Differential Inhibition of Human Nav1.2 Resurgent and Persistent Sodium Currents by Cannabidiol and GS967

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
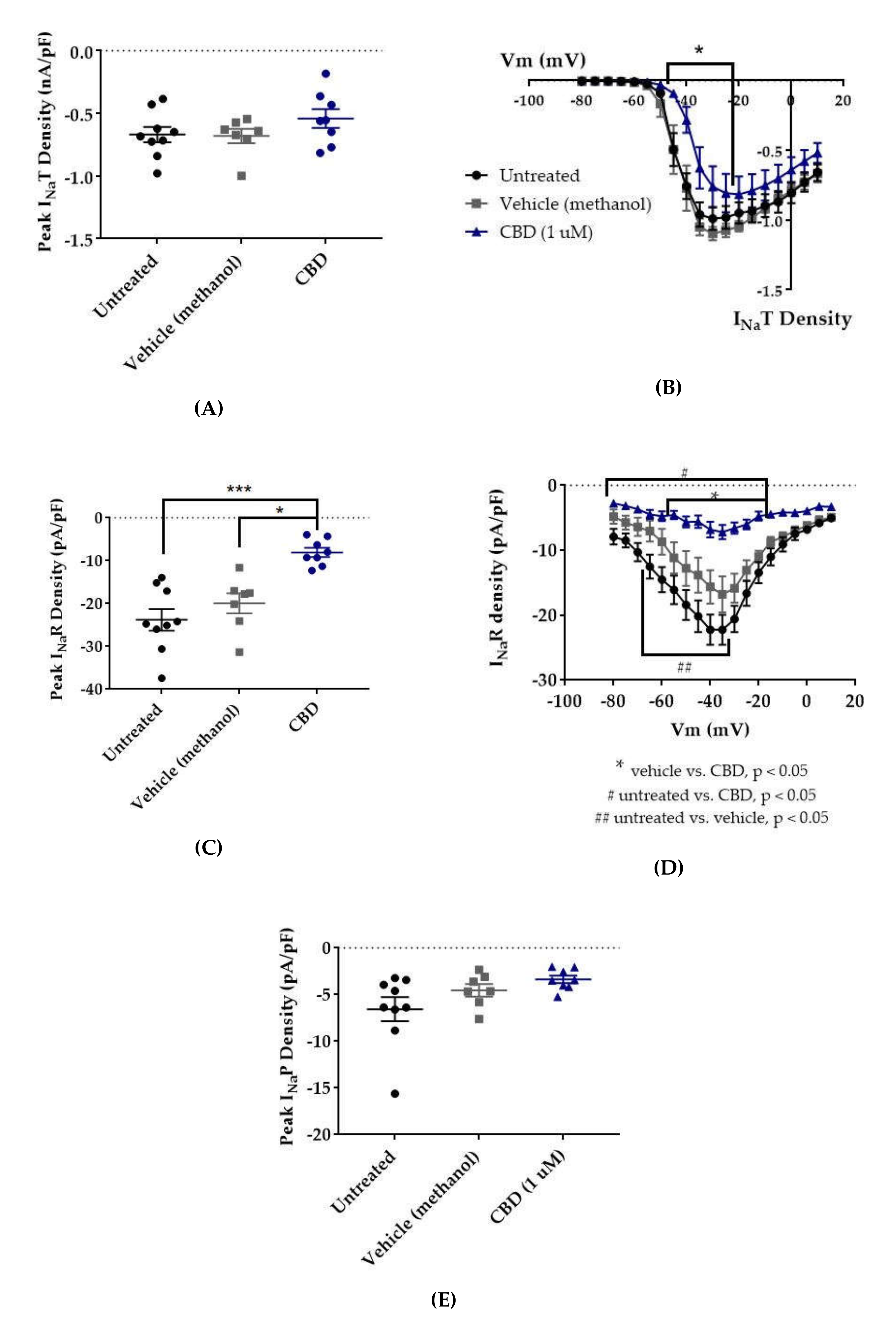
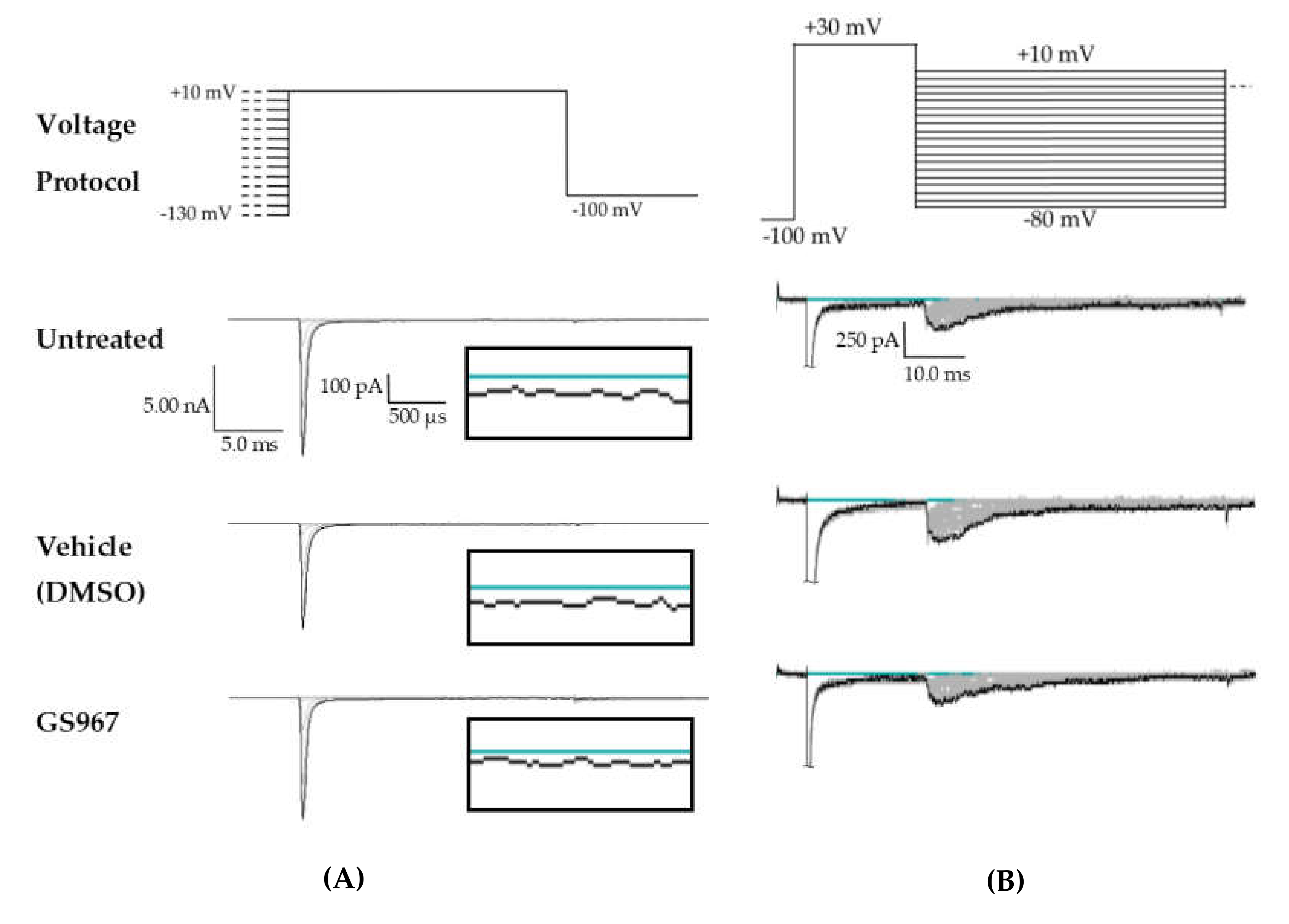
2.1. Cannabidiol (CBD) Effects on INaT, INaR, and INaP
2.2. GS967 Effects on INaT, INaR, and INaP
2.3. Cannabidiol (CBD) Effects on hNav1.2 Gating
2.4. GS967 Effects on hNav1.2 Gating
3. Discussion
4. Materials and Methods
4.1. DNA Constructs
4.2. HEK293 Cell Culture
4.3. Generation of Stable Cell Lines
4.4. HEK Electrophysiology
4.5. HEK Voltage Protocols
4.6. Drug Application
4.7. Statistics and Analysis
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AED | Antiepileptic Drug |
| CBD | Cannabidiol |
| DMSO | Dimethyl Sulfoxide |
| HEK | Human Embryonic Kidney (cells) |
| INaP | Persistent Sodium Current |
| INaR | Resurgent Sodium Current |
| INaT | Transient Sodium Current |
| Nav | Voltage-Gated Sodium (channel) |
| WT | Wild-Type |
References
- French, J.A. Refractory epilepsy: clinical overview. Epilepsia 2007, 48, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Beleza, P. Refractory epilepsy: a clinically oriented review. Eur. Neurol. 2009, 62, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Löscher, W.; Klitgaard, H.; Twyman, R.E.; Schmidt, D. New avenues for anti-epileptic drug discovery and development. Nat. Rev. Drug Disc. 2013, 12, 757–776. [Google Scholar] [CrossRef]
- Laxer, K.D.; Trinka, E.; Hirsch, L.J.; Cendes, F.; Langfitt, J.; Delanty, N.; Resnick, T.; Benbadis, S.R. The consequences of refractory epilepsy and its treatment. Epilepsy Behav. 2014, 37, 59–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moshe, S.L.; Perucca, E.; Ryvlin, P.; Tomson, T. Epilepsy: new advances. Lancet 2015, 385, 884–898. [Google Scholar] [CrossRef]
- UpToDate. Available online: http://www.uptodate.com/contents/evaluation-and-management-of-drug-resistant-epilepsy (accessed on 27 February 2020).
- Nicita, F.; De Liso, P.; Danti, F.R.; Papetti, L.; Ursitti, F.; Castronovo, A.; Allemand, F.; Gennaro, E.; Zara, F.; Striano, P.; et al. The genetics of monogenic idiopathic epilepsies and epileptic encephalopathies. Seizure 2012, 21, 3–11. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Anttonen, A.K.; Liukkonen, E.; Gaily, E.; Maljevic, S.; Schubert, S.; Bellan-Koch, A.; Petrou, S.; Ahonen, V.E.; Lerche, H.; et al. SCN2A mutation associated with neonatal epilepsy, late-onset episodic ataxia, myoclonus, and pain. Neurology 2010, 75, 1454–1458. [Google Scholar] [CrossRef]
- Lauxmann, S.; Boutry-Kryza, N.; Rivier, C.; Mueller, S.; Hedrich, U.B.; Maljevic, S.; Szepetowski, P.; Lerche, H.; Lesca, G. An SCN2A mutation in a family with infantile seizures from Madagascar reveals an increased subthreshold Na(+) current. Epilepsia 2013, 54, e117–e121. [Google Scholar] [CrossRef]
- Parrini, E.; Marini, C.; Mei, D.; Galuppi, A.; Cellini, E.; Pucatti, D.; Chiti, L.; Rutigliano, D.; Bianchini, C.; Virdo, S.; et al. Diagnostic Targeted Resequencing in 349 Patients with Drug-Resistant Pediatric Epilepsies Identifies Causative Mutations in 30 Different Genes. Hum. Mutat. 2017, 38, 216–225. [Google Scholar] [CrossRef] [Green Version]
- Wolff, M.; Johannesen, K.M.; Hedrich, U.B.S.; Masnada, S.; Rubboli, G.; Gardella, E.; Lesca, G.; Ville, D.; Milh, M.; Villard, L.; et al. Genetic and phenotypic heterogeneity suggest therapeutic implications in SCN2A-related disorders. Brain 2017, 140, 1316–1336. [Google Scholar] [CrossRef]
- Mason, E.R.; Wu, F.; Patel, R.R.; Xiao, Y.; Cannon, S.C.; Cummins, T.R. Resurgent and Gating Pore Currents Induced by De Novo SCN2A Epilepsy Mutations. eNeuro 2019, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, B.; Rhodes, K.J.; Bekele-Arcuri, Z.; Trimmer, J.S. Type I and type II Na+ channel α-subunit polypeptides exhibit distinct spatial and temporal patterning, and association with auxiliary subunits in rat brain. J. Comparative Neurol. 1999, 412, 342–352. [Google Scholar] [CrossRef]
- Yu, F.H.; Westenbroek, R.E.; Silos-Santiago, I.; McCormick, K.A.; Lawson, D.; Ge, P.; Ferriera, H.; Lilly, J.; DiStefano, P.S.; Catterall, W.A.; et al. Sodium channel beta4, a new disulfide-linked auxiliary subunit with similarity to beta2. The J. Neurosci. 2003, 23, 7577–7585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mercer, J.N.; Chan, C.S.; Tkatch, T.; Held, J.; Surmeier, D.J. Nav1.6 Sodium Channels Are Critical to Pacemaking and Fast Spiking in Globus Pallidus Neurons. J. Neurosci. 2007, 27, 13552–13566. [Google Scholar] [CrossRef]
- Whitaker, W.R.; Faull, R.L.; Waldvogel, H.J.; Plumpton, C.J.; Emson, P.C.; Clare, J.J. Comparative distribution of voltage-gated sodium channel proteins in human brain. Brain Res. Mol. Brain Res. 2001, 88, 37–53. [Google Scholar] [CrossRef]
- Castelli, L.; Biella, G.; Toselli, M.; Magistretti, J. Resurgent Na+ current in pyramidal neurones of rat perirhinal cortex: axonal location of channels and contribution to depolarizing drive during repetitive firing. J. Physiol 2007, 582, 1179–1193. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Deprez, L.; Maljevic, S.; Pitsch, J.; Claes, L.; Hristova, D.; Jordanova, A.; Ala-Mello, S.; Bellan-Koch, A.; Blazevic, D.; et al. Molecular correlates of age-dependent seizures in an inherited neonatal-infantile epilepsy. Brain 2010, 133, 1403–1414. [Google Scholar] [CrossRef] [Green Version]
- Buffington, S.A.; Rasband, M.N. Na+ channel-dependent recruitment of Navbeta4 to axon initial segments and nodes of Ranvier. J. Neurosci. 2013, 33, 6191–6202. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Bardgett, M.E.; Wong, M.; Wozniak, D.F.; Lou, J.; McNeil, B.D.; Chen, C.; Nardi, A.; Reid, D.C.; Yamada, K.; et al. Ataxia and paroxysmal dyskinesia in mice lacking axonally transported FGF14. Neuron 2002, 35, 25–38. [Google Scholar] [CrossRef] [Green Version]
- Grieco, T.M.; Malhotra, J.D.; Chen, C.; Isom, L.L.; Raman, I.M. Open-channel block by the cytoplasmic tail of sodium channel beta4 as a mechanism for resurgent sodium current. Neuron 2005, 45, 233–244. [Google Scholar] [CrossRef] [Green Version]
- Bant, J.S.; Raman, I.M. Control of transient, resurgent, and persistent current by open-channel block by Na channel beta4 in cultured cerebellar granule neurons. Proc. Natl. Acad. Sci. USA 2010, 107, 12357–12362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, H.; Pablo, J.L.; Wang, C.; Pitt, G.S. FGF14 modulates resurgent sodium current in mouse cerebellar Purkinje neurons. eLife 2014, 3, e04193. [Google Scholar] [CrossRef] [PubMed]
- Raman, I.M.; Sprunger, L.K.; Meisler, M.H.; Bean, B.P. Altered subthreshold sodium currents and disrupted firing patterns in Purkinje neurons of Scn8a mutant mice. Neuron 1997, 19, 881–891. [Google Scholar] [CrossRef] [Green Version]
- Cummins, T.R.; Dib-Hajj, S.D.; Herzog, R.I.; Waxman, S.G. Nav1.6 channels generate resurgent sodium currents in spinal sensory neurons. FEBS Lett. 2005, 579, 2166–2170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aman, T.K.; Raman, I.M. Subunit dependence of Na channel slow inactivation and open channel block in cerebellar neurons. Biophys. J. 2007, 92, 1938–1951. [Google Scholar] [CrossRef] [Green Version]
- Enomoto, A.; Han, J.M.; Hsiao, C.F.; Chandler, S.H. Sodium currents in mesencephalic trigeminal neurons from Nav1.6 null mice. J. Neurophysiol. 2007, 98, 710–719. [Google Scholar] [CrossRef] [Green Version]
- Osorio, N.; Cathala, L.; Meisler, M.H.; Crest, M.; Magistretti, J.; Delmas, P. Persistent Nav1. 6 current at axon initial segments tunes spike timing of cerebellar granule cells. J. Physiol. 2010, 588, 651–670. [Google Scholar] [CrossRef]
- Rush, A.M.; Dib-Hajj, S.D.; Waxman, S.G. Electrophysiological properties of two axonal sodium channels, Nav1.2 and Nav1.6, expressed in mouse spinal sensory neurones. J. Physiol. 2005, 564, 803–815. [Google Scholar] [CrossRef]
- Vreugdenhil, M.; Hoogland, G.; van Veelen, C.W.; Wadman, W.J. Persistent sodium current in subicular neurons isolated from patients with temporal lobe epilepsy. Eur. J. Neurosci. 2004, 19, 2769–2778. [Google Scholar] [CrossRef]
- Yue, C.; Remy, S.; Su, H.; Beck, H.; Yaari, Y. Proximal persistent Na+ channels drive spike afterdepolarizations and associated bursting in adult CA1 pyramidal cells. J. Neurosci. 2005, 25, 9704–9720. [Google Scholar] [CrossRef] [Green Version]
- Raman, I.M.; Bean, B.P. Resurgent sodium current and action potential formation in dissociated cerebellar Purkinje neurons. J. Neurosci. 1997, 17, 4517–4526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khaliq, Z.M.; Gouwens, N.W.; Raman, I.M. The contribution of resurgent sodium current to high-frequency firing in Purkinje neurons: an experimental and modeling study. J. Neurosci. 2003, 23, 4899–4912. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, C.; Tan, Z.Y.; Wang, R.; Xie, W.; Strong, J.A.; Patel, R.R.; Vasko, M.R.; Zhang, J.M.; Cummins, T.R. Navbeta4 regulates fast resurgent sodium currents and excitability in sensory neurons. Mol. Pain 2015, 11, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, Y.; Barbosa, C.; Pei, Z.; Xie, W.; Strong, J.A.; Zhang, J.M.; Cummins, T.R. Increased resurgent sodium currents in Nav1.8 contribute to nociceptive sensory neuron hyperexcitability associated with peripheral neuropathies. J. Neurosci. 2019. [Google Scholar] [CrossRef] [PubMed]
- Van Drongelen, W.; Koch, H.; Elsen, F.P.; Lee, H.C.; Mrejeru, A.; Doren, E.; Marcuccilli, C.J.; Hereld, M.; Stevens, R.L.; Ramirez, J.M. Role of persistent sodium current in bursting activity of mouse neocortical networks in vitro. J. Neurophysiol 2006, 96, 2564–2577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afshari, F.S.; Ptak, K.; Khaliq, Z.M.; Grieco, T.M.; Slater, N.T.; McCrimmon, D.R.; Raman, I.M. Resurgent Na currents in four classes of neurons of the cerebellum. J. Neurophysiol. 2004, 92, 2831–2843. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Kushmerick, C.; von Gersdorff, H. Presynaptic resurgent Na+ currents sculpt the action potential waveform and increase firing reliability at a CNS nerve terminal. J. Neurosci. 2010, 30, 15479–15490. [Google Scholar] [CrossRef] [Green Version]
- Hargus, N.J.; Nigam, A.; Bertram, E.H., 3rd; Patel, M.K. Evidence for a role of Nav1.6 in facilitating increases in neuronal hyperexcitability during epileptogenesis. J. Neurophysiol. 2013, 110, 1144–1157. [Google Scholar] [CrossRef] [Green Version]
- Barker, B.S.; Nigam, A.; Ottolini, M.; Gaykema, R.P.; Hargus, N.J.; Patel, M.K. Pro-excitatory alterations in sodium channel activity facilitate subiculum neuron hyperexcitability in temporal lobe epilepsy. Neurobiol. Dis. 2017, 108, 183–194. [Google Scholar] [CrossRef]
- Ottolini, M.; Barker, B.S.; Gaykema, R.P.; Meisler, M.H.; Patel, M.K. Aberrant Sodium Channel Currents and Hyperexcitability of Medial Entorhinal Cortex Neurons in a Mouse Model of SCN8A Encephalopathy. J. Neurosci. 2017, 37, 7643–7655. [Google Scholar] [CrossRef] [Green Version]
- Shao, H.; Yang, Y.; Qi, A.P.; Hong, P.; Zhu, G.X.; Cao, X.Y.; Ji, W.G.; Zhu, Z.R. Gastrodin Reduces the Severity of Status Epilepticus in the Rat Pilocarpine Model of Temporal Lobe Epilepsy by Inhibiting Nav1.6 Sodium Currents. Neurochem. Res. 2017, 42, 360–374. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, N.; Hahn, A.; Bast, T.; Muller, S.; Loffler, H.; Maljevic, S.; Gaily, E.; Prehl, I.; Biskup, S.; Joensuu, T.; et al. Mutations in the sodium channel gene SCN2A cause neonatal epilepsy with late-onset episodic ataxia. J. Neurol. 2016, 263, 334–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarecki, B.W.; Piekarz, A.D.; Jackson, J.O.; Cummins, T.R. Human voltage-gated sodium channel mutations that cause inherited neuronal and muscle channelopathies increase resurgent sodium currents. J. Clin. Invest. 2010, 120, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.R.; Barbosa, C.; Brustovetsky, T.; Brustovetsky, N.; Cummins, T.R. Aberrant epilepsy-associated mutant Nav1.6 sodium channel activity can be targeted with cannabidiol. Brain 2016, 139, 2164–2181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berecki, G.; Howell, K.B.; Deerasooriya, Y.H.; Cilio, M.R.; Oliva, M.K.; Kaplan, D.; Scheffer, I.E.; Berkovic, S.F.; Petrou, S. Dynamic action potential clamp predicts functional separation in mild familial and severe de novo forms of SCN2A epilepsy. Proc. Natl. Acad. Sci. USA. 2018, 115, E5516–e5525. [Google Scholar] [CrossRef] [Green Version]
- Kearney, J.A.; Plummer, N.W.; Smith, M.R.; Kapur, J.; Cummins, T.R.; Waxman, S.G.; Goldin, A.L.; Meisler, M.H. A gain-of-function mutation in the sodium channel gene Scn2a results in seizures and behavioral abnormalities. Neuroscience 2001, 102, 307–317. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-first-drug-comprised-active-ingredient-derived-marijuana-treat-rare-severe-forms (accessed on 27 February 2020).
- Hill, A.J.; Jones, N.A.; Smith, I.; Hill, C.L.; Williams, C.M.; Stephens, G.J.; Whalley, B.J. Voltage-gated sodium (NaV) channel blockade by plant cannabinoids does not confer anticonvulsant effects per se. Neurosci. Lett. 2014, 566, 269–274. [Google Scholar] [CrossRef]
- Mao, K.; You, C.; Lei, D.; Zhang, H. High dosage of cannabidiol (CBD) alleviates pentylenetetrazole-induced epilepsy in rats by exerting an anticonvulsive effect. Int. J. Clin. Exp. Med. 2015, 8, 8820–8827. [Google Scholar]
- Jones, N.A.; Hill, A.J.; Smith, I.; Bevan, S.A.; Williams, C.M.; Whalley, B.J.; Stephens, G.J. Cannabidiol displays antiepileptiform and antiseizure properties in vitro and in vivo. J. Pharmacol. Exp. Tther. 2010, 332, 569–577. [Google Scholar] [CrossRef] [Green Version]
- Devinsky, O.; Marsh, E.; Friedman, D.; Thiele, E.; Laux, L.; Sullivan, J.; Miller, I.; Flamini, R.; Wilfong, A.; Filloux, F.; et al. Cannabidiol in patients with treatment-resistant epilepsy: an open-label interventional trial. Lancet Neurol. 2016, 15, 270–278. [Google Scholar] [CrossRef]
- Devinsky, O.; Cross, J.H.; Laux, L.; Marsh, E.; Miller, I.; Nabbout, R.; Scheffer, I.E.; Thiele, E.A.; Wright, S. Trial of Cannabidiol for Drug-Resistant Seizures in the Dravet Syndrome. N. Engl. J. Med. 2017, 376, 2011–2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devinsky, O.; Patel, A.D.; Cross, J.H.; Villanueva, V.; Wirrell, E.C.; Privitera, M.; Greenwood, S.M.; Roberts, C.; Checketts, D.; VanLandingham, K.E.; et al. Effect of Cannabidiol on Drop Seizures in the Lennox-Gastaut Syndrome. N. Engl. J. Med. 2018, 378, 1888–1897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Press, C.A.; Knupp, K.G.; Chapman, K.E. Parental reporting of response to oral cannabis extracts for treatment of refractory epilepsy. Epilepsy Behav. 2015, 45, 49–52. [Google Scholar] [CrossRef] [PubMed]
- Thiele, E.A.; Marsh, E.D.; French, J.A.; Mazurkiewicz-Beldzinska, M.; Benbadis, S.R.; Joshi, C.; Lyons, P.D.; Taylor, A.; Roberts, C. Cannabidiol in patients with seizures associated with Lennox-Gastaut syndrome (GWPCARE4): A randomised, double-blind, placebo-controlled phase 3 trial. Lancet 2018, 391, 1085–1096. [Google Scholar] [CrossRef]
- Szaflarski, J.P.; Bebin, E.M.; Comi, A.M.; Patel, A.D.; Joshi, C.; Checketts, D.; Beal, J.C.; Laux, L.C.; De Boer, L.M.; Wong, M.H.; et al. Long-term safety and treatment effects of cannabidiol in children and adults with treatment-resistant epilepsies: Expanded access program results. Epilepsia 2018, 59(8), 1540–1548. [Google Scholar] [CrossRef] [Green Version]
- Szaflarski, J.P.; Bebin, E.M.; Cutter, G.; DeWolfe, J.; Dure, L.S.; Gaston, T.E.; Kankirawatana, P.; Liu, Y.; Singh, R.; Standaert, D.G.; et al. Cannabidiol improves frequency and severity of seizures and reduces adverse events in an open-label add-on prospective study. Epilepsy Behav. 2018, 87, 131–136. [Google Scholar] [CrossRef] [Green Version]
- Savage, T.E.; Sourbron, J.; Bruno, P.L.; Skirvin, L.A.; Wolper, E.S.; Anagnos, C.J.; Thiele, E.A. Efficacy of cannabidiol in subjects with refractory epilepsy relative to concomitant use of clobazam. Epilepsy Res. 2019, 160, 106263. [Google Scholar] [CrossRef]
- Ghovanloo, M.R.; Shuart, N.G.; Mezeyova, J.; Dean, R.A.; Ruben, P.C.; Goodchild, S.J. Inhibitory effects of cannabidiol on voltage-dependent sodium currents. J. Biol. Chem. 2018. [Google Scholar] [CrossRef] [Green Version]
- Premoli, M.; Aria, F.; Bonini, S.A.; Maccarinelli, G.; Gianoncelli, A.; Pina, S.D.; Tambaro, S.; Memo, M.; Mastinu, A. Cannabidiol: Recent advances and new insights for neuropsychiatric disorders treatment. Life Sci. 2019, 224, 120–127. [Google Scholar] [CrossRef]
- Baker, E.M.; Thompson, C.H.; Hawkins, N.A.; Wagnon, J.L.; Wengert, E.R.; Patel, M.K.; George, A.L., Jr.; Meisler, M.H.; Kearney, J.A. The novel sodium channel modulator GS-458967 (GS967) is an effective treatment in a mouse model of SCN8A encephalopathy. Epilepsia 2018, 59, 1166–1176. [Google Scholar] [CrossRef] [Green Version]
- Wengert, E.R.; Saga, A.U.; Panchal, P.S.; Barker, B.S.; Patel, M.K. Prax330 reduces persistent and resurgent sodium channel currents and neuronal hyperexcitability of subiculum neurons in a mouse model of SCN8A epileptic encephalopathy. Neuropharmacology 2019, 107699. [Google Scholar] [CrossRef] [PubMed]
- Anderson, L.L.; Thompson, C.H.; Hawkins, N.A.; Nath, R.D.; Petersohn, A.A.; Rajamani, S.; Bush, W.S.; Frankel, W.N.; Vanoye, C.G.; Kearney, J.A.; et al. Antiepileptic activity of preferential inhibitors of persistent sodium current. Epilepsia 2014, 55, 1274–1283. [Google Scholar] [CrossRef] [PubMed]
- Anderson, L.L.; Hawkins, N.A.; Thompson, C.H.; Kearney, J.A.; George, A.L., Jr. Unexpected Efficacy of a Novel Sodium Channel Modulator in Dravet Syndrome. Sci. Rep. 2017, 7, 1682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bunton-Stasyshyn, R.K.A.; Wagnon, J.L.; Wengert, E.R.; Barker, B.S.; Faulkner, A.; Wagley, P.K.; Bhatia, K.; Jones, J.M.; Maniaci, M.R.; Parent, J.M.; et al. Prominent role of forebrain excitatory neurons in SCN8A encephalopathy. Brain 2019, 142, 362–375. [Google Scholar] [CrossRef] [Green Version]
- DuBridge, R.B.; Tang, P.; Hsia, H.C.; Leong, P.M.; Miller, J.H.; Calos, M.P. Analysis of mutation in human cells by using an Epstein-Barr virus shuttle system. Mol. Cell Biol. 1987, 7, 379–387. [Google Scholar] [CrossRef]
- Aman, T.K.; Grieco-Calub, T.M.; Chen, C.; Rusconi, R.; Slat, E.A.; Isom, L.L.; Raman, I.M. Regulation of persistent Na current by interactions between beta subunits of voltage-gated Na channels. J. Neurosci. 2009, 29, 2027–2042. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Yu, F.H.; Sharp, E.M.; Beacham, D.; Scheuer, T.; Catterall, W.A. Functional properties and differential neuromodulation of Na(v)1.6 channels. Mol. Cell Neurosci. 2008, 38, 607–615. [Google Scholar] [CrossRef] [Green Version]
- Potet, F.; Vanoye, C.G.; George, A.L., Jr. Use-Dependent Block of Human Cardiac Sodium Channels by GS967. Mol. Pharmacol. 2016, 90, 52–60. [Google Scholar] [CrossRef] [Green Version]
- Cummins, T.R.; Xia, Y.; Haddad, G.G. Functional properties of rat and human neocortical voltage-sensitive sodium currents. J. Neurophysiol. 1994, 71, 1052–1064. [Google Scholar] [CrossRef]









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mason, E.R.; Cummins, T.R. Differential Inhibition of Human Nav1.2 Resurgent and Persistent Sodium Currents by Cannabidiol and GS967. Int. J. Mol. Sci. 2020, 21, 2454. https://doi.org/10.3390/ijms21072454
Mason ER, Cummins TR. Differential Inhibition of Human Nav1.2 Resurgent and Persistent Sodium Currents by Cannabidiol and GS967. International Journal of Molecular Sciences. 2020; 21(7):2454. https://doi.org/10.3390/ijms21072454
Chicago/Turabian StyleMason, Emily R., and Theodore R. Cummins. 2020. "Differential Inhibition of Human Nav1.2 Resurgent and Persistent Sodium Currents by Cannabidiol and GS967" International Journal of Molecular Sciences 21, no. 7: 2454. https://doi.org/10.3390/ijms21072454
APA StyleMason, E. R., & Cummins, T. R. (2020). Differential Inhibition of Human Nav1.2 Resurgent and Persistent Sodium Currents by Cannabidiol and GS967. International Journal of Molecular Sciences, 21(7), 2454. https://doi.org/10.3390/ijms21072454