 ratio as a cause for elevated MMP-13 expression in osteoarthritis in humans and mice. J. Immunol. 2009, 182, 7937–7945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferguson, C.M.; Schwarz, E.M.; Reynolds, P.R.; Puzas, J.E.; Rosier, R.N.; O’Keefe, R.J. Smad2 and 3 mediate transforming growth factor-beta1-induced inhibition of chondrocyte maturation. Endocrinology 2000, 141, 4728–4735. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Chen, L.; Xu, X.; Li, C.; Huang, C.; Deng, C.X. TGF-beta/Smad3 signals repress chondrocyte hypertrophic differentiation and are required for maintaining articular cartilage. J. Cell Biol. 2001, 153, 35–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goumans, M.J.; Mummery, C. Functional analysis of the TGFbeta receptor/Smad pathway through gene ablation in mice. Int. J. Dev. Biol. 2000, 44, 253–265. [Google Scholar] [PubMed]
- Ito, H.; Akiyama, H.; Shigeno, C.; Nakamura, T. Noggin and bone morphogenetic protein-4 coordinately regulate the progression of chondrogenic differentiation in mouse clonal EC cells, ATDC5. Biochem. Biophys. Res. Commun. 1999, 260, 240–244. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhang, C.; Luo, C.; Zhan, Y.; Zhong, B. Design, cyclization, and optimization of MMP13-TIMP1 interaction-derived self-inhibitory peptides against chondrocyte senescence in osteoarthritis. Int. J. Biol. Macromol. 2019, 121, 921–929. [Google Scholar] [CrossRef]
- Forsyth, C.B.; Cole, A.; Murphy, G.; Bienias, J.L.; Im, H.J.; Loeser, R.F., Jr. Increased matrix metalloproteinase-13 production with aging by human articular chondrocytes in response to catabolic stimuli. J. Gerontol. A Biol. Sci. Med. Sci. 2005, 60, 1118–1124. [Google Scholar] [CrossRef] [Green Version]
- Hecht, J.; Seitz, V.; Urban, M.; Wagner, F.; Robinson, P.N.; Stiege, A.; Dieterich, C.; Kornak, U.; Wilkening, U.; Brieske, N.; et al. Detection of novel skeletogenesis target genes by comprehensive analysis of a Runx2(-/-) mouse model. Gene Expr. Patterns 2007, 7, 102–112. [Google Scholar] [CrossRef]
- Chen, D.; Shen, J.; Zhao, W.; Wang, T.; Han, L.; Hamilton, J.L.; Im, H.J. Osteoarthritis: Toward a comprehensive understanding of pathological mechanism. Bone Res. 2017, 5, 16044. [Google Scholar] [CrossRef]
- Kilbey, A.; Blyth, K.; Wotton, S.; Terry, A.; Jenkins, A.; Bell, M.; Hanlon, L.; Cameron, E.R.; Neil, J.C. Runx2 disruption promotes immortalization and confers resistance to oncogene-induced senescence in primary murine fibroblasts. Cancer Res. 2007, 67, 11263–11271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlevaro, M.F.; Cermelli, S.; Cancedda, R.; Descalzi Cancedda, F. Vascular endothelial growth factor (VEGF) in cartilage neovascularization and chondrocyte differentiation: Auto-paracrine role during endochondral bone formation. J. Cell Sci. 2000, 113 (Pt 1), 59–69. [Google Scholar]
- Enomoto, H.; Inoki, I.; Komiya, K.; Shiomi, T.; Ikeda, E.; Obata, K.; Matsumoto, H.; Toyama, Y.; Okada, Y. Vascular endothelial growth factor isoforms and their receptors are expressed in human osteoarthritic cartilage. Am. J. Pathol. 2003, 162, 171–181. [Google Scholar] [CrossRef] [Green Version]
- Beckmann, R.; Houben, A.; Tohidnezhad, M.; Kweider, N.; Fragoulis, A.; Wruck, C.J.; Brandenburg, L.O.; Hermanns-Sachweh, B.; Goldring, M.B.; Pufe, T.; et al. Mechanical forces induce changes in VEGF and VEGFR-1/sFlt-1 expression in human chondrocytes. Int. J. Mol. Sci. 2014, 15, 15456–15474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagao, M.; Hamilton, J.L.; Kc, R.; Berendsen, A.D.; Duan, X.; Cheong, C.W.; Li, X.; Im, H.J.; Olsen, B.R. Vascular Endothelial Growth Factor in Cartilage Development and Osteoarthritis. Sci. Rep. 2017, 7, 13027. [Google Scholar] [CrossRef] [PubMed]
- Wei, F.; Zhou, J.; Wei, X.; Zhang, J.; Fleming, B.C.; Terek, R.; Pei, M.; Chen, Q.; Liu, T.; Wei, L. Activation of Indian hedgehog promotes chondrocyte hypertrophy and upregulation of MMP-13 in human osteoarthritic cartilage. Osteoarthr. Cartil. 2012, 20, 755–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, A.C.; Seeto, B.L.; Bartoszko, J.M.; Khoury, M.A.; Whetstone, H.; Ho, L.; Hsu, C.; Ali, S.A.; Alman, B.A. Modulating hedgehog signaling can attenuate the severity of osteoarthritis. Nat. Med. 2009, 15, 1421–1425. [Google Scholar] [CrossRef] [PubMed]
- Beaupre, G.S.; Stevens, S.S.; Carter, D.R. Mechanobiology in the development, maintenance, and degeneration of articular cartilage. J. Rehabil. Res. Dev. 2000, 37, 145–151. [Google Scholar]
- Mak, K.K.; Kronenberg, H.M.; Chuang, P.T.; Mackem, S.; Yang, Y. Indian hedgehog signals independently of PTHrP to promote chondrocyte hypertrophy. Development 2008, 135, 1947–1956. [Google Scholar] [CrossRef] [Green Version]
- St-Jacques, B.; Hammerschmidt, M.; McMahon, A.P. Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation. Genes Dev. 1999, 13, 2072–2086. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, S.; Nishiyama, T.; Hayashi, S.; Fujishiro, T.; Takebe, K.; Kanzaki, N.; Kuroda, R.; Kurosaka, M. Role of p53 in human chondrocyte apoptosis in response to shear strain. Arthritis Rheum. 2009, 60, 2340–2349. [Google Scholar] [CrossRef] [PubMed]
- Diekman, B.O.; Sessions, G.A.; Collins, J.A.; Knecht, A.K.; Strum, S.L.; Mitin, N.K.; Carlson, C.S.; Loeser, R.F.; Sharpless, N.E. Expression of p16(INK) (4a) is a biomarker of chondrocyte aging but does not cause osteoarthritis. Aging Cell 2018, 17, e12771. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Park, S.R.; Park, H.J.; Choi, B.H.; Min, B.H. Potential predictive markers for proliferative capacity of cultured human articular chondrocytes: PCNA and p21. Artif. Organs 2005, 29, 393–398. [Google Scholar] [CrossRef] [PubMed]
- De la Fuente, M.; Miquel, J. An update of the oxidation-inflammation theory of aging: The involvement of the immune system in oxi-inflamm-aging. Curr. Pharm. Des. 2009, 15, 3003–3026. [Google Scholar] [CrossRef]
- Minguzzi, M.; Cetrullo, S.; D’Adamo, S.; Silvestri, Y.; Flamigni, F.; Borzi, R.M. Emerging Players at the Intersection of Chondrocyte Loss of Maturational Arrest, Oxidative Stress, Senescence and Low-Grade Inflammation in Osteoarthritis. Oxid. Med. Cell Longev. 2018, 2018, 3075293. [Google Scholar] [CrossRef] [Green Version]
- Yudoh, K.; Nguyen v, T.; Nakamura, H.; Hongo-Masuko, K.; Kato, T.; Nishioka, K. Potential involvement of oxidative stress in cartilage senescence and development of osteoarthritis: Oxidative stress induces chondrocyte telomere instability and downregulation of chondrocyte function. Arthritis Res. Ther. 2005, 7, R380–R391. [Google Scholar] [CrossRef] [Green Version]
- Brandl, A.; Hartmann, A.; Bechmann, V.; Graf, B.; Nerlich, M.; Angele, P. Oxidative stress induces senescence in chondrocytes. J. Orthop. Res. 2011, 29, 1114–1120. [Google Scholar] [CrossRef]
- Nakase, T.; Miyaji, T.; Tomita, T.; Kaneko, M.; Kuriyama, K.; Myoui, A.; Sugamoto, K.; Ochi, T.; Yoshikawa, H. Localization of bone morphogenetic protein-2 in human osteoarthritic cartilage and osteophyte. Osteoarthr. Cartil. 2003, 11, 278–284. [Google Scholar] [CrossRef] [Green Version]
- Retting, K.N.; Song, B.; Yoon, B.S.; Lyons, K.M. BMP canonical Smad signaling through Smad1 and Smad5 is required for endochondral bone formation. Development 2009, 136, 1093–1104. [Google Scholar] [CrossRef] [Green Version]
- McNulty, A.L.; Estes, B.T.; Wilusz, R.E.; Weinberg, J.B.; Guilak, F. Dynamic loading enhances integrative meniscal repair in the presence of interleukin-1. Osteoarthr. Cartil. 2010, 18, 830–838. [Google Scholar] [CrossRef] [Green Version]
- Clancy, R.; Rediske, J.; Koehne, C.; Stoyanovsky, D.; Amin, A.; Attur, M.; Iyama, K.; Abramson, S.B. Activation of stress-activated protein kinase in osteoarthritic cartilage: Evidence for nitric oxide dependence. Osteoarthr. Cartil. 2001, 9, 294–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, D.; Blake, S.; Song, X.Y.; Lark, M.; Loeser, R.F. Human articular chondrocytes produce IL-7 and respond to IL-7 with increased production of matrix metalloproteinase-13. Arthritis Res. Ther. 2008, 10, R23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Randell, E.W.; Sun, G.; Likhodii, S.; Liu, M.; Furey, A.; Zhai, G. Hyperglycemia-related advanced glycation end-products is associated with the altered phosphatidylcholine metabolism in osteoarthritis patients with diabetes. PLoS ONE 2017, 12, e0184105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uribarri, J.; Woodruff, S.; Goodman, S.; Cai, W.; Chen, X.; Pyzik, R.; Yong, A.; Striker, G.E.; Vlassara, H. Advanced glycation end products in foods and a practical guide to their reduction in the diet. J. Am. Diet. Assoc. 2010, 110, 911–916 e912. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.Y.; Lai, K.Y.; Hung, L.F.; Wu, W.L.; Liu, F.C.; Ho, L.J. Advanced glycation end products cause collagen II reduction by activating Janus kinase/signal transducer and activator of transcription 3 pathway in porcine chondrocytes. Rheumatology (Oxford) 2011, 50, 1379–1389. [Google Scholar] [CrossRef] [Green Version]
- Nah, S.S.; Choi, I.Y.; Lee, C.K.; Oh, J.S.; Kim, Y.G.; Moon, H.B.; Yoo, B. Effects of advanced glycation end products on the expression of COX-2, PGE2 and NO in human osteoarthritic chondrocytes. Rheumatology (Oxford) 2008, 47, 425–431. [Google Scholar] [CrossRef] [Green Version]
- Nah, S.S.; Choi, I.Y.; Yoo, B.; Kim, Y.G.; Moon, H.B.; Lee, C.K. Advanced glycation end products increases matrix metalloproteinase-1, -3, and -13, and TNF-alpha in human osteoarthritic chondrocytes. FEBS Lett. 2007, 581, 1928–1932. [Google Scholar] [CrossRef] [Green Version]
- Chen, A.C.; Temple, M.M.; Ng, D.M.; Verzijl, N.; DeGroot, J.; TeKoppele, J.M.; Sah, R.L. Induction of advanced glycation end products and alterations of the tensile properties of articular cartilage. Arthritis Rheum. 2002, 46, 3212–3217. [Google Scholar] [CrossRef] [Green Version]
- Bank, R.A.; Bayliss, M.T.; Lafeber, F.P.; Maroudas, A.; Tekoppele, J.M. Ageing and zonal variation in post-translational modification of collagen in normal human articular cartilage. The age-related increase in non-enzymatic glycation affects biomechanical properties of cartilage. Biochem. J. 1998, 330 (Pt 1), 345–351. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.C.; Hung, L.F.; Wu, W.L.; Chang, D.M.; Huang, C.Y.; Lai, J.H.; Ho, L.J. Chondroprotective effects and mechanisms of resveratrol in advanced glycation end products-stimulated chondrocytes. Arthritis Res. Ther. 2010, 12, R167. [Google Scholar] [CrossRef] [Green Version]
- Ballock, R.T.; Heydemann, A.; Wakefield, L.M.; Flanders, K.C.; Roberts, A.B.; Sporn, M.B. TGF-beta 1 prevents hypertrophy of epiphyseal chondrocytes: Regulation of gene expression for cartilage matrix proteins and metalloproteases. Dev. Biol. 1993, 158, 414–429. [Google Scholar] [CrossRef]
- Tschan, T.; Bohme, K.; Conscience-Egli, M.; Zenke, G.; Winterhalter, K.H.; Bruckner, P. Autocrine or paracrine transforming growth factor-beta modulates the phenotype of chick embryo sternal chondrocytes in serum-free agarose culture. J. Biol. Chem. 1993, 268, 5156–5161. [Google Scholar] [PubMed]
- Serra, R.; Johnson, M.; Filvaroff, E.H.; LaBorde, J.; Sheehan, D.M.; Derynck, R.; Moses, H.L. Expression of a truncated, kinase-defective TGF-beta type II receptor in mouse skeletal tissue promotes terminal chondrocyte differentiation and osteoarthritis. J. Cell. Biol. 1997, 139, 541–552. [Google Scholar] [CrossRef] [PubMed]
- Rim, Y.A.; Nam, Y.; Park, N.; Jung, H.; Lee, K.; Lee, J.; Ju, J.H. Chondrogenic Differentiation from Induced Pluripotent Stem Cells Using Non-Viral Minicircle Vectors. Cells 2020, 9, 582. [Google Scholar] [CrossRef] [PubMed]
- Mueller, M.B.; Fischer, M.; Zellner, J.; Berner, A.; Dienstknecht, T.; Prantl, L.; Kujat, R.; Nerlich, M.; Tuan, R.S.; Angele, P. Hypertrophy in mesenchymal stem cell chondrogenesis: Effect of TGF-beta isoforms and chondrogenic conditioning. Cells Tissues Organs 2010, 192, 158–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pei, M.; He, F.; Li, J.; Tidwell, J.E.; Jones, A.C.; McDonough, E.B. Repair of large animal partial-thickness cartilage defects through intraarticular injection of matrix-rejuvenated synovium-derived stem cells. Tissue Eng. Part A 2013, 19, 1144–1154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cals, F.L.; Hellingman, C.A.; Koevoet, W.; Baatenburg de Jong, R.J.; van Osch, G.J. Effects of transforming growth factor-beta subtypes on in vitro cartilage production and mineralization of human bone marrow stromal-derived mesenchymal stem cells. J. Tissue Eng. Regen. Med. 2012, 6, 68–76. [Google Scholar] [CrossRef]
- Shintani, N.; Siebenrock, K.A.; Hunziker, E.B. TGF-ss1 enhances the BMP-2-induced chondrogenesis of bovine synovial explants and arrests downstream differentiation at an early stage of hypertrophy. PLoS ONE 2013, 8, e53086. [Google Scholar] [CrossRef] [Green Version]
- Frieling, J.S.; Lynch, C.C. Proteolytic Regulation of Parathyroid Hormone-Related Protein: Functional Implications for Skeletal Malignancy. Int. J. Mol. Sci. 2019, 20, 2814. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.J.; Kim, H.J.; Im, G.I. PTHrP promotes chondrogenesis and suppresses hypertrophy from both bone marrow-derived and adipose tissue-derived MSCs. Biochem. Biophys. Res. Commun. 2008, 373, 104–108. [Google Scholar] [CrossRef]
- Fischer, J.; Aulmann, A.; Dexheimer, V.; Grossner, T.; Richter, W. Intermittent PTHrP(1-34) exposure augments chondrogenesis and reduces hypertrophy of mesenchymal stromal cells. Stem Cells Dev. 2014, 23, 2513–2523. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.M.; Im, G.I. PTHrP isoforms have differing effect on chondrogenic differentiation and hypertrophy of mesenchymal stem cells. Biochem. Biophys. Res. Commun. 2012, 421, 819–824. [Google Scholar] [CrossRef] [PubMed]
- Li, N.G.; Shi, Z.H.; Tang, Y.P.; Wang, Z.J.; Song, S.L.; Qian, L.H.; Qian, D.W.; Duan, J.A. New hope for the treatment of osteoarthritis through selective inhibition of MMP-13. Curr. Med. Chem. 2011, 18, 977–1001. [Google Scholar] [CrossRef]
- Peterson, J.T. The importance of estimating the therapeutic index in the development of matrix metalloproteinase inhibitors. Cardiovasc. Res. 2006, 69, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Bertram, H.; Boeuf, S.; Wachters, J.; Boehmer, S.; Heisel, C.; Hofmann, M.W.; Piecha, D.; Richter, W. Matrix metalloprotease inhibitors suppress initiation and progression of chondrogenic differentiation of mesenchymal stromal cells in vitro. Stem Cells Dev. 2009, 18, 881–892. [Google Scholar] [CrossRef] [PubMed]
- Levitt, N.C.; Eskens, F.A.; O’Byrne, K.J.; Propper, D.J.; Denis, L.J.; Owen, S.J.; Choi, L.; Foekens, J.A.; Wilner, S.; Wood, J.M.; et al. Phase I and pharmacological study of the oral matrix metalloproteinase inhibitor, MMI270 (CGS27023A), in patients with advanced solid cancer. Clin. Cancer Res. 2001, 7, 1912–1922. [Google Scholar]
- Collins, J.A.; Diekman, B.O.; Loeser, R.F. Targeting aging for disease modification in osteoarthritis. Curr. Opin. Rheumatol. 2018, 30, 101–107. [Google Scholar] [CrossRef]
- Baker, D.J.; Childs, B.G.; Durik, M.; Wijers, M.E.; Sieben, C.J.; Zhong, J.; Saltness, R.A.; Jeganathan, K.B.; Verzosa, G.C.; Pezeshki, A.; et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature 2016, 530, 184–189. [Google Scholar] [CrossRef] [Green Version]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rim, Y.A.; Nam, Y.; Ju, J.H. The Role of Chondrocyte Hypertrophy and Senescence in Osteoarthritis Initiation and Progression. Int. J. Mol. Sci. 2020, 21, 2358. https://doi.org/10.3390/ijms21072358
Rim YA, Nam Y, Ju JH. The Role of Chondrocyte Hypertrophy and Senescence in Osteoarthritis Initiation and Progression. International Journal of Molecular Sciences. 2020; 21(7):2358. https://doi.org/10.3390/ijms21072358
Chicago/Turabian StyleRim, Yeri Alice, Yoojun Nam, and Ji Hyeon Ju. 2020. "The Role of Chondrocyte Hypertrophy and Senescence in Osteoarthritis Initiation and Progression" International Journal of Molecular Sciences 21, no. 7: 2358. https://doi.org/10.3390/ijms21072358
APA StyleRim, Y. A., Nam, Y., & Ju, J. H. (2020). The Role of Chondrocyte Hypertrophy and Senescence in Osteoarthritis Initiation and Progression. International Journal of Molecular Sciences, 21(7), 2358. https://doi.org/10.3390/ijms21072358