
Mapping of Microglial Brain Region, Sex and Age Heterogeneity in Obesity



{kind=link}
Abstract
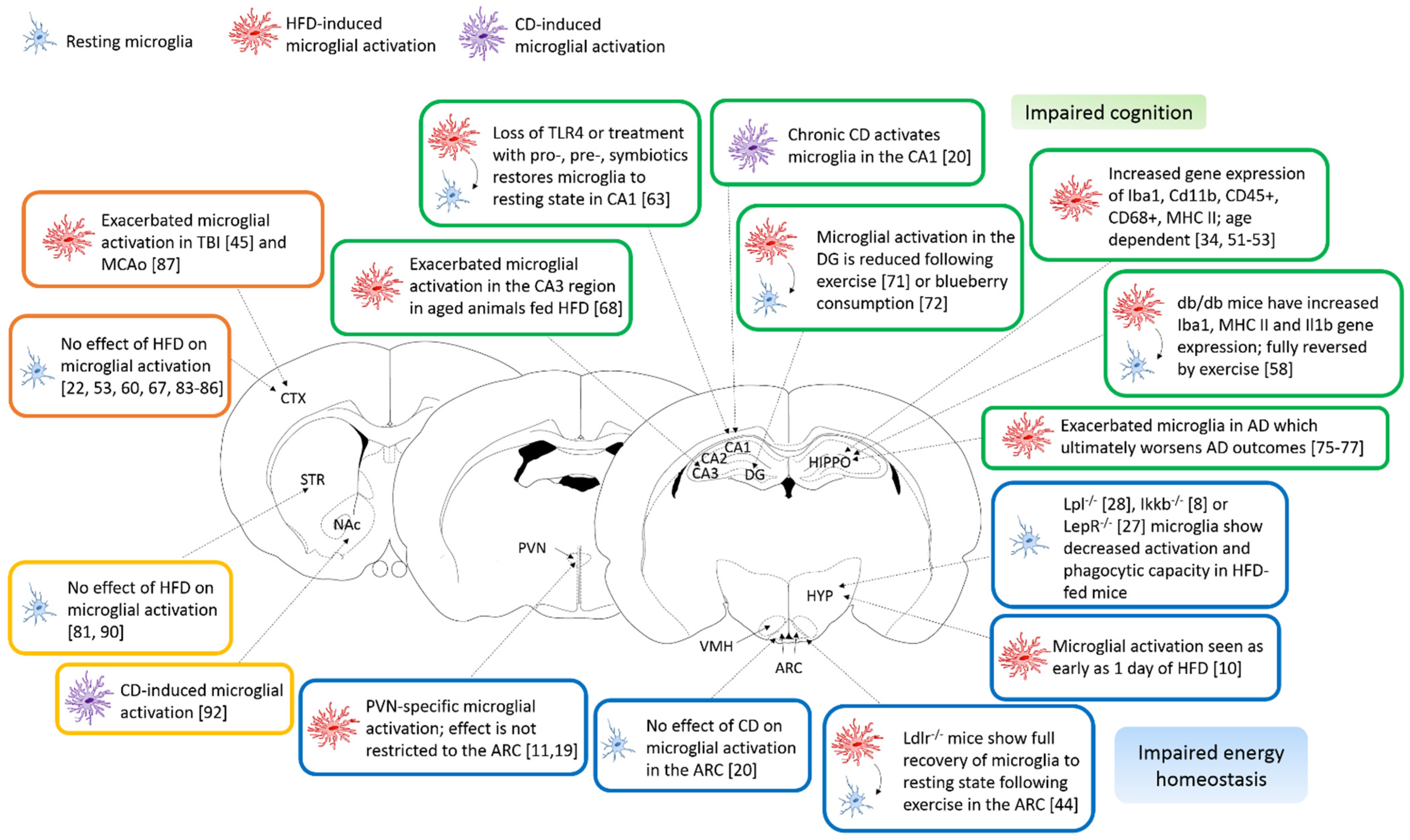
:1. Introduction
2. Hypothalamus
3. Hippocampus
3.1. CA1-CA3
3.2. DG
3.3. Hippocampus and Alzheimer’s Disease in Obesity
4. Cerebral Cortex
5. Striatum
6. Other Brain Regions
7. Effect of Overnutrition on Microglial Function in the pre- and Neonatal Stages of Life
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Janda, E.; Boi, L.; Carta, A.R. Microglial Phagocytosis and Its Regulation: A Therapeutic Target in Parkinson’s Disease? Front. Mol. Neurosci. 2018, 11, 144. [Google Scholar] [CrossRef] [Green Version]
- Valdearcos, M.; Robblee, M.M.; Benjamin, D.I.; Nomura, D.K.; Xu, A.W.; Koliwad, S.K. Microglia Dictate the Impact of Saturated Fat Consumption on Hypothalamic Inflammation and Neuronal Function. Cell Rep. 2014, 9, 2124–2138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.; Ottaway, N.; Schriever, S.C.; Legutko, B.; García-Cáceres, C.; De La Fuente, E.; Mergen, C.; Bour, S.; Thaler, J.P.; Seeley, R.J.; et al. Hormones and diet, but not body weight, control hypothalamic microglial activity. Glia 2014, 62, 17–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.; Bielohuby, M.; Fleming, T.; Grabner, G.F.; Foppen, E.; Bernhard, W.; Guzmán-Ruiz, M.; Layritz, C.; Legutko, B.; Zinser, E.; et al. Dietary sugars, not lipids, drive hypothalamic inflammation. Mol. Metab. 2017, 6, 897–908. [Google Scholar] [CrossRef] [PubMed]
- Orihuela, R.; McPherson, C.A.; Harry, G.J. Microglial M1/M2 polarization and metabolic states. Br. J. Pharmacol. 2016, 173, 649–665. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Proenca, R.; Maffei, M.; Barone, M.; Leopold, L.; Friedman, J.M. Positional cloning of the mouse obese gene and its human homologue. Nature 1994, 372, 425–432. [Google Scholar] [CrossRef]
- Schwartz, M.W.; Woods, S.C.; Porte, D., Jr.; Seeley, R.J.; Baskin, D.G. Central nervous system control of food intake. Nature 2000, 404, 661–671. [Google Scholar] [CrossRef]
- Valdearcos, M.; Douglass, J.D.; Robblee, M.M.; Dorfman, M.D.; Stifler, D.R.; Bennett, M.L.; Gerritse, I.; Fasnacht, R.; Barres, B.A.; Thaler, J.P.; et al. Microglial Inflammatory Signaling Orchestrates the Hypothalamic Immune Response to Dietary Excess and Mediates Obesity Susceptibility. Cell Metab. 2017, 26, 185–197. [Google Scholar] [CrossRef] [Green Version]
- Morelli, A.; Sarchielli, E.; Comeglio, P.; Filippi, S.; Vignozzi, L.; Marini, M.; Rastrelli, G.; Maneschi, E.; Cellai, I.; Persani, L.; et al. Metabolic syndrome induces inflammation and impairs gonadotropin-releasing hormone neurons in the preoptic area of the hypothalamus in rabbits. Mol. Cell. Endocrinol. 2014, 382, 107–119. [Google Scholar] [CrossRef]
- Thaler, J.P.; Yi, C.X.; Schur, E.A.; Guyenet, S.J.; Hwang, B.H.; Dietrich, M.O.; Zhao, X.; Sarruf, D.A.; Izgur, V.; Maravilla, K.R.; et al. Obesity is associated with hypothalamic injury in rodents and humans. J. Clin. Investig. 2012, 122, 153–162. [Google Scholar] [CrossRef] [Green Version]
- de Kloet, A.D.; Pioquinto, D.J.; Nguyen, D.; Wang, L.; Smith, J.A.; Hiller, H.; Sumners, C. Obesity induces neuroinflammation mediated by altered expression of the renin-angiotensin system in mouse forebrain nuclei. Physiol. Behav. 2014, 136, 31–38. [Google Scholar] [CrossRef] [Green Version]
- Faivre, E.; Holscher, C. D-Ala2GIP facilitated synaptic plasticity and reduces plaque load in aged wild type mice and in an Alzheimer’s disease mouse model. J. Alzheimer Dis. JAD 2013, 35, 267–283. [Google Scholar] [CrossRef]
- Lee, D.; Thaler, J.P.; Berkseth, K.E.; Melhorn, S.J.; Schwartz, M.W.; Schur, E.A. Longer T2relaxation time is a marker of hypothalamic gliosis in mice with diet-induced obesity. Am. J. Physiol. Endocrinol. Metab. 2013, 304, E1245–E1250. [Google Scholar] [CrossRef] [Green Version]
- Berkseth, K.E.; Guyenet, S.J.; Melhorn, S.J.; Lee, D.; Thaler, J.P.; Schur, E.A.; Schwartz, M.W. Hypothalamic Gliosis Associated with High-Fat Diet Feeding Is Reversible in Mice: A Combined Immunohistochemical and Magnetic Resonance Imaging Study. Endocrinology 2014, 155, 2858–2867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naznin, F.; Toshinai, K.; Waise, T.M.Z.; Namkoong, C.; Moin, A.S.M.; Sakoda, H.; Nakazato, M. Diet-induced obesity causes peripheral and central ghrelin resistance by promoting inflammation. J. Endocrinol. 2015, 226, 81–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fonken, L.K.; Lieberman, R.A.; Weil, Z.M.; Nelson, R.J. Dim Light at Night Exaggerates Weight Gain and Inflammation Associated with a High-Fat Diet in Male Mice. Endocrinology 2013, 154, 3817–3825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sapolsky, R.; Rivier, C.; Yamamoto, G.; Plotsky, P.; Vale, W. Interleukin-1 stimulates the secretion of hypothalamic corticotropin-releasing factor. Science 1987, 238, 522–524. [Google Scholar] [CrossRef]
- Berkenbosch, F.; Van Oers, J.; Del Rey, A.; Tilders, F.; Besedovsky, H. Corticotropin-releasing factor-producing neurons in the rat activated by interleukin-1. Science 1987, 238, 524–526. [Google Scholar] [CrossRef]
- Xue, B.; Yu, Y.; Zhang, Z.; Guo, F.; Beltz, T.G.; Thunhorst, R.L.; Felder, R.B.; Johnson, A.K. Leptin Mediates High-Fat Diet Sensitization of Angiotensin II-Elicited Hypertension by Upregulating the Brain Renin-Angiotensin System and Inflammation. Hypertension 2016, 67, 970–976. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Smith, M.; Karthikeyan, S.; Jeffers, M.S.; Janik, R.; Thomason, L.A.; Stefanovic, B.; Corbett, D. A physiological characterization of the Cafeteria diet model of metabolic syndrome in the rat. Physiol. Behav. 2016, 167, 382–391. [Google Scholar] [CrossRef]
- Yi, C.X.; Walter, M.; Gao, Y.; Pitra, S.; Legutko, B.; Kalin, S.; Layritz, C.; García-Cáceres, C.; Bielohuby, M.; Bidlingmaier, M.; et al. TNFalpha drives mitochondrial stress in POMC neurons in obesity. Nat. Commun. 2017, 8, 15143. [Google Scholar] [CrossRef] [Green Version]
- Yin, Z.; Raj, D.D.; Schaafsma, W.; Van Der Heijden, R.A.; Kooistra, S.M.; Reijne, A.C.; Zhang, X.; Moser, J.; Brouwer, N.; Heeringa, P.; et al. Low-Fat Diet with Caloric Restriction Reduces White Matter Microglia Activation during Aging. Front. Mol. Neurosci. 2018, 11, 65. [Google Scholar] [CrossRef] [PubMed]
- Waise, T.M.Z.; Toshinai, K.; Naznin, F.; Namkoong, C.; Moin, A.S.M.; Sakoda, H.; Nakazato, M. One-day high-fat diet induces inflammation in the nodose ganglion and hypothalamus of mice. Biochem. Biophys. Res. Commun. 2015, 464, 1157–1162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baufeld, C.; Osterloh, A.; Prokop, S.; Miller, K.R.; Heppner, F.L. High-fat diet-induced brain region-specific phenotypic spectrum of CNS resident microglia. Acta Neuropathol. 2016, 132, 361–375. [Google Scholar] [CrossRef] [Green Version]
- Naznin, F.; Toshinai, K.; Waise, T.M.Z.; Okada, T.; Sakoda, H.; Nakazato, M. Restoration of metabolic inflammation-related ghrelin resistance by weight loss. J. Mol. Endocrinol. 2018, 60, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Farooqi, I.S.; Jebb, S.A.; Langmack, G.; Lawrence, E.; Cheetham, C.H.; Prentice, A.M.; Hughes, I.A.; McCamish, M.A.; O’Rahilly, S. Effects of Recombinant Leptin Therapy in a Child with Congenital Leptin Deficiency. N. Engl. J. Med. 1999, 341, 879–884. [Google Scholar] [CrossRef]
- Gao, Y.; Vidal-Itriago, A.; Milanova, I.; Korpel, N.L.; Kalsbeek, M.J.; Tom, R.Z.; Kalsbeek, A.; Hofmann, S.M.; Yi, C.-X. Deficiency of leptin receptor in myeloid cells disrupts hypothalamic metabolic circuits and causes body weight increase. Mol. Metab. 2017, 7, 155–160. [Google Scholar] [CrossRef]
- Gao, Y.; Vidal-Itriago, A.; Kalsbeek, M.J.; Layritz, C.; García-Cáceres, C.; Tom, R.Z.; Eichmann, T.O.; Vaz, F.M.; Houtkooper, R.H.; Van Der Wel, N.; et al. Lipoprotein Lipase Maintains Microglial Innate Immunity in Obesity. Cell Rep. 2017, 20, 3034–3042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ignacio-Souza, L.M.; Bombassaro, B.; Pascoal, L.B.; Portovedo, M.A.; Razolli, D.S.; Coope, A.; Victorio, S.C.; de Moura, R.F.; Nascimento, L.F.; Arruda, A.P.; et al. Defective regulation of the ubiquitin/proteasome system in the hypothalamus of obese male mice. Endocrinology 2014, 155, 2831–2844. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.-K.; Shin, M.-S.; Youn, B.-S.; Namkoong, C.; Gil, S.Y.; Kang, G.M.; Yu, J.H.; Kim, M.-S. Involvement of Progranulin in Hypothalamic Glucose Sensing and Feeding Regulation. Endocrinology 2011, 152, 4672–4682. [Google Scholar] [CrossRef] [Green Version]
- Yi, C.-X.; Tschöp, M.H.; Woods, S.C.; Hofmann, S.M. High-fat-diet exposure induces IgG accumulation in hypothalamic microglia. Dis. Models Mech. 2012, 5, 686–690. [Google Scholar] [CrossRef] [Green Version]
- Vidarsson, G.; Dekkers, G.; Rispens, T. IgG Subclasses and Allotypes: From Structure to Effector Functions. Front. Immunol. 2014, 5, 520. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Kwon, Y.H.; Kim, C.S.; Tu, T.H.; Kim, B.S.; Joe, Y.; Chung, H.T.; Goto, T.; Kawada, T.; Park, T.; et al. The involvement of 4-1BB/4-1BBL signaling in glial cell-mediated hypothalamic inflammation in obesity. FEBS Open Bio. 2018, 8, 843–853. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.-L.; Xia, J.-L.; Gao, X. Suppressing Irf2bp2 expressions accelerates metabolic syndrome-associated brain injury and hepatic dyslipidemia. Biochem. Biophys. Res. Commun. 2018, 503, 1651–1658. [Google Scholar] [CrossRef]
- Chen, H.H.; Keyhanian, K.; Zhou, X.; Vilmundarson, R.O.; Almontashiri, N.A.; Cruz, S.A.; Pandey, N.R.; Lerma Yap, N.; Ho, T.; Stewart, C.A.; et al. IRF2BP2 Reduces Macrophage Inflammation and Susceptibility to Atherosclerosis. Circ. Res. 2015, 117, 671–683. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Kim, C.-S.; Choe, S.-Y.; Lee, J.H.; Joe, Y.; Choi, H.-S.; Back, S.H.; Chung, H.T.; Yu, R.; Tu, T.H.; et al. Quercetin Protects Obesity-Induced Hypothalamic Inflammation by Reducing Microglia-Mediated Inflammatory Responses via HO-1 Induction. Nutrients 2017, 9, 650. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Sarasua, S.; Moustafa, S.; Garcia-Aviles, A.; Lopez-Climent, M.F.; Gomez-Cadenas, A.; Olucha-Bordonau, F.E.; Sánchez-Pérez, A.M. The effect of abscisic acid chronic treatment on neuroinflammatory markers and memory in a rat model of high-fat diet induced neuroinflammation. Nutr. Metab. 2016, 13, 73. [Google Scholar] [CrossRef] [Green Version]
- Naznin, F.; Sakoda, H.; Okada, T.; Tsubouchi, H.; Waise, T.Z.; Arakawa, K.; Nakazato, M. Canagliflozin, a sodium glucose cotransporter 2 inhibitor, attenuates obesity-induced inflammation in the nodose ganglion, hypothalamus, and skeletal muscle of mice. Eur. J. Pharmacol. 2017, 794, 37–44. [Google Scholar] [CrossRef]
- Jeon, B.T.; Kim, K.E.; Heo, R.W.; Shin, H.J.; Yi, C.-O.; Hah, Y.-S.; Kim, W.-H.; Lee, S.-I.; Roh, G.S. Myeloid-specific deletion of SIRT1 increases hepatic steatosis and hypothalamic inflammation in mice fed a high-fat diet. Metab. Brain Dis. 2014, 29, 635–643. [Google Scholar] [CrossRef]
- Ye, J.; Liu, Z.; Wei, J.; Lu, L.; Huang, Y.; Luo, L.; Xie, H. Protective effect of SIRT1 on toxicity of microglial-derived factors induced by LPS to PC12 cells via the p53-caspase-3-dependent apoptotic pathway. Neurosci. Lett. 2013, 553, 72–77. [Google Scholar] [CrossRef]
- André, C.; Guzman-Quevedo, O.; Rey, C.; Rémus-Borel, J.; Clark, S.; Castellanos-Jankiewicz, A.; Ladeveze, E.; Leste-Lasserre, T.; Nadjar, A.; Abrous, D.N.; et al. Inhibiting Microglia Expansion Prevents Diet-Induced Hypothalamic and Peripheral Inflammation. Diabetes 2016, 66, 908–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barreto-Vianna, A.R.; Aguila, M.B.; Mandarim-De-Lacerda, C.A. Effects of liraglutide in hypothalamic arcuate nucleus of obese mice. Obesity 2016, 24, 626–633. [Google Scholar] [CrossRef] [Green Version]
- Baquedano, E.; Ruiz-Lopez, A.M.; Sustarsic, E.G.; Herpy, J.; List, E.O.; Chowen, J.A.; Frago, L.M.; Kopchick, J.J.; Argente, J. The Absence of GH Signaling Affects the Susceptibility to High-Fat Diet-Induced Hypothalamic Inflammation in Male Mice. Endocrinology 2014, 155, 4856–4867. [Google Scholar] [CrossRef] [Green Version]
- Yi, C.-X.; Al-Massadi, O.; Donelan, E.; Lehti, M.; Weber, J.; Ress, C.; Trivedi, C.; Müller, T.D.; Woods, S.C.; Hofmann, S.M. Exercise protects against high-fat diet-induced hypothalamic inflammation. Physiol. Behav. 2012, 106, 485–490. [Google Scholar] [CrossRef] [Green Version]
- Sherman, M.; Liu, M.-M.; Birnbaum, S.; Wolf, S.E.; Minei, J.P.; Gatson, J.W. Adult obese mice suffer from chronic secondary brain injury after mild TBI. J. Neuroinflamm. 2016, 13, 171. [Google Scholar] [CrossRef] [Green Version]
- Dorfman, M.D.; Krull, J.E.; Douglass, J.D.; Fasnacht, R.; Lara-Lince, F.; Meek, T.H.; Shi, X.; Damian, V.; Nguyen, H.T.; Matsen, M.E.; et al. Sex differences in microglial CX3CR1 signalling determine obesity susceptibility in mice. Nat. Commun. 2017, 8, 14556. [Google Scholar] [CrossRef] [Green Version]
- Fang, X.; Ge, K.; Song, C.; Ge, Y.; Zhang, J. Effects of n-3PUFAs on autophagy and inflammation of hypothalamus and body weight in mice. Biochem. Biophys. Res. Commun. 2018, 501, 927–932. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, P.; Shou, Q.; Lu, Y.; Wang, G.; Qiu, J.; Wang, J.; He, L.; Chen, J.; Jiao, J.; Zhang, Y. Arachidonic acid sex-dependently affects obesity through linking gut microbiota-driven inflammation to hypothalamus-adipose-liver axis. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2017, 1863, 2715–2726. [Google Scholar] [CrossRef]
- Bochukova, E.G.; Lawler, K.; Croizier, S.; Keogh, J.M.; Patel, N.; Strohbehn, G.; Lo, K.K.; Humphrey, J.; Hokken-Koelega, A.; Damen, L.; et al. A Transcriptomic Signature of the Hypothalamic Response to Fasting and BDNF Deficiency in Prader-Willi Syndrome. Cell Rep. 2018, 22, 3401–3408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, J.C.D.; Killcross, A.S.; Jenkins, T.A. Obesity and cognitive decline: Role of inflammation and vascular changes. Front. Neurosci. 2014, 8, 375. [Google Scholar] [CrossRef]
- Thirumangalakudi, L.; Prakasam, A.; Zhang, R.; Bimonte-Nelson, H.; Sambamurti, K.; Kindy, M.S.; Bhat, N.R. High cholesterol-induced neuroinflammation and amyloid precursor protein processing correlate with loss of working memory in mice. J. Neurochem. 2008, 106, 475–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spencer, S.J.; D’Angelo, H.; Soch, A.; Watkins, L.R.; Maier, S.F.; Barrientos, R.M. High-fat diet and aging interact to produce neuroinflammation and impair hippocampal- and amygdalar-dependent memory. Neurobiol. Aging 2017, 58, 88–101. [Google Scholar] [CrossRef] [PubMed]
- Tramullas, M.; Finger, B.C.; Dinan, T.G.; Cryan, J.F. Obesity Takes Its Toll on Visceral Pain: High-Fat Diet Induces Toll-Like Receptor 4-Dependent Visceral Hypersensitivity. PLoS ONE 2016, 11, e0155367. [Google Scholar] [CrossRef] [Green Version]
- Tucsek, Z.; Toth, P.; Sosnowska, D.; Gautam, T.; Mitschelen, M.; Koller, A.; Szalai, G.; Sonntag, W.E.; Ungvari, Z.; Csiszar, A. Obesity in aging exacerbates blood-brain barrier disruption, neuroinflammation, and oxidative stress in the mouse hippocampus: Effects on expression of genes involved in beta-amyloid generation and Alzheimer’s disease. J. Gerontol. Ser. A Biol. Sci. Med Sci. 2014, 69, 1212–1226. [Google Scholar] [CrossRef]
- Valcarcel-Ares, M.N.; Tucsek, Z.; Kiss, T.; Giles, C.B.; Tarantini, S.; Yabluchanskiy, A.; Balasubramanian, P.; Gautam, T.; Galvan, V.; Ballabh, P.; et al. Obesity in Aging Exacerbates Neuroinflammation, Dysregulating Synaptic Function-Related Genes and Altering Eicosanoid Synthesis in the Mouse Hippocampus: Potential Role in Impaired Synaptic Plasticity and Cognitive Decline. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2018, 74, 290–298. [Google Scholar] [CrossRef] [PubMed]
- Tarantini, S.; Valcarcel-Ares, M.N.; Yabluchanskiy, A.; Tucsek, Z.; Hertelendy, P.; Kiss, T.; Gautam, T.; Zhang, X.A.; Sonntag, W.E.; De Cabo, R.; et al. Nrf2 Deficiency Exacerbates Obesity-Induced Oxidative Stress, Neurovascular Dysfunction, Blood-Brain Barrier Disruption, Neuroinflammation, Amyloidogenic Gene Expression, and Cognitive Decline in Mice, Mimicking the Aging Phenotype. J. Gerontol. Ser. A Biol. Sci. Med Sci. 2018, 73, 853–863. [Google Scholar] [CrossRef]
- Asatryan, L.; Khoja, S.; Rodgers, K.E.; Alkana, R.L.; Tsukamoto, H.; Davies, D.L. Chronic ethanol exposure combined with high fat diet up-regulates P2X7 receptors that parallels neuroinflammation and neuronal loss in C57BL/6J mice. J. Neuroimmunol. 2015, 285, 169–179. [Google Scholar] [CrossRef] [Green Version]
- Erion, J.R.; Wosiski-Kuhn, M.; Dey, A.; Hao, S.; Davis, C.L.; Pollock, N.K.; Stranahan, A.M. Obesity elicits interleukin 1-mediated deficits in hippocampal synaptic plasticity. J. Neurosci. 2014, 34, 2618–2631. [Google Scholar] [CrossRef] [Green Version]
- White, K.A.; Hutton, S.R.; Weimer, J.M.; Sheridan, P.A. Diet-induced obesity prolongs neuroinflammation and recruits CCR2(+) monocytes to the brain following herpes simplex virus (HSV)-1 latency in mice. Brain Behav. Immun. 2016, 57, 68–78. [Google Scholar] [CrossRef] [Green Version]
- Bocarsly, M.E.; Fasolino, M.; Kane, G.A.; LaMarca, E.A.; Kirschen, G.W.; Karatsoreos, I.N.; McEwen, B.S.; Gould, E. Obesity diminishes synaptic markers, alters microglial morphology, and impairs cognitive function. Proc. Natl. Acad. Sci. USA 2015, 112, 15731–15736. [Google Scholar] [CrossRef] [Green Version]
- Gzielo, K.; Kielbinski, M.; Ploszaj, J.; Janeczko, K.; Gazdzinski, S.P.; Setkowicz, Z. Long-Term Consumption of High-Fat Diet in Rats: Effects on Microglial and Astrocytic Morphology and Neuronal Nitric Oxide Synthase Expression. Cell. Mol. Neurobiol. 2016, 37, 783–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, I.K.; Kim, I.Y.; Na Kim, Y.; Yi, S.S.; Park, I.-S.; Min, B.-H.; Doo, H.-K.; Ahn, S.-Y.; Kim, Y.-S.; Lee, I.S.; et al. Comparative Study on High Fat Diet-induced 4-Hydroxy-2E-nonenal Adducts in the Hippocampal CA1 Region of C57BL/6N and C3H/HeN Mice. Neurochem. Res. 2008, 34, 964–972. [Google Scholar] [CrossRef] [PubMed]
- Chunchai, T.; Thunapong, W.; Yasom, S.; Wanchai, K.; Eaimworawuthikul, S.; Metzler, G.; Lungkaphin, A.; Pongchaidecha, A.; Sirilun, S.; Chaiyasut, C.; et al. Decreased microglial activation through gut-brain axis by prebiotics, probiotics, or synbiotics effectively restored cognitive function in obese-insulin resistant rats. J. Neuroinflamm. 2018, 15, 11. [Google Scholar] [CrossRef] [PubMed]
- Moon, S.M.; Choi, G.-M.; Yoo, D.Y.; Jung, H.Y.; Yim, H.S.; Kim, D.W.; Hwang, I.K.; Cho, B.M.; Chang, I.B.; Cho, S.-M.; et al. Differential Effects of Pioglitazone in the Hippocampal CA1 Region Following Transient Forebrain Ischemia in Low- and High-Fat Diet-Fed Gerbils. Neurochem. Res. 2015, 40, 1063–1073. [Google Scholar] [CrossRef] [PubMed]
- Cope, E.C.; LaMarca, E.A.; Monari, P.K.; Olson, L.B.; Martinez, S.; Zych, A.D.; Katchur, N.J.; Gould, E. Microglia Play an Active Role in Obesity-Associated Cognitive Decline. J. Neurosci. 2018, 38, 8889–8904. [Google Scholar] [CrossRef] [Green Version]
- Calvo-Ochoa, E.; Hernández-Ortega, K.; Ferrera, P.; Morimoto, S.; Arias, C. Short-Term High-Fat-and-Fructose Feeding Produces Insulin Signaling Alterations Accompanied by Neurite and Synaptic Reduction and Astroglial Activation in the Rat Hippocampus. Br. J. Pharmacol. 2014, 34, 1001–1008. [Google Scholar] [CrossRef]
- Auer, M.K.; Sack, M.; Lenz, J.N.; Jakovcevski, M.; Biedermann, S.V.; Falfan-Melgoza, C.; Deussing, J.; Steinle, J.; Bielohuby, M.; Bidlingmaier, M.; et al. Effects of a high-caloric diet and physical exercise on brain metabolite levels: A combined proton MRS and histologic study. J. Cereb. Blood Flow Metab. 2015, 35, 554–564. [Google Scholar] [CrossRef] [Green Version]
- Ledreux, A.; Wang, X.; Schultzberg, M.; Granholm, A.-C.; Freeman, L.R. Detrimental effects of a high fat/high cholesterol diet on memory and hippocampal markers in aged rats. Behav. Brain Res. 2016, 312, 294–304. [Google Scholar] [CrossRef]
- Vinuesa, A.; Pomilio, C.; Menafra, M.; Bonaventura, M.M.; Garay, L.; Mercogliano, M.F.; Schillaci, R.; Lantos, R.S.V.L.; Brites, F.; Beauquis, J.; et al. Juvenile exposure to a high fat diet promotes behavioral and limbic alterations in the absence of obesity. Psychoneuroendocrinology 2016, 72, 22–33. [Google Scholar] [CrossRef]
- Hao, S.; Dey, A.; Yu, X.; Stranahan, A.M. Dietary obesity reversibly induces synaptic stripping by microglia and impairs hippocampal plasticity. Brain Behav. Immun. 2016, 51, 230–239. [Google Scholar] [CrossRef] [Green Version]
- Kang, E.; Koo, J.; Jang, Y.; Yang, C.; Lee, Y.; Cosio-Lima, L.M.; Cho, J. Neuroprotective Effects of Endurance Exercise Against High-Fat Diet-Induced Hippocampal Neuroinflammation. J. Neuroendocr. 2016, 28. [Google Scholar] [CrossRef] [PubMed]
- Carey, A.N.; Gildawie, K.R.; Rovnak, A.; Thangthaeng, N.; Fisher, D.R.; Shukitt-Hale, B. Blueberry supplementation attenuates microglia activation and increases neuroplasticity in mice consuming a high-fat diet. Nutr. Neurosci. 2017, 22, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Alford, S.; Patel, D.; Perakakis, N.; Mantzoros, C.S. Obesity as a risk factor for Alzheimer’s disease: Weighing the evidence. Obes. Rev. 2018, 19, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Solito, E.; Sastre, M. Microglia function in Alzheimer’s disease. Front. Pharmacol. 2012, 3, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knight, E.M.; Martins, I.V.; Gumusgoz, S.; Allan, S.M.; Lawrence, C.B. High-fat diet-induced memory impairment in triple-transgenic Alzheimer’s disease (3xTgAD) mice is independent of changes in amyloid and tau pathology. Neurobiol. Aging 2014, 35, 1821–1832. [Google Scholar] [CrossRef]
- Park, S.H.; Kim, J.H.; Choi, K.H.; Jang, Y.J.; Bae, S.S.; Choi, B.T.; Shin, H.K. Hypercholesterolemia accelerates amyloid beta-induced cognitive deficits. Int. J. Mol. Med. 2013, 31, 577–582. [Google Scholar] [CrossRef] [Green Version]
- Herculano, B.; Tamura, M.; Ohba, A.; Shimatani, M.; Kutsuna, N.; Hisatsune, T. Beta-alanyl-L-histidine rescues cognitive deficits caused by feeding a high fat diet in a transgenic mouse model of Alzheimer’s disease. J. Alzheimer Dis. JAD 2013, 33, 983–997. [Google Scholar] [CrossRef] [Green Version]
- Christensen, A.; Pike, C.J. Age-dependent regulation of obesity and Alzheimer-related outcomes by hormone therapy in female 3xTg-AD mice. PLoS ONE 2017, 12, e0178490. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Dasuri, K.; Fernandez-Kim, S.O.; Bruce-Keller, A.J.; Freeman, L.R.; Pepping, J.K.; Beckett, T.L.; Murphy, M.P.; Keller, J.N. Prolonged diet induced obesity has minimal effects towards brain pathology in mouse model of cerebral amyloid angiopathy: Implications for studying obesity-brain interactions in mice. Biochim. Biophys. Acta 2013, 1832, 1456–1462. [Google Scholar] [CrossRef] [Green Version]
- Drummond, E.; Wisniewski, T. Alzheimer’s disease: Experimental models and reality. Acta Neuropathol. 2017, 133, 155–175. [Google Scholar] [CrossRef]
- Décarie-Spain, L.; Sharma, S.; Hryhorczuk, C.; Issa-Garcia, V.; Barker, P.A.; Arbour, N.; Alquier, T.; Fulton, S. Nucleus accumbens inflammation mediates anxiodepressive behavior and compulsive sucrose seeking elicited by saturated dietary fat. Mol. Metab. 2018, 10, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Dasuri, K.; Zhang, L.; Kim, S.O.; Bruce-Keller, A.J.; Keller, J.N. Dietary and donepezil modulation of mTOR signaling and neuroinflammation in the brain. Biochim. Biophys. Acta 2016, 1862, 274–283. [Google Scholar] [CrossRef] [PubMed]
- Pistell, P.J.; Morrison, C.D.; Gupta, S.; Knight, A.G.; Keller, J.N.; Ingram, D.K.; Bruce-Keller, A.J. Cognitive impairment following high fat diet consumption is associated with brain inflammation. J. Neuroimmunol. 2010, 219, 25–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guillemot-Legris, O.; Masquelier, J.; Everard, A.; Cani, P.D.; Alhouayek, M.; Muccioli, G.G. High-fat diet feeding differentially affects the development of inflammation in the central nervous system. J. Neuroinflamm. 2016, 13, 206. [Google Scholar] [CrossRef] [Green Version]
- Taga, M.; Mouton-Liger, F.; Sadoune, M.; Gourmaud, S.; Norman, J.; Tible, M.; Thomasseau, S.; Paquet, C.; Nicoll, J.A.R.; Boche, D.; et al. PKR modulates abnormal brain signaling in experimental obesity. PLoS ONE 2018, 13, e0196983. [Google Scholar] [CrossRef]
- Meireles, M.; Marques, C.; Norberto, S.; Fernandes, I.; Mateus, N.; Rendeiro, C.; Spencer, J.P.; Faria, A.; Calhau, C. The impact of chronic blackberry intake on the neuroinflammatory status of rats fed a standard or high-fat diet. J. Nutr. Biochem. 2015, 26, 1166–1173. [Google Scholar] [CrossRef]
- Maysami, S.; Haley, M.J.; Gorenkova, N.; Krishnan, S.; McColl, B.W.; Lawrence, C.B. Prolonged diet-induced obesity in mice modifies the inflammatory response and leads to worse outcome after stroke. J. Neuroinflamm. 2015, 12, 140. [Google Scholar] [CrossRef] [Green Version]
- Nam, K.N.; Wolfe, C.M.; Fitz, N.F.; Letronne, F.; Castranio, E.L.; Mounier, A.; Schug, J.; Lefterov, I.; Koldamova, R. Integrated approach reveals diet, APOE genotype and sex affect immune response in APP mice. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2018, 1864, 152–161. [Google Scholar] [CrossRef]
- Contreras-Rodríguez, O.; Martín-Pérez, C.; Vilar-López, R.; Verdejo-Garcia, A. Ventral and Dorsal Striatum Networks in Obesity: Link to Food Craving and Weight Gain. Biol. Psychiatry 2017, 81, 789–796. [Google Scholar] [CrossRef] [Green Version]
- Gomes, J.A.S.; Oliveira, M.C.; Gobira, P.H.; Silva, G.C.; Marçal, A.P.; Gomes, G.F.; Ferrari, C.Z.; Lemos, V.S.; De Oliveira, A.C.P.; Vieira, L.B.; et al. A high-refined carbohydrate diet facilitates compulsive-like behavior in mice through the nitric oxide pathway. Nitric Oxide 2018, 80, 61–69. [Google Scholar] [CrossRef]
- Drake, C.; Boutin, H.; Jones, M.S.; Denes, A.; McColl, B.W.; Selvarajah, J.R.; Hulme, S.; Georgiou, R.F.; Hinz, R.; Gerhard, A.; et al. Brain inflammation is induced by co-morbidities and risk factors for stroke. Brain Behav. Immun. 2011, 25, 1113–1122. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Martos, M.; Girard, B.; Mendonça-Netto, S.; Perroy, J.; Valjent, E.; Maldonado, R.; Martin, M. Cafeteria diet induces neuroplastic modifications in the nucleus accumbens mediated by microglia activation. Addict. Biol. 2017, 23, 735–749. [Google Scholar] [CrossRef] [PubMed]
- Kenny, P.J. Reward Mechanisms in Obesity: New Insights and Future Directions. Neuron 2011, 69, 664–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, C.B.; MacKenzie, K.C.; Gahagan, S. The Effect of Maternal Obesity on the Offspring. Clin. Obstet. Gynecol. 2014, 57, 508–515. [Google Scholar] [CrossRef]
- Godfrey, K.M.; Reynolds, R.M.; Prescott, S.L.; Nyirenda, M.; Jaddoe, V.W.V.; Eriksson, J.G.; Broekman, B.F.P. Influence of maternal obesity on the long-term health of offspring. Lancet Diabetes Endocrinol. 2017, 5, 53–64. [Google Scholar] [CrossRef] [Green Version]
- Glavas, M.M.; Kirigiti, M.A.; Xiao, X.Q.; Enriori, P.J.; Fisher, S.K.; Evans, A.E.; Grayson, B.E.; Cowley, M.A.; Smith, M.S.; Grove, K.L. Early Overnutrition Results in Early-Onset Arcuate Leptin Resistance and Increased Sensitivity to High-Fat Diet. Endocrinology 2010, 151, 1598–1610. [Google Scholar] [CrossRef] [Green Version]
- Habbout, A.; Li, N.; Rochette, L.; Vergely, C. Postnatal Overfeeding in Rodents by Litter Size Reduction Induces Major Short- and Long-Term Pathophysiological Consequences. J. Nutr. 2013, 143, 553–562. [Google Scholar] [CrossRef]
- Carvalho, A.L.O.; Ferri, B.G.; Sousa, F.A.L.D.; Vilela, F.C.; Giusti-Paiva, A. Early life overnutrition induced by litter size manipulation decreases social play behavior in adolescent male rats. Int. J. Dev. Neurosci. 2016, 53, 75–82. [Google Scholar] [CrossRef]
- Argente-Arizón, P.; Díaz, F.; Ros, P.; Barrios, V.; Tena-Sempere, M.; García-Segura, L.M.; Argente, J.; Chowen, J.A. The Hypothalamic Inflammatory/Gliosis Response to Neonatal Overnutrition Is Sex and Age Dependent. Endocrinology 2017, 159, 368–387. [Google Scholar] [CrossRef] [Green Version]
- Ziko, I.; De Luca, S.; Dinan, T.; Barwood, J.M.; Sominsky, L.; Cai, G.; Kenny, R.; Stokes, L.; Jenkins, T.A.; Spencer, S.J. Neonatal overfeeding alters hypothalamic microglial profiles and central responses to immune challenge long-term. Brain Behav. Immun. 2014, 41, 32–43. [Google Scholar] [CrossRef]
- Tapia-Gonzalez, S.; Garcia-Segura, L.M.; Tena-Sempere, M.; Frago, L.M.; Castellano, J.M.; Fuente-Martin, E.; García-Cáceres, C.; Argente, J.; Chowen, J.A. Activation of microglia in specific hypothalamic nuclei and the cerebellum of adult rats exposed to neonatal overnutrition. J. Neuroendocrinol. 2011, 23, 365–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, G.; Dinan, T.; Barwood, J.M.; De Luca, S.N.; Soch, A.; Ziko, I.; Chan, S.M.; Zeng, X.Y.; Li, S.; Molero, J.; et al. Neonatal overfeeding attenuates acute central pro-inflammatory effects of short-term high fat diet. Front. Neurosci. 2015, 8, 446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuente-Martín, E.; García-Cáceres, C.; Díaz, F.; Argente-Arizón, P.; Granado, M.; Barrios, V.; Argente, J.; Chowen, J.A. Hypothalamic Inflammation Without Astrogliosis in Response to High Sucrose Intake Is Modulated by Neonatal Nutrition in Male Rats. Endocrinology 2013, 154, 2318–2330. [Google Scholar] [CrossRef]
- Grayson, B.E.; Levasseur, P.R.; Williams, S.M.; Smith, M.S.; Marks, D.L.; Grove, K.L. Changes in melanocortin expression and inflammatory pathways in fetal offspring of nonhuman primates fed a high-fat diet. Endocrinology 2010, 151, 1622–1632. [Google Scholar] [CrossRef]
- Bilbo, S.D.; Tsang, V. Enduring consequences of maternal obesity for brain inflammation and behavior of offspring. FASEB J. 2010, 24, 2104–2115. [Google Scholar] [CrossRef]
- De Luca, S.N.; Ziko, I.; Sominsky, L.; Nguyen, J.C.D.; Dinan, T.; Miller, A.A.; Jenkins, T.A.; Spencer, S.J. Early life overfeeding impairs spatial memory performance by reducing microglial sensitivity to learning. J. Neuroinflamm. 2016, 13, 112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Luca, S.N.; Ziko, I.; Dhuna, K.; Sominsky, L.; Tolcos, M.; Stokes, L.; Spencer, S.J. Neonatal overfeeding by small-litter rearing sensitises hippocampal microglial responses to immune challenge: Reversal with neonatal repeated injections of saline or minocycline. J. Neuroendocrinol. 2017, 29, e12540. [Google Scholar] [CrossRef]
- Tu, Y.-F.; Tsai, Y.-S.; Wang, L.-W.; Wu, H.-C.; Huang, C.-C.; Ho, C.-J. Overweight worsens apoptosis, neuroinflammation and blood-brain barrier damage after hypoxic ischemia in neonatal brain through JNK hyperactivation. J. Neuroinflamm. 2011, 8, 40. [Google Scholar] [CrossRef] [Green Version]
- Hirosumi, J.; Tuncman, G.; Chang, L.; Gorgun, C.Z.; Uysal, K.T.; Maeda, K.; Karin, M.; Hotamisligil, G.S. A central role for JNK in obesity and insulin resistance. Nature 2002, 420, 333–336. [Google Scholar] [CrossRef]
- Teo, J.D.; Morris, M.J.; Jones, N.M. Maternal obesity increases inflammation and exacerbates damage following neonatal hypoxic-ischaemic brain injury in rats. Brain Behav. Immun. 2017, 63, 186–196. [Google Scholar] [CrossRef]
- Bolton, J.L.; Auten, R.L.; Bilbo, S.D. Prenatal air pollution exposure induces sexually dimorphic fetal programming of metabolic and neuroinflammatory outcomes in adult offspring. Brain Behav. Immun. 2014, 37, 30–44. [Google Scholar] [CrossRef] [PubMed]
- Bolton, J.L.; Smith, S.H.; Huff, N.C.; Gilmour, M.I.; Foster, W.M.; Auten, R.L.; Bilbo, S.D. Prenatal air pollution exposure induces neuroinflammation and predisposes offspring to weight gain in adulthood in a sex-specific manner. FASEB J. 2012, 26, 4743–4754. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Milanova, I.V.; Correa-da-Silva, F.; Kalsbeek, A.; Yi, C.-X. Mapping of Microglial Brain Region, Sex and Age Heterogeneity in Obesity. Int. J. Mol. Sci. 2021, 22, 3141. https://doi.org/10.3390/ijms22063141
Milanova IV, Correa-da-Silva F, Kalsbeek A, Yi C-X. Mapping of Microglial Brain Region, Sex and Age Heterogeneity in Obesity. International Journal of Molecular Sciences. 2021; 22(6):3141. https://doi.org/10.3390/ijms22063141
Chicago/Turabian StyleMilanova, Irina V., Felipe Correa-da-Silva, Andries Kalsbeek, and Chun-Xia Yi. 2021. "Mapping of Microglial Brain Region, Sex and Age Heterogeneity in Obesity" International Journal of Molecular Sciences 22, no. 6: 3141. https://doi.org/10.3390/ijms22063141
APA StyleMilanova, I. V., Correa-da-Silva, F., Kalsbeek, A., & Yi, C. -X. (2021). Mapping of Microglial Brain Region, Sex and Age Heterogeneity in Obesity. International Journal of Molecular Sciences, 22(6), 3141. https://doi.org/10.3390/ijms22063141