
Epigenetic Age Acceleration Is Not Associated with Age-Related Macular Degeneration

 and
and
Abstract
:1. Introduction
2. Results
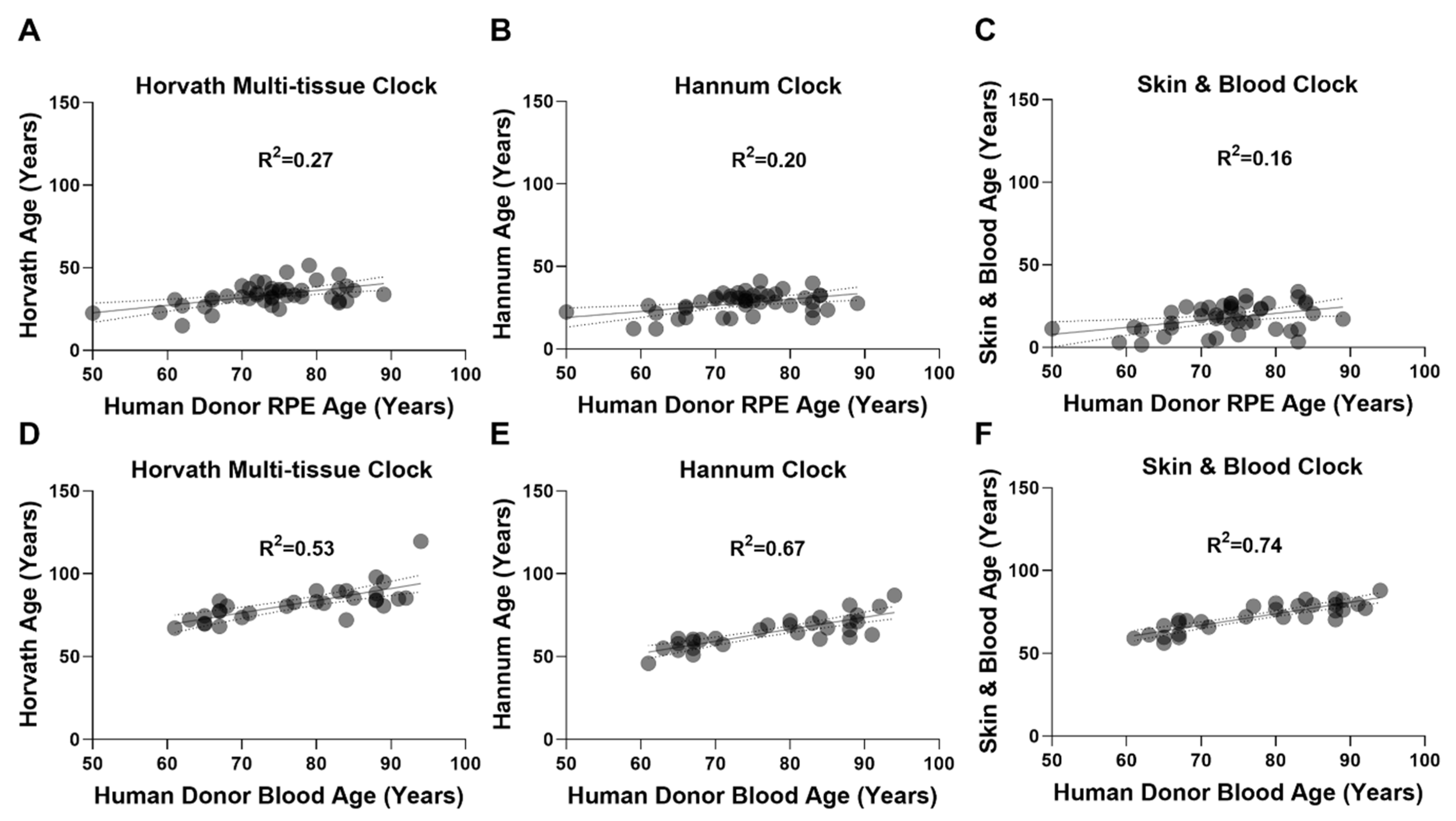
2.1. Epigenetic Clocks Display Poor Performance in the RPE
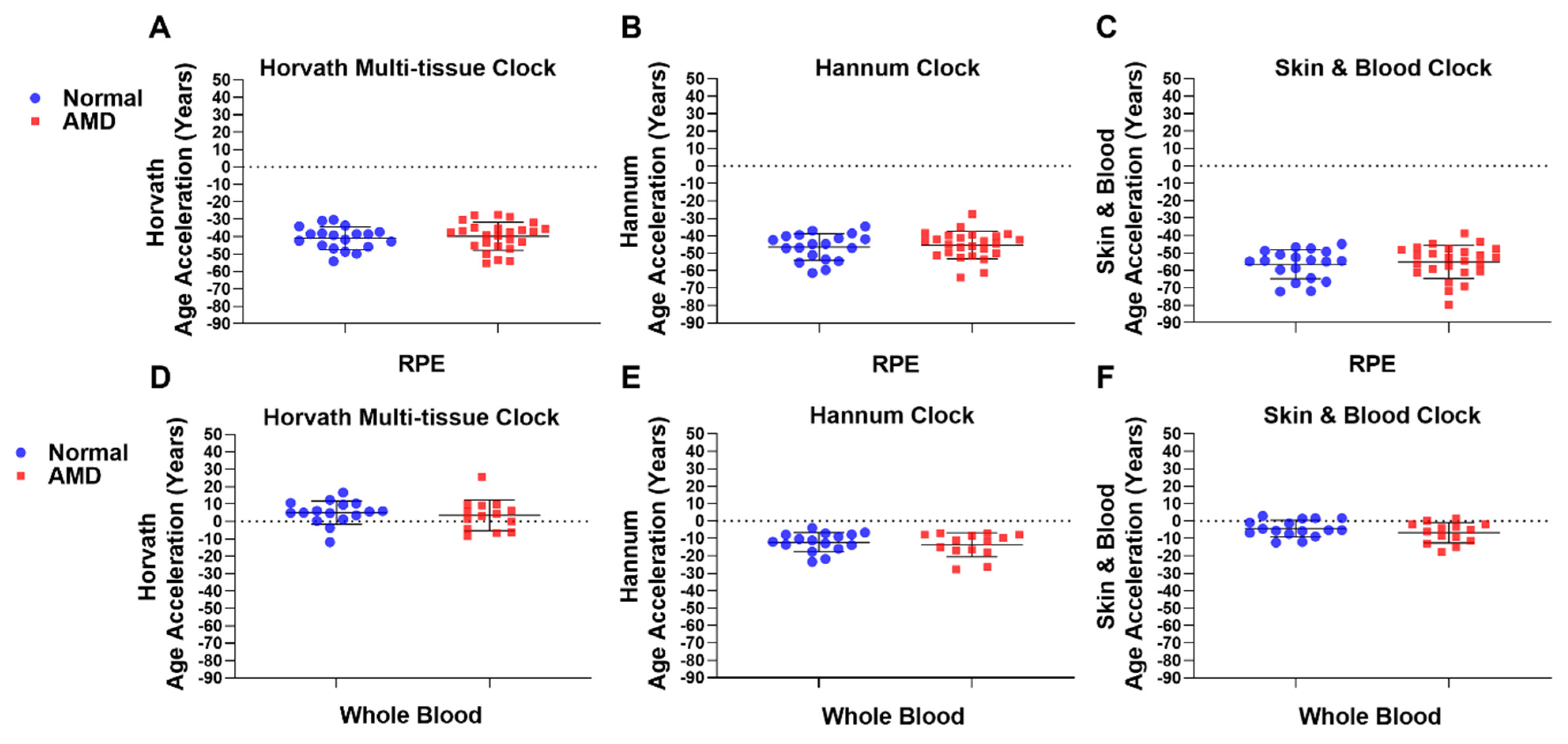
2.2. Epigenetic Age Acceleration Is Not Associated with AMD
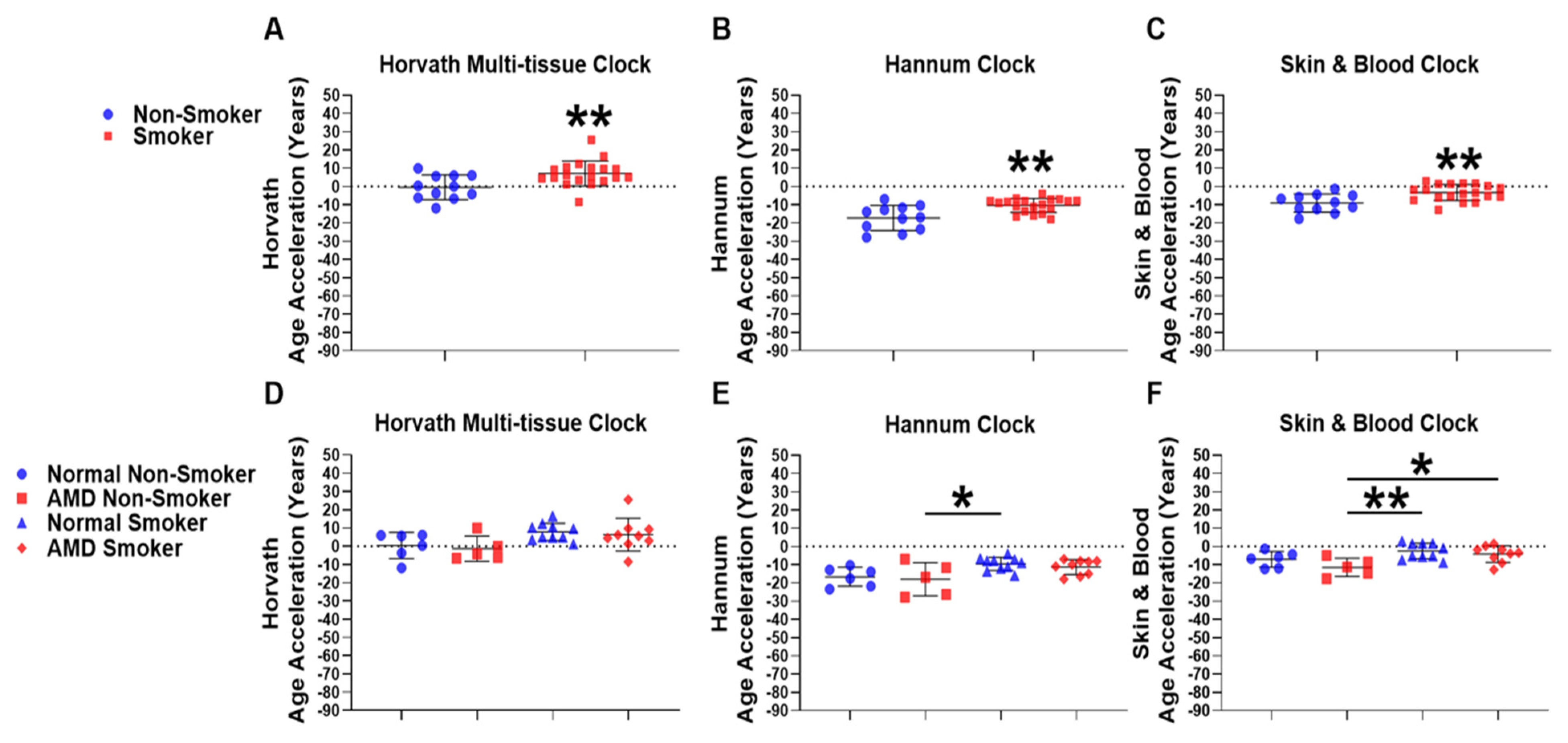
2.3. Smokers Display Increased Epigenetic Age Acceleration Compared to Non-Smokers
2.4. AMD Smokers Display Positive Epigenetic Age Acceleration Using the Skin & Blood Epigenetic Clock
2.5. Differentially Methylated CpG Probes Identified in Whole Blood gDNA from AMD Patients
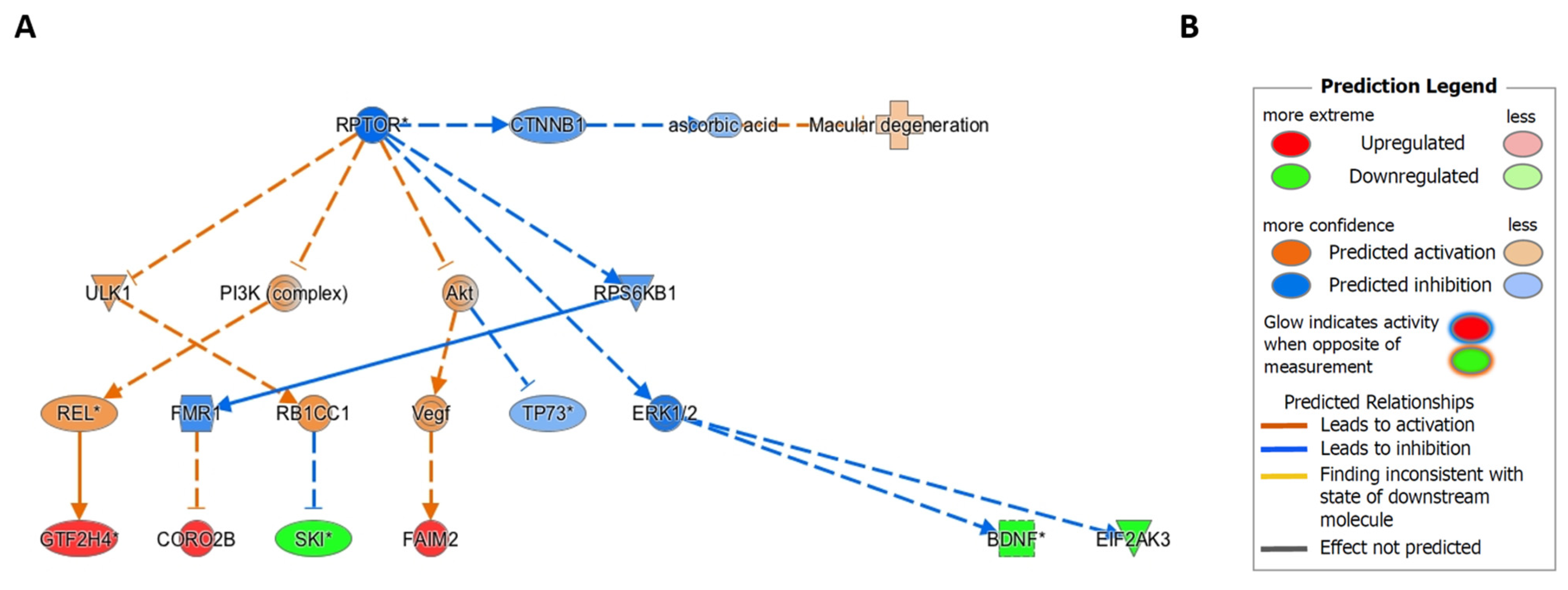
2.6. Ingenuity Pathway Causal Network Analysis Identified RPTOR as a Master Regulator of Methylation Changes in RPE
3. Discussion
4. Materials and Methods
4.1. Sample Collection, Grading and DNA Extraction
4.2. Illumina Infinum MethylationEPIC BeadChip Array
4.3. Pre-Processing and Normalisation
4.4. Epigenetic Clock Analyses
4.5. Differentially Methylated CpG Probe and Region Analysis
4.6. Histone Modification Enrichment and GeneHancer Analysis
4.7. Ingenuity Pathway Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 450K-array | Illumina Infinium HumanMethylation450 BeadChip Array |
| AREDS | Age-related Eye Disease Study |
| AMD | Age-related Macular Degeneration |
| DNAm | DNA methylation |
| DNAm Age | DNA methylation age |
| EAA | Epigenetic Age Acceleration |
| EMS | Epigenetic Maintenance System |
| EPIC-array | Illumina Infinium MethylationEPIC BeadChip Array |
| FDR | False Discovery Rate |
| gDNA | Genomic DNA |
| IPA CNA | Ingenuity Pathway Analysis; Causal Network Analysis |
| mTORC1 | Mammalian Target of Rapamycin Complex 1 |
| PERK | Protein Kinase ER-like Kinase |
| RAPTOR | Regulatory Associated Protein of mTOR Complex 1 |
| RPE | Retinal Pigment Epithelium |
| SWAN | Subset-quantile Within Array Normalisation |
References
- Niccoli, T.; Partridge, L. Ageing as a Risk Factor for Disease. Curr. Biol. 2012, 22, R741–R752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, M.; Pfeifer, G.P. Aging and DNA methylation. BMC Biol. 2015, 13, 7. [Google Scholar] [CrossRef] [Green Version]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, R115. [Google Scholar] [CrossRef] [Green Version]
- Hannum, G.; Guinney, J.; Zhao, L.; Zhang, L.; Hughes, G.; Sadda, S.; Klotzle, B.; Bibikova, M.; Fan, J.-B.; Gao, Y.; et al. Genome-wide Methylation Profiles Reveal Quantitative Views of Human Aging Rates. Mol. Cell 2012, 49, 359–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, M.E.; Lu, A.T.; Quach, A.; Chen, B.H.; Assimes, T.L.; Bandinelli, S.; Hou, L.; Baccarelli, A.A.; Stewart, J.D.; Li, Y.; et al. An epigenetic biomarker of aging for lifespan and healthspan. Aging 2018, 10, 573–591. [Google Scholar] [CrossRef] [Green Version]
- Bergsma, T.; Rogaeva, E. DNA Methylation Clocks and Their Predictive Capacity for Aging Phenotypes and Healthspan. Neurosci. Insights 2020, 15, 2633105520942221. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S.; Raj, K. DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat. Rev. Genet. 2018, 19, 371–384. [Google Scholar] [CrossRef]
- Horvath, S.; Oshima, J.; Martin, G.M.; Lu, A.T.; Quach, A.; Cohen, H.; Felton, S.; Matsuyama, M.; Lowe, D.; Kabacik, S.; et al. Epigenetic clock for skin and blood cells applied to Hutchinson Gilford Progeria Syndrome and ex vivo studies. Aging 2018, 10, 1758–1775. [Google Scholar] [CrossRef]
- Levine, M.E.; Lu, A.T.; Bennett, D.A.; Horvath, S. Epigenetic age of the pre-frontal cortex is associated with neuritic plaques, amyloid load, and Alzheimer’s disease related cognitive functioning. Aging 2015, 7, 1198–1211. [Google Scholar] [CrossRef]
- Horvath, S.; Ritz, B.R. Increased epigenetic age and granulocyte counts in the blood of Parkinson’s disease patients. Aging 2015, 7, 1130–1142. [Google Scholar] [CrossRef] [Green Version]
- Xiao, C.; Beitler, J.J.; Peng, G.; Levine, M.E.; Conneely, K.N.; Zhao, H.; Felger, J.C.; Bs, E.C.W.; Bs, C.E.C.; Jeon, S.; et al. Epigenetic age acceleration, fatigue, and inflammation in patients undergoing radiation therapy for head and neck cancer: A longitudinal study. Cancer 2021, 127, 3361–3371. [Google Scholar] [CrossRef]
- Horvath, S.; Mah, V.; Lu, A.T.; Woo, J.S.; Choi, O.-W.; Jasinska, A.J.; Riancho, J.A.; Tung, S.; Coles, N.S.; Braun, J.; et al. The cerebellum ages slowly according to the epigenetic clock. Aging 2015, 7, 294–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, C.; Li, L.; Xu, R. Association of Epigenetic Clock with Consensus Molecular Subtypes and Overall Survival of Colorectal Cancer. Cancer Epidemiol. Biomark. Prev. 2019, 28, 1720–1724. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Maden, S.; Luebeck, G.E.; Li, C.I.; Newcomb, P.A.; Ulrich, C.M.; Joo, J.-H.E.; Buchanan, D.D.; Milne, R.L.; Southey, M.C.; et al. Dysfunctional epigenetic aging of the normal colon and colorectal cancer risk. Clin. Epigenet. 2020, 12, 5. [Google Scholar] [CrossRef]
- Ren, J.-T.; Wang, M.-X.; Su, Y.; Tang, L.-Y.; Ren, Z.-F. Decelerated DNA methylation age predicts poor prognosis of breast cancer. BMC Cancer 2018, 18, 989. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, P.; Liew, G.; Gopinath, B.; Wong, T.Y. Age-related macular degeneration. Lancet 2018, 392, 1147–1159. [Google Scholar] [CrossRef]
- Bourne, R.; Steinmetz, J.D.; Flaxman, S.; Briant, P.S.; Taylor, H.R.; Resnikoff, S.; Casson, R.J.; Abdoli, A.; Abu-Gharbieh, E.; Afshin, A.; et al. Trends in prevalence of blindness and distance and near vision impairment over 30 years: An analysis for the Global Burden of Disease Study. Lancet Glob. Heath 2021, 9, e130–e143. [Google Scholar] [CrossRef]
- Wong, W.L.; Su, X.; Li, X.; Cheung, C.M.G.; Klein, R.; Cheng, C.Y.; Wong, T.Y. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: A systematic review and meta-analysis. Lancet Glob. Health 2014, 2, e106–e116. [Google Scholar] [CrossRef] [Green Version]
- Khan, J.C.; Thurlby, D.A.; Shahid, H.; Clayton, D.; Yates, J.R.W.; Bradley, M.M.; Moore, A.T.; Bird, A.C. Smoking and age related macular degeneration: The number of pack years of cigarette smoking is a major determinant of risk for both geographic atrophy and choroidal neovascularisation. Br. J. Ophthalmol. 2006, 90, 75–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, J.S.L.; Mitchell, P.; Kifley, A.; Flood, V.; Smith, W.; Wang, J.J. Smoking and the Long-term Incidence of Age-Related Macular Degeneration: The blue mountains eye study. Arch. Ophthalmol. 2007, 125, 1089–1095. [Google Scholar] [CrossRef] [Green Version]
- Vingerling, J.R.; Hofman, A.; Grobbee, D.E.; De Jong, P.T.V.M. Age-Related Macular Degeneration and Smoking: The Rotterdam study. Arch. Ophthalmol. 1996, 114, 1193–1196. [Google Scholar] [CrossRef] [Green Version]
- Klein, R.; Klein, B.E.K.; Moss, S.E. Relation of Smoking to the Incidence of Age-related Maculopathy: The Beaver Dam Eye Study. Am. J. Epidemiol. 1998, 147, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Hunter, A.; Spechler, P.A.; Cwanger, A.; Song, Y.; Zhang, Z.; Ying, G.-S.; Hunter, A.K.; Dezoeten, E.; Dunaief, J.L. DNA Methylation Is Associated with Altered Gene Expression in AMD. Investig. Opthalmol. Vis. Sci. 2012, 53, 2089–2105. [Google Scholar] [CrossRef]
- Wei, L.; Liu, B.; Tuo, J.; Shen, D.; Chen, P.; Li, Z.; Liu, X.; Ni, J.; Dagur, P.; Sen, H.N.; et al. Hypomethylation of the IL17RC Promoter Associates with Age-Related Macular Degeneration. Cell Rep. 2012, 2, 1151–1158. [Google Scholar] [CrossRef] [Green Version]
- Oliver, V.F.; Franchina, M.; Jaffe, A.E.; Branham, K.E.; Othman, M.; Heckenlively, J.R.; Swaroop, A.; Campochiaro, B.; Vote, B.J.; Craig, J.E.; et al. Hypomethylation of the IL17RC Promoter in Peripheral Blood Leukocytes Is Not A Hallmark of Age-Related Macular Degeneration. Cell Rep. 2013, 5, 1527–1535. [Google Scholar] [CrossRef] [Green Version]
- Oliver, V.F.; Jaffe, A.; Song, J.; Wang, G.; Zhang, P.; Branham, K.; Swaroop, A.; Eberhart, C.G.; Zack, D.J.; Qian, J.; et al. Differential DNA methylation identified in the blood and retina of AMD patients. Epigenetics 2015, 10, 698–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porter, L.F.; Saptarshi, N.; Fang, Y.; Rathi, S.; Hollander, A.I.D.; De Jong, E.K.; Clark, S.J.; Bishop, P.N.; Olsen, T.W.; Liloglou, T.; et al. Whole-genome methylation profiling of the retinal pigment epithelium of individuals with age-related macular degeneration reveals differential methylation of the SKI, GTF2H4, and TNXB genes. Clin. Epigenet. 2019, 11, 6. [Google Scholar] [CrossRef]
- Kõks, G.; Uudelepp, M.-L.; Limbach, M.; Peterson, P.; Reimann, E.; Kõks, S. Smoking-Induced Expression of the GPR15 Gene Indicates Its Potential Role in Chronic Inflammatory Pathologies. Am. J. Pathol. 2015, 185, 2898–2906. [Google Scholar] [CrossRef] [Green Version]
- Ambatipudi, S.; Cuenin, C.; Hernandez-Vargas, H.; Ghantous, A.; Le Calvez-Kelm, F.; Kaaks, R.; Barrdahl, M.; Boeing, H.; Aleksandrova, K.; Trichopoulou, A.; et al. Tobacco smoking-associated genome-wide DNA methylation changes in the EPIC study. Epigenomics 2016, 8, 599–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Huang, Q.; Javed, R.; Zhong, J.; Gao, H.; Liang, H. Effect of tobacco smoking on the epigenetic age of human respiratory organs. Clin. Epigenet. 2019, 11, 183. [Google Scholar] [CrossRef]
- Hewitt, A.W.; Januar, V.; Sexton-Oates, A.; Joo, J.E.; Franchina, M.; Wang, J.J.; Liang, H.; Craig, J.; Saffery, R. DNA methylation landscape of ocular tissue relative to matched peripheral blood. Sci. Rep. 2017, 7, 46330. [Google Scholar] [CrossRef] [Green Version]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- El Khoury, L.Y.; Gorrie-Stone, T.; Smart, M.; Hughes, A.; Bao, Y.; Andrayas, A.; Burrage, J.; Hannon, E.; Kumari, M.; Mill, J.; et al. Systematic underestimation of the epigenetic clock and age acceleration in older subjects. Genome Biol. 2019, 20, 283. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Chao, D.L.; Rocha, L.; Kolar, M.; Huu, V.A.N.; Krawczyk, M.; Dasyani, M.; Wang, T.; Jafari, M.; Jabari, M.; et al. The lipid elongation enzyme ELOVL2 is a molecular regulator of aging in the retina. Aging Cell 2020, 19, e13100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voisin, S.; Harvey, N.R.; Haupt, L.M.; Griffiths, L.; Ashton, K.; Coffey, V.G.; Doering, T.; Thompson, J.-L.; Benedict, C.; Cedernaes, J.; et al. An epigenetic clock for human skeletal muscle. J. Cachex-Sarcopenia Muscle 2020, 11, 887–898. [Google Scholar] [CrossRef] [Green Version]
- Castle, J.R.; Lin, N.; Liu, J.; Storniolo, A.M.V.; Shendre, A.; Hou, L.; Horvath, S.; Liu, Y.; Wang, C.; He, C. Estimating breast tissue-specific DNA methylation age using next-generation sequencing data. Clin. Epigenet. 2020, 12, 45. [Google Scholar] [CrossRef] [PubMed]
- Ecker, S.; Beck, S. The epigenetic clock: A molecular crystal ball for human aging? Aging 2019, 11, 833–835. [Google Scholar] [CrossRef]
- Breton, C.V.; Marsit, C.; Faustman, E.; Nadeau, K.; Goodrich, J.; Dolinoy, D.C.; Herbstman, J.; Holland, N.; LaSalle, J.; Schmidt, R.; et al. Small-Magnitude Effect Sizes in Epigenetic End Points are Important in Children’s Environmental Health Studies: The Children’s Environmental Health and Disease Prevention Research Center’s Epigenetics Working Group. Environ. Health Perspect. 2017, 125, 511–526. [Google Scholar] [CrossRef]
- Krämer, A.; Green, J.; Pollard, J.; Tugendreich, S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef]
- Geeleher, P.; Hartnett, L.; Egan, L.J.; Golden, A.; Ali, R.A.R.; Seoighe, C. Gene-set analysis is severely biased when applied to genome-wide methylation data. Bioinformatics 2013, 29, 1851–1857. [Google Scholar] [CrossRef] [Green Version]
- Foster, K.G.; Acosta-Jaquez, H.A.; Romeo, Y.; Ekim, B.; Soliman, G.A.; Carriere, A.; Roux, P.P.; Ballif, B.A.; Fingar, D.C. Regulation of mTOR Complex 1 (mTORC1) by Raptor Ser863 and Multisite Phosphorylation. J. Biol. Chem. 2010, 285, 80–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Jiang, N.; Chu, Y.; Postnikova, O.; Varghese, R.; Horvath, A.; Cheema, A.K.; Golestaneh, N. Dysregulated metabolic pathways in age-related macular degeneration. Sci. Rep. 2020, 10, 2464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papadopoli, D.; Boulay, K.; Kazak, L.; Pollak, M.; Mallette, F.A.; Topisirovic, I.; Hulea, L. mTOR as a central regulator of lifespan and aging. F1000Research 2019, 8, 998. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Gu, S.; Chen, M.; Zhang, S.-J.; Jiang, Z.; Chen, X.; Jiang, C.; Liu, G.; Radu, R.A.; Sun, X.; et al. Abnormal mTORC1 signaling leads to retinal pigment epithelium degeneration. Theranostics 2019, 9, 1170–1180. [Google Scholar] [CrossRef]
- Krishnamoorthy, J.; Tenkerian, C.; Gupta, J.; Ghaddar, N.; Wang, S.; Darini, C.; Staschke, K.A.; Ghosh, A.; Gandin, V.; Topisirovic, I.; et al. Downregulation of PERK activity and eIF2α serine 51 phosphorylation by mTOR complex 1 elicits pro-oxidant and pro-death effects in tuberous sclerosis-deficient cells article. Cell Death Dis. 2018, 9, 254. [Google Scholar] [CrossRef] [Green Version]
- Scholl, H.P.N.; Issa, P.C.; Walier, M.; Janzer, S.; Pollok-Kopp, B.; Börncke, F.; Fritsche, L.; Chong, V.; Fimmers, R.; Wienker, T.; et al. Systemic Complement Activation in Age-Related Macular Degeneration. PLoS ONE 2008, 3, e2593. [Google Scholar] [CrossRef]
- Lu, Y.; Brommer, B.; Tian, X.; Krishnan, A.; Meer, M.; Wang, C.; Vera, D.L.; Zeng, Q.; Yu, D.; Bonkowski, M.S.; et al. Reprogramming to recover youthful epigenetic information and restore vision. Nat. Cell Biol. 2020, 588, 124–129. [Google Scholar] [CrossRef]
- Ennis, S.; Jomary, C.; Mullins, R.; Cree, A.; Chen, X.; MacLeod, A.; Jones, S.; Collins, A.; Stone, E.; Lotery, A. Association between the SERPING1 gene and age-related macular degeneration: A two-stage case-control study. Lancet 2008, 372, 1828–1834. [Google Scholar] [CrossRef] [Green Version]
- Age-Related Eye Disease Study Research Group The age-related eye disease study system for classifying age-related macular degeneration from stereoscopic color fundus photographs: The age-related eye disease study report number 6. Am. J. Ophthalmol. 2001, 132, 668–681. [CrossRef]
- Aryee, M.J.; Jaffe, A.E.; Corrada-Bravo, H.; Ladd-Acosta, C.; Feinberg, A.P.; Hansen, K.D.; Irizarry, R.A. Minfi: A flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 2014, 30, 1363–1369. [Google Scholar] [CrossRef] [Green Version]
- Koestler, D.C.; Jones, M.J.; Usset, J.; Christensen, B.C.; Butler, R.A.; Kobor, M.S.; Wiencke, J.K.; Kelsey, K.T. Improving cell mixture deconvolution by identifying optimal DNA methylation libraries (IDOL). BMC Bioinform. 2016, 17, 120. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-A.; Lemire, M.; Choufani, S.; Butcher, D.T.; Grafodatskaya, D.; Zanke, B.W.; Gallinger, S.; Hudson, T.J.; Weksberg, R. Discovery of cross-reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray. Epigenetics 2013, 8, 203–209. [Google Scholar] [CrossRef] [Green Version]
- Pidsley, R.; Zotenko, E.; Peters, T.J.; Lawrence, M.G.; Risbridger, G.P.; Molloy, P.; Van Djik, S.; Muhlhausler, B.; Stirzaker, C.; Clark, S.J. Critical evaluation of the Illumina MethylationEPIC BeadChip microarray for whole-genome DNA methylation profiling. Genome Biol. 2016, 17, 208. [Google Scholar] [CrossRef] [Green Version]
- Maksimovic, J.; Gordon, L.; Oshlack, A. SWAN: Subset-quantile within array normalization for illumina infinium HumanMethylation450 BeadChips. Genome Biol. 2012, 13, R44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Peters, T.J.; Buckley, M.J.; Statham, A.L.; Pidsley, R.; Samaras, K.; Lord, R.V.; Clark, S.J.; Molloy, P.L. De novo identification of differentially methylated regions in the human genome. Epigenet. Chromatin 2015, 8, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Probe I.D. | Gene I.D. | logFC | Unadjusted p-Value | Δβ (* ±≥10%) | Chromosome:Position (Hg19) | Relation to CpG Island | Relation to Gene |
|---|---|---|---|---|---|---|---|
| cg24522809 | UPP2 | −0.472041082 | 1.01 × 10−6 | −3.35 | chr2:158851594 | Open Sea | TSS200 |
| cg15985873 | NOX5 | 0.41292444 | 1.40 × 10−5 | 2.82 | chr15:69264323 | Open Sea | Body (Intron) |
| cg21175976 | BLK | −0.922913398 | 1.53 × 10−5 | −9.49 | chr8:11421337 | Island | Body (Intron) |
| cg07212053 | UBE4A | 0.480522948 | 1.87 × 10−5 | 1.1 | chr11:118230307 | Island | Body (1st Exon) |
| cg14426911 | SEMA5A | −0.637685904 | 2.07 × 10−5 | −4.36 | chr5:9363104 | Open Sea | Body (Intron) |
| cg05306123 | INTS7 | 0.52368943 | 2.60 × 10−5 | 4.33 | chr1:212159068 | Open Sea | Body (Intron) |
| cg06569202 | DBP | 0.511049469 | 4.21 × 10−5 | 1.42 | chr19:49140842 | Island | TSS200 |
| cg12917056 | PDC | 0.545557387 | 4.38 × 10−5 | 4.38 | chr1:186416576 | OpenSea | Body (Intron) |
| cg12855166 | MYO1D | −0.955212746 | 4.56 × 10−5 | −0.88 | chr17:30846586 | Island | Body (Intron) |
| cg23282837 | CSMD3 | −0.554156229 | 5.06 × 10−5 | −2.92 | chr8:114449418 | Open Sea | TSS200 |
| cg17303711 | ZSCAN22 | 0.472499092 | 5.27 × 10−5 | 1.68 | chr19:58838235 | Island | TSS200 |
| cg17303822 | - | 1.107838488 | 5.40 × 10−5 | 8.89 | chr4:120992550 | Open Sea | Intergenic |
| cg22945982 | VWA8 | 0.827121348 | 5.85 × 10−5 | 4.52 | chr13:42443309 | Open Sea | Body (Intron) |
| cg04953735 | RPTOR | −0.731183406 | 6.51 × 10−5 | −11.95 * | chr17:78652628 | Open Sea | Body (Intron) |
| cg08636246 | RSBN1 | 0.400242281 | 7.37 × 10−5 | 1.14 | chr1:114354993 | Island | Body (1st Exon) |
| cg17247365 | WWOX | −0.787476578 | 7.97 × 10−5 | −9.56 | chr16:78275151 | Open Sea | Body (Intron) |
| cg03380182 | - | 0.777809274 | 8.36 × 10−5 | 10.68 * | chr1:244065456 | Open Sea | Intergenic |
| cg13955747 | TMEM18 | 0.301564284 | 8.38 × 10−5 | 1.54 | chr2:677585 | Island | TSS200 |
| cg22541572 | LRCH1 | 0.662406112 | 8.71 × 10−5 | 4.64 | chr13:47237047 | Open Sea | Body (Intron) |
| cg07642595 | LAMP1 | −0.545207974 | 9.06 × 10−5 | −6.63 | chr13:113952548 | South Shore | Body (Intron) |
| cg09228785 | CD82 | 0.574387376 | 9.91 × 10−5 | 8.33 | chr11:44630602 | Open Sea | Body (Intron) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saptarshi, N.; Green, D.; Cree, A.; Lotery, A.; Paraoan, L.; Porter, L.F. Epigenetic Age Acceleration Is Not Associated with Age-Related Macular Degeneration. Int. J. Mol. Sci. 2021, 22, 13457. https://doi.org/10.3390/ijms222413457
Saptarshi N, Green D, Cree A, Lotery A, Paraoan L, Porter LF. Epigenetic Age Acceleration Is Not Associated with Age-Related Macular Degeneration. International Journal of Molecular Sciences. 2021; 22(24):13457. https://doi.org/10.3390/ijms222413457
Chicago/Turabian StyleSaptarshi, Neil, Daniel Green, Angela Cree, Andrew Lotery, Luminita Paraoan, and Louise F. Porter. 2021. "Epigenetic Age Acceleration Is Not Associated with Age-Related Macular Degeneration" International Journal of Molecular Sciences 22, no. 24: 13457. https://doi.org/10.3390/ijms222413457
APA StyleSaptarshi, N., Green, D., Cree, A., Lotery, A., Paraoan, L., & Porter, L. F. (2021). Epigenetic Age Acceleration Is Not Associated with Age-Related Macular Degeneration. International Journal of Molecular Sciences, 22(24), 13457. https://doi.org/10.3390/ijms222413457