
Transcriptional Regulation of Postnatal Cardiomyocyte Maturation and Regeneration




Abstract
:1. Introduction
2. Overview of Postnatal Cardiomyocyte Maturation
2.1. Cell Cycle Arrest and Multinucleation
2.2. Switch to Hypertrophic Cardiomyocyte Growth
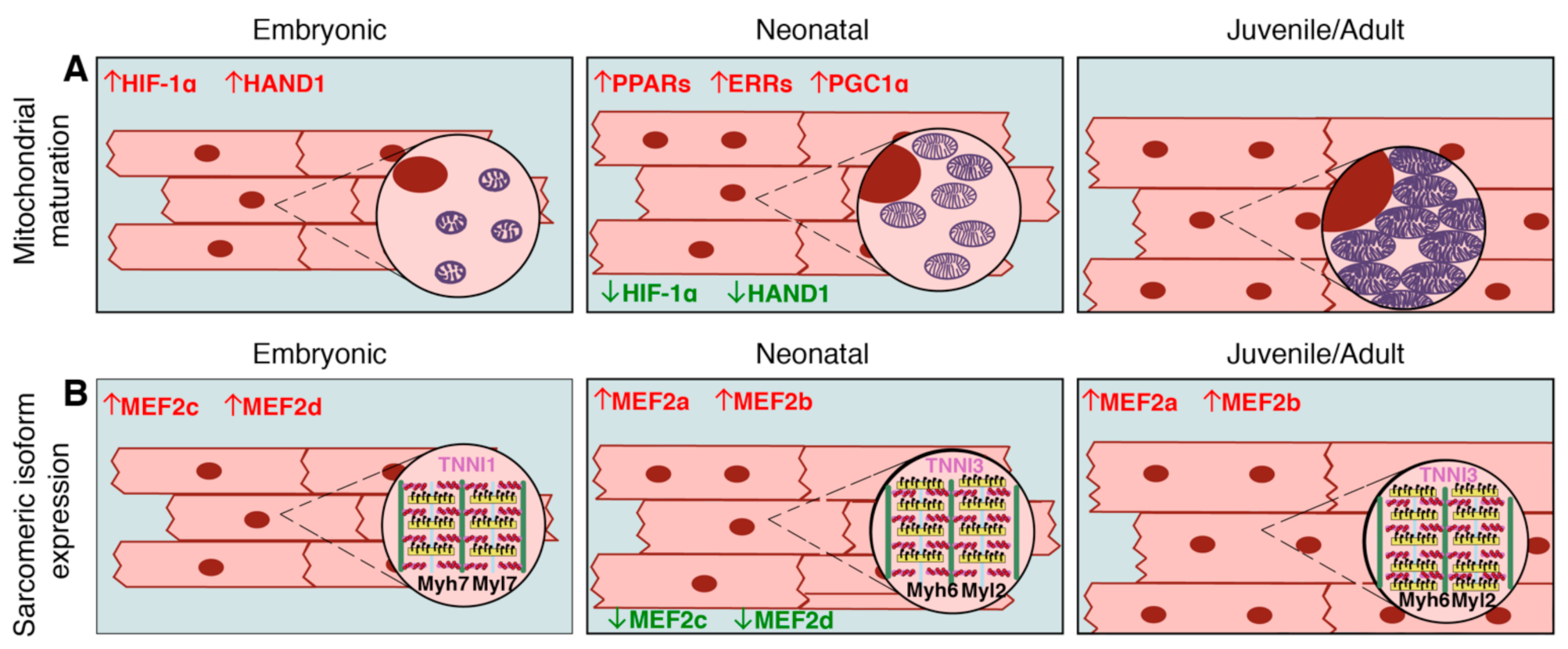
2.3. Transition to Oxidative Metabolism
2.4. Fetal to Adult Contractile Protein Isoform Switching
3. Transcriptional Regulation of Postnatal Cardiomyocyte Maturation
3.1. Transcriptional Regulation of Prenatal Versus Postnatal Cardiomyocyte Cell Cycling
3.2. Transcriptional Regulation of the Postnatal Induction of Hypertrophic Growth in Cardiomyocytes
3.3. Transcriptional Regulation of Fetal and Adult Sarcomeric Isoform Gene Expression
3.4. Transcription Factor Regulation of Mitochondrial Maturation in Cardiomyocytes
4. Chromatin Remodeling and Epigenetic Control of Cardiomyocyte Maturation
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Porrello, E.R.; Mahmoud, A.I.; Simpson, E.; Hill, J.A.; Richardson, J.A.; Olson, E.N.; Sadek, H.A. Transient regenerative potential of the neonatal mouse heart. Science 2011, 331, 1078–1080. [Google Scholar]
- Ye, L.; D’Agostino, G.; Loo, S.J.; Wang, C.X.; Su, L.P.; Tan, S.H.; Tee, G.Z.; Pua, C.J.; Pena, E.M.; Cheng, R.B.; et al. Early Regenerative Capacity in the Porcine Heart. Circulation 2018, 138, 2798–2808. [Google Scholar]
- Zhu, W.; Zhang, E.; Zhao, M.; Chong, Z.; Fan, C.; Tang, Y.; Hunter, J.D.; Borovjagin, A.V.; Walcott, G.P.; Chen, J.Y.; et al. Regenerative Potential of Neonatal Porcine Hearts. Circulation 2018, 138, 2809–2816. [Google Scholar]
- Porrello, E.R.; Olson, E.N. A neonatal blueprint for cardiac regeneration. Stem Cell Res. 2014, 13 Pt B, 556–570. [Google Scholar]
- Bergmann, O.; Bhardwaj, R.D.; Bernard, S.; Zdunek, S.; Barnabe-Heider, F.; Walsh, S.; Zupicich, J.; Alkass, K.; Buchholz, B.A.; Druid, H.; et al. Evidence for cardiomyocyte renewal in humans. Science 2009, 324, 98–102. [Google Scholar]
- Bergmann, O.; Zdunek, S.; Felker, A.; Salehpour, M.; Alkass, K.; Bernard, S.; Sjostrom, S.L.; Szewczykowska, M.; Jackowska, T.; Dos Remedios, C.; et al. Dynamics of Cell Generation and Turnover in the Human Heart. Cell 2015, 161, 1566–1575. [Google Scholar]
- Gan, P.; Patterson, M.; Sucov, H.M. Cardiomyocyte Polyploidy and Implications for Heart Regeneration. Annu. Rev. Physiol. 2020, 82, 45–61. [Google Scholar]
- Senyo, S.E.; Steinhauser, M.L.; Pizzimenti, C.L.; Yang, V.K.; Cai, L.; Wang, M.; Wu, T.D.; Guerquin-Kern, J.L.; Lechene, C.P.; Lee, R.T. Mammalian heart renewal by pre-existing cardiomyocytes. Nature 2013, 493, 433–436. [Google Scholar]
- Velayutham, N.; Alfieri, C.M.; Agnew, E.J.; Riggs, K.W.; Baker, R.S.; Ponny, S.R.; Zafar, F.; Yutzey, K.E. Cardiomyocyte cell cycling, maturation, and growth by multinucleation in postnatal swine. J. Mol. Cell. Cardiol. 2020, 146, 95–108. [Google Scholar]
- Mollova, M.; Bersell, K.; Walsh, S.; Savla, J.; Das, L.T.; Park, S.Y.; Silberstein, L.E.; Dos Remedios, C.G.; Graham, D.; Colan, S.; et al. Cardiomyocyte proliferation contributes to heart growth in young humans. Proc. Natl. Acad. Sci. USA 2013, 110, 1446–1451. [Google Scholar]
- Lazar, E.; Sadek, H.A.; Bergmann, O. Cardiomyocyte renewal in the human heart: Insights from the fall-out. Eur. Heart J. 2017, 38, 2333–2342. [Google Scholar]
- Gunthel, M.; Barnett, P.; Christoffels, V.M. Development, Proliferation, and Growth of the Mammalian Heart. Mol. Ther. 2018, 26, 1599–1609. [Google Scholar]
- Patterson, M.; Barske, L.; Van Handel, B.; Rau, C.D.; Gan, P.; Sharma, A.; Parikh, S.; Denholtz, M.; Huang, Y.; Yamaguchi, Y.; et al. Frequency of mononuclear diploid cardiomyocytes underlies natural variation in heart regeneration. Nat. Genet. 2017, 49, 1346–1353. [Google Scholar]
- Ascuitto, R.J.; Ross-Ascuitto, N.T. Substrate metabolism in the developing heart. Semin. Perinatol. 1996, 20, 542–563. [Google Scholar]
- Yin, Z.; Ren, J.; Guo, W. Sarcomeric protein isoform transitions in cardiac muscle: A journey to heart failure. Biochim. Biophys. Acta 2015, 1852, 47–52. [Google Scholar]
- Saggin, L.; Gorza, L.; Ausoni, S.; Schiaffino, S. Troponin I switching in the developing heart. J. Biol. Chem. 1989, 264, 16299–16302. [Google Scholar]
- Warren, C.M.; Krzesinski, P.R.; Campbell, K.S.; Moss, R.L.; Greaser, M.L. Titin isoform changes in rat myocardium during development. Mech. Dev. 2004, 121, 1301–1312. [Google Scholar]
- Velayutham, N.; Agnew, E.J.; Yutzey, K.E. Postnatal Cardiac Development and Regenerative Potential in Large Mammals. Pediatr. Cardiol. 2019, 40, 1345–1358. [Google Scholar]
- Quaife-Ryan, G.A.; Sim, C.B.; Ziemann, M.; Kaspi, A.; Rafehi, H.; Ramialison, M.; El-Osta, A.; Hudson, J.E.; Porrello, E.R. Multicellular Transcriptional Analysis of Mammalian Heart Regeneration. Circulation 2017, 136, 1123–1139. [Google Scholar]
- Hirose, K.; Payumo, A.Y.; Cutie, S.; Hoang, A.; Zhang, H.; Guyot, R.; Lunn, D.; Bigley, R.B.; Yu, H.; Wang, J.; et al. Evidence for hormonal control of heart regenerative capacity during endothermy acquisition. Science 2019, 364, 184–188. [Google Scholar]
- Soonpaa, M.H.; Kim, K.K.; Pajak, L.; Franklin, M.; Field, L.J. Cardiomyocyte DNA synthesis and binucleation during murine development. Am. J. Physiol. 1996, 271 Pt 2, H2183–H2189. [Google Scholar]
- Liu, H.; Zhang, C.H.; Ammanamanchi, N.; Suresh, S.; Lewarchik, C.; Rao, K.; Uys, G.M.; Han, L.; Abrial, M.; Yimlamai, D.; et al. Control of cytokinesis by beta-adrenergic receptors indicates an approach for regulating cardiomyocyte endowment. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef]
- Li, F.; Wang, X.; Capasso, J.M.; Gerdes, A.M. Rapid transition of cardiac myocytes from hyperplasia to hypertrophy during postnatal development. J. Mol. Cell. Cardiol. 1996, 28, 1737–1746. [Google Scholar]
- Soonpaa, M.H.; Zebrowski, D.C.; Platt, C.; Rosenzweig, A.; Engel, F.B.; Field, L.J. Cardiomyocyte Cell-Cycle Activity during Preadolescence. Cell 2015, 163, 781–782. [Google Scholar]
- Alkass, K.; Panula, J.; Westman, M.; Wu, T.D.; Guerquin-Kern, J.L.; Bergmann, O. No Evidence for Cardiomyocyte Number Expansion in Preadolescent Mice. Cell 2015, 163, 1026–1036. [Google Scholar]
- Gonzalez-Rosa, J.M.; Sharpe, M.; Field, D.; Soonpaa, M.H.; Field, L.J.; Burns, C.E.; Burns, C.G. Myocardial Polyploidization Creates a Barrier to Heart Regeneration in Zebrafish. Dev. Cell 2018, 44, 433–446.e7. [Google Scholar]
- Jean, M.J.; Deverteuil, P.; Lopez, N.H.; Tapia, J.D.; Schoffstall, B. Adult zebrafish hearts efficiently compensate for excessive forced overload cardiac stress with hyperplastic cardiomegaly. Biores. Open Access 2012, 1, 88–91. [Google Scholar]
- Fisher, D.J.; Heymann, M.A.; Rudolph, A.M. Myocardial oxygen and carbohydrate consumption in fetal lambs in utero and in adult sheep. Am. J. Physiol. 1980, 238, H399–H405. [Google Scholar]
- Fukuda, R.; Marin-Juez, R.; El-Sammak, H.; Beisaw, A.; Ramadass, R.; Kuenne, C.; Guenther, S.; Konzer, A.; Bhagwat, A.M.; Graumann, J.; et al. Stimulation of glycolysis promotes cardiomyocyte proliferation after injury in adult zebrafish. EMBO Rep. 2020, 21, e49752. [Google Scholar]
- Piquereau, J.; Ventura-Clapier, R. Maturation of Cardiac Energy Metabolism During Perinatal Development. Front. Physiol. 2018, 9, 959. [Google Scholar]
- Puente, B.N.; Kimura, W.; Muralidhar, S.A.; Moon, J.; Amatruda, J.F.; Phelps, K.L.; Grinsfelder, D.; Rothermel, B.A.; Chen, R.; Garcia, J.A.; et al. The oxygen-rich postnatal environment induces cardiomyocyte cell-cycle arrest through DNA damage response. Cell 2014, 157, 565–579. [Google Scholar]
- Nakada, Y.; Canseco, D.C.; Thet, S.; Abdisalaam, S.; Asaithamby, A.; Santos, C.X.; Shah, A.M.; Zhang, H.; Faber, J.E.; Kinter, M.T.; et al. Hypoxia induces heart regeneration in adult mice. Nature 2017, 541, 222–227. [Google Scholar]
- Dorn, G.W., 2nd; Vega, R.B.; Kelly, D.P. Mitochondrial biogenesis and dynamics in the developing and diseased heart. Genes Dev. 2015, 29, 1981–1991. [Google Scholar]
- Chung, S.; Dzeja, P.P.; Faustino, R.S.; Perez-Terzic, C.; Behfar, A.; Terzic, A. Mitochondrial oxidative metabolism is required for the cardiac differentiation of stem cells. Nat. Clin. Pract. Cardiovasc. Med. 2007, 4 (Suppl. 1), S60–S67. [Google Scholar]
- Folmes, C.D.; Dzeja, P.P.; Nelson, T.J.; Terzic, A. Mitochondria in control of cell fate. Circ. Res. 2012, 110, 526–529. [Google Scholar]
- Schiff, M.; Ogier de Baulny, H.; Lombes, A. Neonatal cardiomyopathies and metabolic crises due to oxidative phosphorylation defects. Semin. Fetal Neonatal Med. 2011, 16, 216–221. [Google Scholar]
- Ryzhkova, A.I.; Sazonova, M.A.; Sinyov, V.V.; Galitsyna, E.V.; Chicheva, M.M.; Melnichenko, A.A.; Grechko, A.V.; Postnov, A.Y.; Orekhov, A.N.; Shkurat, T.P. Mitochondrial diseases caused by mtDNA mutations: A mini-review. Ther. Clin. Risk Manag. 2018, 14, 1933–1942. [Google Scholar]
- Finsterer, J.; Kothari, S. Cardiac manifestations of primary mitochondrial disorders. Int. J. Cardiol. 2014, 177, 754–763. [Google Scholar]
- Cretoiu, D.; Pavelescu, L.; Duica, F.; Radu, M.; Suciu, N.; Cretoiu, S.M. Myofibers. Adv. Exp. Med. Biol. 2018, 1088, 23–46. [Google Scholar]
- Wilson, A.J.; Schoenauer, R.; Ehler, E.; Agarkova, I.; Bennett, P.M. Cardiomyocyte growth and sarcomerogenesis at the intercalated disc. Cell. Mol. Life Sci. 2014, 71, 165–181. [Google Scholar]
- DeLaughter, D.M.; Bick, A.G.; Wakimoto, H.; McKean, D.; Gorham, J.M.; Kathiriya, I.S.; Hinson, J.T.; Homsy, J.; Gray, J.; Pu, W.; et al. Single-Cell Resolution of Temporal Gene Expression during Heart Development. Dev. Cell 2016, 39, 480–490. [Google Scholar]
- England, J.; Loughna, S. Heavy and light roles: Myosin in the morphogenesis of the heart. Cell. Mol. Life Sci. 2013, 70, 1221–1239. [Google Scholar]
- Ferrante, M.I.; Kiff, R.M.; Goulding, D.A.; Stemple, D.L. Troponin T is essential for sarcomere assembly in zebrafish skeletal muscle. J. Cell. Sci. 2011, 124, 565–577. [Google Scholar]
- Hsiao, C.D.; Tsai, W.Y.; Horng, L.S.; Tsai, H.J. Molecular structure and developmental expression of three muscle-type troponin T genes in zebrafish. Dev. Dyn. 2003, 227, 266–279. [Google Scholar]
- Paige, S.L.; Plonowska, K.; Xu, A.; Wu, S.M. Molecular regulation of cardiomyocyte differentiation. Circ. Res. 2015, 116, 341–353. [Google Scholar]
- Vagnozzi, R.J.; Molkentin, J.D.; Houser, S.R. New Myocyte Formation in the Adult Heart: Endogenous Sources and Therapeutic Implications. Circ. Res. 2018, 123, 159–176. [Google Scholar]
- Galdos, F.X.; Guo, Y.; Paige, S.L.; VanDusen, N.J.; Wu, S.M.; Pu, W.T. Cardiac Regeneration: Lessons From Development. Circ. Res. 2017, 120, 941–959. [Google Scholar]
- Zebrowski, D.C.; Vergarajauregui, S.; Wu, C.C.; Piatkowski, T.; Becker, R.; Leone, M.; Hirth, S.; Ricciardi, F.; Falk, N.; Giessl, A.; et al. Developmental alterations in centrosome integrity contribute to the post-mitotic state of mammalian cardiomyocytes. Elife 2015, 4, e05563. [Google Scholar]
- Xin, M.; Kim, Y.; Sutherland, L.B.; Qi, X.; McAnally, J.; Schwartz, R.J.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. Regulation of insulin-like growth factor signaling by Yap governs cardiomyocyte proliferation and embryonic heart size. Sci. Signal. 2011, 4, ra70. [Google Scholar]
- von Gise, A.; Lin, Z.; Schlegelmilch, K.; Honor, L.B.; Pan, G.M.; Buck, J.N.; Ma, Q.; Ishiwata, T.; Zhou, B.; Camargo, F.D.; et al. YAP1, the nuclear target of Hippo signaling, stimulates heart growth through cardiomyocyte proliferation but not hypertrophy. Proc. Natl. Acad. Sci. USA 2012, 109, 2394–2399. [Google Scholar]
- Heallen, T.; Zhang, M.; Wang, J.; Bonilla-Claudio, M.; Klysik, E.; Johnson, R.L.; Martin, J.F. Hippo pathway inhibits Wnt signaling to restrain cardiomyocyte proliferation and heart size. Science 2011, 332, 458–461. [Google Scholar]
- Liu, R.; Jagannathan, R.; Li, F.; Lee, J.; Balasubramanyam, N.; Kim, B.S.; Yang, P.; Yechoor, V.K.; Moulik, M. Tead1 is required for perinatal cardiomyocyte proliferation. PLoS ONE 2019, 14, e0212017. [Google Scholar]
- Lee, K.F.; Simon, H.; Chen, H.; Bates, B.; Hung, M.C.; Hauser, C. Requirement for neuregulin receptor erbB2 in neural and cardiac development. Nature 1995, 378, 394–398. [Google Scholar]
- Zhao, Y.Y.; Sawyer, D.R.; Baliga, R.R.; Opel, D.J.; Han, X.; Marchionni, M.A.; Kelly, R.A. Neuregulins promote survival and growth of cardiac myocytes. Persistence of ErbB2 and ErbB4 expression in neonatal and adult ventricular myocytes. J. Biol. Chem. 1998, 273, 10261–10269. [Google Scholar]
- D’Uva, G.; Aharonov, A.; Lauriola, M.; Kain, D.; Yahalom-Ronen, Y.; Carvalho, S.; Weisinger, K.; Bassat, E.; Rajchman, D.; Yifa, O.; et al. ERBB2 triggers mammalian heart regeneration by promoting cardiomyocyte dedifferentiation and proliferation. Nat. Cell Biol. 2015, 17, 627–638. [Google Scholar]
- Bersell, K.; Arab, S.; Haring, B.; Kuhn, B. Neuregulin1/ErbB4 signaling induces cardiomyocyte proliferation and repair of heart injury. Cell 2009, 138, 257–270. [Google Scholar]
- Mori, A.D.; Zhu, Y.; Vahora, I.; Nieman, B.; Koshiba-Takeuchi, K.; Davidson, L.; Pizard, A.; Seidman, J.G.; Seidman, C.E.; Chen, X.J.; et al. Tbx5-dependent rheostatic control of cardiac gene expression and morphogenesis. Dev. Biol. 2006, 297, 566–586. [Google Scholar]
- Misra, C.; Chang, S.W.; Basu, M.; Huang, N.; Garg, V. Disruption of myocardial Gata4 and Tbx5 results in defects in cardiomyocyte proliferation and atrioventricular septation. Hum. Mol. Genet. 2014, 23, 5025–5035. [Google Scholar]
- Nadadur, R.D.; Broman, M.T.; Boukens, B.; Mazurek, S.R.; Yang, X.; van den Boogaard, M.; Bekeny, J.; Gadek, M.; Ward, T.; Zhang, M.; et al. Pitx2 modulates a Tbx5-dependent gene regulatory network to maintain atrial rhythm. Sci. Transl. Med. 2016, 8, 354ra115. [Google Scholar]
- Cai, C.L.; Zhou, W.; Yang, L.; Bu, L.; Qyang, Y.; Zhang, X.; Li, X.; Rosenfeld, M.G.; Chen, J.; Evans, S. T-box genes coordinate regional rates of proliferation and regional specification during cardiogenesis. Development 2005, 132, 2475–2487. [Google Scholar]
- Singh, M.K.; Christoffels, V.M.; Dias, J.M.; Trowe, M.O.; Petry, M.; Schuster-Gossler, K.; Burger, A.; Ericson, J.; Kispert, A. Tbx20 is essential for cardiac chamber differentiation and repression of Tbx2. Development 2005, 132, 2697–2707. [Google Scholar]
- Stennard, F.A.; Costa, M.W.; Lai, D.; Biben, C.; Furtado, M.B.; Solloway, M.J.; McCulley, D.J.; Leimena, C.; Preis, J.I.; Dunwoodie, S.L.; et al. Murine T-box transcription factor Tbx20 acts as a repressor during heart development, and is essential for adult heart integrity, function and adaptation. Development 2005, 132, 2451–2462. [Google Scholar]
- Chakraborty, S.; Yutzey, K.E. Tbx20 regulation of cardiac cell proliferation and lineage specialization during embryonic and fetal development in vivo. Dev. Biol. 2012, 363, 234–246. [Google Scholar]
- Boogerd, C.J.; Zhu, X.; Aneas, I.; Sakabe, N.; Zhang, L.; Sobreira, D.R.; Montefiori, L.; Bogomolovas, J.; Joslin, A.C.; Zhou, B.; et al. Tbx20 Is Required in Mid-Gestation Cardiomyocytes and Plays a Central Role in Atrial Development. Circ. Res. 2018, 123, 428–442. [Google Scholar]
- Xiang, F.L.; Guo, M.; Yutzey, K.E. Overexpression of Tbx20 in Adult Cardiomyocytes Promotes Proliferation and Improves Cardiac Function After Myocardial Infarction. Circulation 2016, 133, 1081–1092. [Google Scholar]
- Shen, T.; Aneas, I.; Sakabe, N.; Dirschinger, R.J.; Wang, G.; Smemo, S.; Westlund, J.M.; Cheng, H.; Dalton, N.; Gu, Y.; et al. Tbx20 regulates a genetic program essential to adult mouse cardiomyocyte function. J. Clin. Investig. 2011, 121, 4640–4654. [Google Scholar]
- He, A.; Gu, F.; Hu, Y.; Ma, Q.; Ye, L.Y.; Akiyama, J.A.; Visel, A.; Pennacchio, L.A.; Pu, W.T. Dynamic GATA4 enhancers shape the chromatin landscape central to heart development and disease. Nat. Commun. 2014, 5, 4907. [Google Scholar]
- Oka, T.; Maillet, M.; Watt, A.J.; Schwartz, R.J.; Aronow, B.J.; Duncan, S.A.; Molkentin, J.D. Cardiac-specific deletion of Gata4 reveals its requirement for hypertrophy, compensation, and myocyte viability. Circ. Res. 2006, 98, 837–845. [Google Scholar]
- Sengupta, A.; Kalinichenko, V.V.; Yutzey, K.E. FoxO1 and FoxM1 transcription factors have antagonistic functions in neonatal cardiomyocyte cell-cycle withdrawal and IGF1 gene regulation. Circ. Res. 2013, 112, 267–277. [Google Scholar]
- Ebelt, H.; Hufnagel, N.; Neuhaus, P.; Neuhaus, H.; Gajawada, P.; Simm, A.; Muller-Werdan, U.; Werdan, K.; Braun, T. Divergent siblings: E2F2 and E2F4 but not E2F1 and E2F3 induce DNA synthesis in cardiomyocytes without activation of apoptosis. Circ. Res. 2005, 96, 509–517. [Google Scholar]
- Ebelt, H.; Zhang, Y.; Kampke, A.; Xu, J.; Schlitt, A.; Buerke, M.; Muller-Werdan, U.; Werdan, K.; Braun, T. E2F2 expression induces proliferation of terminally differentiated cardiomyocytes in vivo. Cardiovasc. Res. 2008, 80, 219–226. [Google Scholar]
- Judd, J.; Lovas, J.; Huang, G.N. Defined factors to reactivate cell cycle activity in adult mouse cardiomyocytes. Sci. Rep. 2019, 9, 18830. [Google Scholar]
- Cai, C.L.; Liang, X.; Shi, Y.; Chu, P.H.; Pfaff, S.L.; Chen, J.; Evans, S. Isl1 identifies a cardiac progenitor population that proliferates prior to differentiation and contributes a majority of cells to the heart. Dev. Cell 2003, 5, 877–889. [Google Scholar]
- Tsuchihashi, T.; Maeda, J.; Shin, C.H.; Ivey, K.N.; Black, B.L.; Olson, E.N.; Yamagishi, H.; Srivastava, D. Hand2 function in second heart field progenitors is essential for cardiogenesis. Dev. Biol. 2011, 351, 62–69. [Google Scholar]
- Zeisberg, E.M.; Ma, Q.; Juraszek, A.L.; Moses, K.; Schwartz, R.J.; Izumo, S.; Pu, W.T. Morphogenesis of the right ventricle requires myocardial expression of Gata4. J. Clin. Investig. 2005, 115, 1522–1531. [Google Scholar]
- Gao, R.; Liang, X.; Cheedipudi, S.; Cordero, J.; Jiang, X.; Zhang, Q.; Caputo, L.; Gunther, S.; Kuenne, C.; Ren, Y.; et al. Pioneering function of Isl1 in the epigenetic control of cardiomyocyte cell fate. Cell Res. 2019, 29, 486–501. [Google Scholar]
- Mahmoud, A.I.; Kocabas, F.; Muralidhar, S.A.; Kimura, W.; Koura, A.S.; Thet, S.; Porrello, E.R.; Sadek, H.A. Meis1 regulates postnatal cardiomyocyte cell cycle arrest. Nature 2013, 497, 249–253. [Google Scholar]
- Chakraborty, S.; Sengupta, A.; Yutzey, K.E. Tbx20 promotes cardiomyocyte proliferation and persistence of fetal characteristics in adult mouse hearts. J. Mol. Cell. Cardiol. 2013, 62, 203–213. [Google Scholar]
- Zhang, W.; Chen, H.; Wang, Y.; Yong, W.; Zhu, W.; Liu, Y.; Wagner, G.R.; Payne, R.M.; Field, L.J.; Xin, H.; et al. Tbx20 transcription factor is a downstream mediator for bone morphogenetic protein-10 in regulating cardiac ventricular wall development and function. J. Biol. Chem. 2011, 286, 36820–36829. [Google Scholar]
- Wang, J.; Liu, S.; Heallen, T.; Martin, J.F. The Hippo pathway in the heart: Pivotal roles in development, disease, and regeneration. Nat. Rev. Cardiol. 2018, 15, 672–684. [Google Scholar]
- Xin, M.; Kim, Y.; Sutherland, L.B.; Murakami, M.; Qi, X.; McAnally, J.; Porrello, E.R.; Mahmoud, A.I.; Tan, W.; Shelton, J.M.; et al. Hippo pathway effector Yap promotes cardiac regeneration. Proc. Natl. Acad. Sci. USA 2013, 110, 13839–13844. [Google Scholar]
- Lin, Z.; von Gise, A.; Zhou, P.; Gu, F.; Ma, Q.; Jiang, J.; Yau, A.L.; Buck, J.N.; Gouin, K.A.; van Gorp, P.R.; et al. Cardiac-specific YAP activation improves cardiac function and survival in an experimental murine MI model. Circ. Res. 2014, 115, 354–363. [Google Scholar]
- Monroe, T.O.; Hill, M.C.; Morikawa, Y.; Leach, J.P.; Heallen, T.; Cao, S.; Krijger, P.H.L.; de Laat, W.; Wehrens, X.H.T.; Rodney, G.G.; et al. YAP Partially Reprograms Chromatin Accessibility to Directly Induce Adult Cardiogenesis In Vivo. Dev. Cell 2019, 48, 765–779.e7. [Google Scholar]
- Aharonov, A.; Shakked, A.; Umansky, K.B.; Savidor, A.; Genzelinakh, A.; Kain, D.; Lendengolts, D.; Revach, O.Y.; Morikawa, Y.; Dong, J.; et al. ERBB2 drives YAP activation and EMT-like processes during cardiac regeneration. Nat. Cell Biol. 2020, 22, 1346–1356. [Google Scholar]
- Yekelchyk, M.; Guenther, S.; Preussner, J.; Braun, T. Mono- and multi-nucleated ventricular cardiomyocytes constitute a transcriptionally homogenous cell population. Basic Res. Cardiol. 2019, 114, 36. [Google Scholar]
- Hesse, M.; Bednarz, R.; Carls, E.; Becker, C.; Bondareva, O.; Lother, A.; Geisen, C.; Dressen, M.; Krane, M.; Roell, W.; et al. Proximity to injury, but neither number of nuclei nor ploidy define pathological adaptation and plasticity in cardiomyocytes. J. Mol. Cell. Cardiol. 2020, 152, 95–104. [Google Scholar]
- Heineke, J.; Auger-Messier, M.; Xu, J.; Oka, T.; Sargent, M.A.; York, A.; Klevitsky, R.; Vaikunth, S.; Duncan, S.A.; Aronow, B.J.; et al. Cardiomyocyte GATA4 functions as a stress-responsive regulator of angiogenesis in the murine heart. J. Clin. Investig. 2007, 117, 3198–3210. [Google Scholar]
- van Berlo, J.H.; Elrod, J.W.; van den Hoogenhof, M.M.; York, A.J.; Aronow, B.J.; Duncan, S.A.; Molkentin, J.D. The transcription factor GATA-6 regulates pathological cardiac hypertrophy. Circ. Res. 2010, 107, 1032–1040. [Google Scholar]
- Tanaka, M.; Chen, Z.; Bartunkova, S.; Yamasaki, N.; Izumo, S. The cardiac homeobox gene Csx/Nkx2.5 lies genetically upstream of multiple genes essential for heart development. Development 1999, 126, 1269–1280. [Google Scholar]
- Pikkarainen, S.; Tokola, H.; Majalahti-Palviainen, T.; Kerkela, R.; Hautala, N.; Bhalla, S.S.; Charron, F.; Nemer, M.; Vuolteenaho, O.; Ruskoaho, H. GATA-4 is a nuclear mediator of mechanical stretch-activated hypertrophic program. J. Biol. Chem. 2003, 278, 23807–23816. [Google Scholar]
- Valimaki, M.J.; Tolli, M.A.; Kinnunen, S.M.; Aro, J.; Serpi, R.; Pohjolainen, L.; Talman, V.; Poso, A.; Ruskoaho, H.J. Discovery of Small Molecules Targeting the Synergy of Cardiac Transcription Factors GATA4 and NKX2-5. J. Med. Chem. 2017, 60, 7781–7798. [Google Scholar]
- Malek Mohammadi, M.; Kattih, B.; Grund, A.; Froese, N.; Korf-Klingebiel, M.; Gigina, A.; Schrameck, U.; Rudat, C.; Liang, Q.; Kispert, A.; et al. The transcription factor GATA4 promotes myocardial regeneration in neonatal mice. EMBO Mol. Med. 2017, 9, 265–279. [Google Scholar]
- Chattergoon, N.N.; Giraud, G.D.; Louey, S.; Stork, P.; Fowden, A.L.; Thornburg, K.L. Thyroid hormone drives fetal cardiomyocyte maturation. FASEB J. 2012, 26, 397–408. [Google Scholar]
- Chattergoon, N.N. Thyroid hormone signaling and consequences for cardiac development. J. Endocrinol. 2019, 242, T145–T160. [Google Scholar]
- Gloss, B.; Trost, S.; Bluhm, W.; Swanson, E.; Clark, R.; Winkfein, R.; Janzen, K.; Giles, W.; Chassande, O.; Samarut, J.; et al. Cardiac ion channel expression and contractile function in mice with deletion of thyroid hormone receptor alpha or beta. Endocrinology 2001, 142, 544–550. [Google Scholar]
- Bluhm, W.F.; Meyer, M.; Sayen, M.R.; Swanson, E.A.; Dillmann, W.H. Overexpression of sarcoplasmic reticulum Ca(2+)-ATPase improves cardiac contractile function in hypothyroid mice. Cardiovasc. Res. 1999, 43, 382–388. [Google Scholar]
- Johansson, C.; Gothe, S.; Forrest, D.; Vennstrom, B.; Thoren, P. Cardiovascular phenotype and temperature control in mice lacking thyroid hormone receptor-beta or both alpha1 and beta. Am. J. Physiol. 1999, 276, H2006–H2012. [Google Scholar]
- Ferdous, A.; Wang, Z.V.; Luo, Y.; Li, D.L.; Luo, X.; Schiattarella, G.G.; Altamirano, F.; May, H.I.; Battiprolu, P.K.; Nguyen, A.; et al. FoxO1-Dio2 signaling axis governs cardiomyocyte thyroid hormone metabolism and hypertrophic growth. Nat. Commun. 2020, 11, 2551. [Google Scholar]
- Nguyen, N.U.N.; Canseco, D.C.; Xiao, F.; Nakada, Y.; Li, S.; Lam, N.T.; Muralidhar, S.A.; Savla, J.J.; Hill, J.A.; Le, V.; et al. A calcineurin-Hoxb13 axis regulates growth mode of mammalian cardiomyocytes. Nature 2020, 582, 271–276. [Google Scholar]
- Hinits, Y.; Pan, L.; Walker, C.; Dowd, J.; Moens, C.B.; Hughes, S.M. Zebrafish Mef2ca and Mef2cb are essential for both first and second heart field cardiomyocyte differentiation. Dev. Biol. 2012, 369, 199–210. [Google Scholar]
- Pereira, A.H.; Clemente, C.F.; Cardoso, A.C.; Theizen, T.H.; Rocco, S.A.; Judice, C.C.; Guido, M.C.; Pascoal, V.D.; Lopes-Cendes, I.; Souza, J.R.; et al. MEF2C silencing attenuates load-induced left ventricular hypertrophy by modulating mTOR/S6K pathway in mice. PLoS ONE 2009, 4, e8472. [Google Scholar]
- Morin, S.; Charron, F.; Robitaille, L.; Nemer, M. GATA-dependent recruitment of MEF2 proteins to target promoters. EMBO J. 2000, 19, 2046–2055. [Google Scholar]
- Ghosh, T.K.; Song, F.F.; Packham, E.A.; Buxton, S.; Robinson, T.E.; Ronksley, J.; Self, T.; Bonser, A.J.; Brook, J.D. Physical interaction between TBX5 and MEF2C is required for early heart development. Mol. Cell. Biol. 2009, 29, 2205–2218. [Google Scholar]
- Naya, F.J.; Black, B.L.; Wu, H.; Bassel-Duby, R.; Richardson, J.A.; Hill, J.A.; Olson, E.N. Mitochondrial deficiency and cardiac sudden death in mice lacking the MEF2A transcription factor. Nat. Med. 2002, 8, 1303–1309. [Google Scholar]
- Kolodziejczyk, S.M.; Wang, L.; Balazsi, K.; DeRepentigny, Y.; Kothary, R.; Megeney, L.A. MEF2 is upregulated during cardiac hypertrophy and is required for normal post-natal growth of the myocardium. Curr. Biol. 1999, 9, 1203–1206. [Google Scholar]
- Xu, J.; Gong, N.L.; Bodi, I.; Aronow, B.J.; Backx, P.H.; Molkentin, J.D. Myocyte enhancer factors 2A and 2C induce dilated cardiomyopathy in transgenic mice. J. Biol. Chem. 2006, 281, 9152–9162. [Google Scholar]
- van Oort, R.J.; van Rooij, E.; Bourajjaj, M.; Schimmel, J.; Jansen, M.A.; van der Nagel, R.; Doevendans, P.A.; Schneider, M.D.; van Echteld, C.J.; De Windt, L.J. MEF2 activates a genetic program promoting chamber dilation and contractile dysfunction in calcineurin-induced heart failure. Circulation 2006, 114, 298–308. [Google Scholar]
- Kim, Y.; Phan, D.; van Rooij, E.; Wang, D.Z.; McAnally, J.; Qi, X.; Richardson, J.A.; Hill, J.A.; Bassel-Duby, R.; Olson, E.N. The MEF2D transcription factor mediates stress-dependent cardiac remodeling in mice. J. Clin. Investig. 2008, 118, 124–132. [Google Scholar]
- Zhong, L.; Chiusa, M.; Cadar, A.G.; Lin, A.; Samaras, S.; Davidson, J.M.; Lim, C.C. Targeted inhibition of ANKRD1 disrupts sarcomeric ERK-GATA4 signal transduction and abrogates phenylephrine-induced cardiomyocyte hypertrophy. Cardiovasc. Res. 2015, 106, 261–271. [Google Scholar]
- Qian, L.; Wythe, J.D.; Liu, J.; Cartry, J.; Vogler, G.; Mohapatra, B.; Otway, R.T.; Huang, Y.; King, I.N.; Maillet, M.; et al. Tinman/Nkx2-5 acts via miR-1 and upstream of Cdc42 to regulate heart function across species. J. Cell Biol. 2011, 193, 1181–1196. [Google Scholar]
- Martin, O.J.; Lai, L.; Soundarapandian, M.M.; Leone, T.C.; Zorzano, A.; Keller, M.P.; Attie, A.D.; Muoio, D.M.; Kelly, D.P. A role for peroxisome proliferator-activated receptor gamma coactivator-1 in the control of mitochondrial dynamics during postnatal cardiac growth. Circ. Res. 2014, 114, 626–636. [Google Scholar]
- Lai, L.; Leone, T.C.; Zechner, C.; Schaeffer, P.J.; Kelly, S.M.; Flanagan, D.P.; Medeiros, D.M.; Kovacs, A.; Kelly, D.P. Transcriptional coactivators PGC-1alpha and PGC-lbeta control overlapping programs required for perinatal maturation of the heart. Genes Dev. 2008, 22, 1948–1961. [Google Scholar]
- Kar, D.; Bandyopadhyay, A. Targeting Peroxisome Proliferator Activated Receptor alpha (PPAR alpha) for the Prevention of Mitochondrial Impairment and Hypertrophy in Cardiomyocytes. Cell. Physiol. Biochem. 2018, 49, 245–259. [Google Scholar]
- Lee, W.S.; Kim, J. Peroxisome Proliferator-Activated Receptors and the Heart: Lessons from the Past and Future Directions. PPAR Res. 2015, 2015, 271983. [Google Scholar]
- Watanabe, K.; Fujii, H.; Takahashi, T.; Kodama, M.; Aizawa, Y.; Ohta, Y.; Ono, T.; Hasegawa, G.; Naito, M.; Nakajima, T.; et al. Constitutive regulation of cardiac fatty acid metabolism through peroxisome proliferator-activated receptor alpha associated with age-dependent cardiac toxicity. J. Biol. Chem. 2000, 275, 22293–22299. [Google Scholar]
- Campbell, F.M.; Kozak, R.; Wagner, A.; Altarejos, J.Y.; Dyck, J.R.; Belke, D.D.; Severson, D.L.; Kelly, D.P.; Lopaschuk, G.D. A role for peroxisome proliferator-activated receptor alpha (PPARalpha) in the control of cardiac malonyl-CoA levels: Reduced fatty acid oxidation rates and increased glucose oxidation rates in the hearts of mice lacking PPARalpha are associated with higher concentrations of malonyl-CoA and reduced expression of malonyl-CoA decarboxylase. J. Biol. Chem. 2002, 277, 4098–4103. [Google Scholar]
- Guellich, A.; Damy, T.; Lecarpentier, Y.; Conti, M.; Claes, V.; Samuel, J.L.; Quillard, J.; Hebert, J.L.; Pineau, T.; Coirault, C. Role of oxidative stress in cardiac dysfunction of PPARalpha-/- mice. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H93–H102. [Google Scholar]
- Alaynick, W.A.; Kondo, R.P.; Xie, W.; He, W.; Dufour, C.R.; Downes, M.; Jonker, J.W.; Giles, W.; Naviaux, R.K.; Giguere, V.; et al. ERRgamma directs and maintains the transition to oxidative metabolism in the postnatal heart. Cell Metab. 2007, 6, 13–24. [Google Scholar]
- Bookout, A.L.; Jeong, Y.; Downes, M.; Yu, R.T.; Evans, R.M.; Mangelsdorf, D.J. Anatomical profiling of nuclear receptor expression reveals a hierarchical transcriptional network. Cell 2006, 126, 789–799. [Google Scholar]
- Huss, J.M.; Imahashi, K.; Dufour, C.R.; Weinheimer, C.J.; Courtois, M.; Kovacs, A.; Giguere, V.; Murphy, E.; Kelly, D.P. The nuclear receptor ERRalpha is required for the bioenergetic and functional adaptation to cardiac pressure overload. Cell Metab. 2007, 6, 25–37. [Google Scholar]
- Sakamoto, T.; Matsuura, T.R.; Wan, S.; Ryba, D.M.; Kim, J.U.; Won, K.J.; Lai, L.; Petucci, C.; Petrenko, N.; Musunuru, K.; et al. A Critical Role for Estrogen-Related Receptor Signaling in Cardiac Maturation. Circ. Res. 2020, 126, 1685–1702. [Google Scholar]
- Dufour, C.R.; Wilson, B.J.; Huss, J.M.; Kelly, D.P.; Alaynick, W.A.; Downes, M.; Evans, R.M.; Blanchette, M.; Giguere, V. Genome-wide orchestration of cardiac functions by the orphan nuclear receptors ERRalpha and gamma. Cell Metab. 2007, 5, 345–356. [Google Scholar]
- Neary, M.T.; Ng, K.E.; Ludtmann, M.H.; Hall, A.R.; Piotrowska, I.; Ong, S.B.; Hausenloy, D.J.; Mohun, T.J.; Abramov, A.Y.; Breckenridge, R.A. Hypoxia signaling controls postnatal changes in cardiac mitochondrial morphology and function. J. Mol. Cell. Cardiol. 2014, 74, 340–352. [Google Scholar]
- de Carvalho, A.; Bassaneze, V.; Forni, M.F.; Keusseyan, A.A.; Kowaltowski, A.J.; Krieger, J.E. Early Postnatal Cardiomyocyte Proliferation Requires High Oxidative Energy Metabolism. Sci. Rep. 2017, 7, 15434. [Google Scholar]
- Breckenridge, R.A.; Piotrowska, I.; Ng, K.E.; Ragan, T.J.; West, J.A.; Kotecha, S.; Towers, N.; Bennett, M.; Kienesberger, P.C.; Smolenski, R.T.; et al. Hypoxic regulation of hand1 controls the fetal-neonatal switch in cardiac metabolism. PLoS Biol. 2013, 11, e1001666. [Google Scholar]
- Itoh, K.; Ye, P.; Matsumiya, T.; Tanji, K.; Ozaki, T. Emerging functional cross-talk between the Keap1-Nrf2 system and mitochondria. J. Clin. Biochem. Nutr. 2015, 56, 91–97. [Google Scholar]
- Li, W.; Yu, S.; Liu, T.; Kim, J.H.; Blank, V.; Li, H.; Kong, A.N. Heterodimerization with small Maf proteins enhances nuclear retention of Nrf2 via masking the NESzip motif. Biochim. Biophys. Acta 2008, 1783, 1847–1856. [Google Scholar]
- Alam, J.; Stewart, D.; Touchard, C.; Boinapally, S.; Choi, A.M.; Cook, J.L. Nrf2, a Cap’n’Collar transcription factor, regulates induction of the heme oxygenase-1 gene. J. Biol. Chem. 1999, 274, 26071–26078. [Google Scholar]
- Bartz, R.R.; Suliman, H.B.; Piantadosi, C.A. Redox mechanisms of cardiomyocyte mitochondrial protection. Front. Physiol. 2015, 6, 291. [Google Scholar]
- Leach, J.P.; Heallen, T.; Zhang, M.; Rahmani, M.; Morikawa, Y.; Hill, M.C.; Segura, A.; Willerson, J.T.; Martin, J.F. Hippo pathway deficiency reverses systolic heart failure after infarction. Nature 2017, 550, 260–264. [Google Scholar]
- Shao, D.; Zhai, P.; Del Re, D.P.; Sciarretta, S.; Yabuta, N.; Nojima, H.; Lim, D.S.; Pan, D.; Sadoshima, J. A functional interaction between Hippo-YAP signalling and FoxO1 mediates the oxidative stress response. Nat. Commun. 2014, 5, 3315. [Google Scholar]
- Liu, R.; Lee, J.; Kim, B.S.; Wang, Q.; Buxton, S.K.; Balasubramanyam, N.; Kim, J.J.; Dong, J.; Zhang, A.; Li, S.; et al. Tead1 is required for maintaining adult cardiomyocyte function, and its loss results in lethal dilated cardiomyopathy. JCI Insight 2017, 2, e93343. [Google Scholar]
- Calmettes, G.; John, S.A.; Weiss, J.N.; Ribalet, B. Hexokinase-mitochondrial interactions regulate glucose metabolism differentially in adult and neonatal cardiac myocytes. J. Gen. Physiol. 2013, 142, 425–436. [Google Scholar]
- Hang, C.T.; Yang, J.; Han, P.; Cheng, H.L.; Shang, C.; Ashley, E.; Zhou, B.; Chang, C.P. Chromatin regulation by Brg1 underlies heart muscle development and disease. Nature 2010, 466, 62–67. [Google Scholar]
- Takeuchi, J.K.; Lou, X.; Alexander, J.M.; Sugizaki, H.; Delgado-Olguin, P.; Holloway, A.K.; Mori, A.D.; Wylie, J.N.; Munson, C.; Zhu, Y.; et al. Chromatin remodelling complex dosage modulates transcription factor function in heart development. Nat. Commun. 2011, 2, 187. [Google Scholar]
- May, D.; Blow, M.J.; Kaplan, T.; McCulley, D.J.; Jensen, B.C.; Akiyama, J.A.; Holt, A.; Plajzer-Frick, I.; Shoukry, M.; Wright, C.; et al. Large-scale discovery of enhancers from human heart tissue. Nat. Genet. 2011, 44, 89–93. [Google Scholar]
- Takaya, T.; Kawamura, T.; Morimoto, T.; Ono, K.; Kita, T.; Shimatsu, A.; Hasegawa, K. Identification of p300-targeted acetylated residues in GATA4 during hypertrophic responses in cardiac myocytes. J. Biol. Chem. 2008, 283, 9828–9835. [Google Scholar]
- Sun, H.; Yang, X.; Zhu, J.; Lv, T.; Chen, Y.; Chen, G.; Zhong, L.; Li, Y.; Huang, X.; Huang, G.; et al. Inhibition of p300-HAT results in a reduced histone acetylation and down-regulation of gene expression in cardiac myocytes. Life Sci. 2010, 87, 707–714. [Google Scholar]
- Dickel, D.E.; Barozzi, I.; Zhu, Y.; Fukuda-Yuzawa, Y.; Osterwalder, M.; Mannion, B.J.; May, D.; Spurrell, C.H.; Plajzer-Frick, I.; Pickle, C.S.; et al. Genome-wide compendium and functional assessment of in vivo heart enhancers. Nat. Commun. 2016, 7, 12923. [Google Scholar]
- He, A.; Ma, Q.; Cao, J.; von Gise, A.; Zhou, P.; Xie, H.; Zhang, B.; Hsing, M.; Christodoulou, D.C.; Cahan, P.; et al. Polycomb repressive complex 2 regulates normal development of the mouse heart. Circ. Res. 2012, 110, 406–415. [Google Scholar]
- Ai, S.; Yu, X.; Li, Y.; Peng, Y.; Li, C.; Yue, Y.; Tao, G.; Li, C.; Pu, W.T.; He, A. Divergent Requirements for EZH1 in Heart Development Versus Regeneration. Circ. Res. 2017, 121, 106–112. [Google Scholar]
- Pang, J.K.S.; Phua, Q.H.; Soh, B.S. Applications of miRNAs in cardiac development, disease progression and regeneration. Stem Cell Res. Ther. 2019, 10, 336. [Google Scholar]
- Kalsotra, A.; Xiao, X.; Ward, A.J.; Castle, J.C.; Johnson, J.M.; Burge, C.B.; Cooper, T.A. A postnatal switch of CELF and MBNL proteins reprograms alternative splicing in the developing heart. Proc. Natl. Acad. Sci. USA 2008, 105, 20333–20338. [Google Scholar]
- Wang, E.T.; Ward, A.J.; Cherone, J.M.; Giudice, J.; Wang, T.T.; Treacy, D.J.; Lambert, N.J.; Freese, P.; Saxena, T.; Cooper, T.A.; et al. Antagonistic regulation of mRNA expression and splicing by CELF and MBNL proteins. Genome Res. 2015, 25, 858–871. [Google Scholar]
- Koshelev, M.; Sarma, S.; Price, R.E.; Wehrens, X.H.; Cooper, T.A. Heart-specific overexpression of CUGBP1 reproduces functional and molecular abnormalities of myotonic dystrophy type 1. Hum. Mol. Genet. 2010, 19, 1066–1075. [Google Scholar]
- Hollern, D.P.; Honeysett, J.; Cardiff, R.D.; Andrechek, E.R. The E2F transcription factors regulate tumor development and metastasis in a mouse model of metastatic breast cancer. Mol. Cell. Biol. 2014, 34, 3229–3243. [Google Scholar]
- McBride, K.L.; Zender, G.A.; Fitzgerald-Butt, S.M.; Seagraves, N.J.; Fernbach, S.D.; Zapata, G.; Lewin, M.; Towbin, J.A.; Belmont, J.W. Association of common variants in ERBB4 with congenital left ventricular outflow tract obstruction defects. Birth Defects Res. A Clin. Mol. Teratol. 2011, 91, 162–168. [Google Scholar]
- Kwon, D.H.; Eom, G.H.; Kee, H.J.; Nam, Y.S.; Cho, Y.K.; Kim, D.K.; Koo, J.Y.; Kim, H.S.; Nam, K.I.; Kim, K.K.; et al. Estrogen-related receptor gamma induces cardiac hypertrophy by activating GATA4. J. Mol. Cell. Cardiol. 2013, 65, 88–97. [Google Scholar]
- Hu, X.; Xu, X.; Lu, Z.; Zhang, P.; Fassett, J.; Zhang, Y.; Xin, Y.; Hall, J.L.; Viollet, B.; Bache, R.J.; et al. AMP activated protein kinase-alpha2 regulates expression of estrogen-related receptor-alpha, a metabolic transcription factor related to heart failure development. Hypertension 2011, 58, 696–703. [Google Scholar]
- Sihag, S.; Cresci, S.; Li, A.Y.; Sucharov, C.C.; Lehman, J.J. PGC-1alpha and ERRalpha target gene downregulation is a signature of the failing human heart. J. Mol. Cell. Cardiol. 2009, 46, 201–212. [Google Scholar]
- Fang, T.; Zhu, Y.; Xu, A.; Zhang, Y.; Wu, Q.; Huang, G.; Sheng, W.; Chen, M. Functional analysis of the congenital heart diseaseassociated GATA4 H436Y mutation in vitro. Mol. Med. Rep. 2019, 20, 2325–2331. [Google Scholar]
- Sun, Y.M.; Wang, J.; Qiu, X.B.; Yuan, F.; Li, R.G.; Xu, Y.J.; Qu, X.K.; Shi, H.Y.; Hou, X.M.; Huang, R.T.; et al. A HAND2 Loss-of-Function Mutation Causes Familial Ventricular Septal Defect and Pulmonary Stenosis. G3 Genes Genomes Genom. 2016, 6, 987–992. [Google Scholar]
- Qing, M.; Gorlach, A.; Schumacher, K.; Woltje, M.; Vazquez-Jimenez, J.F.; Hess, J.; Seghaye, M.C. The hypoxia-inducible factor HIF-1 promotes intramyocardial expression of VEGF in infants with congenital cardiac defects. Basic Res. Cardiol. 2007, 102, 224–232. [Google Scholar]
- Ma, L.; Wang, J.; Li, L.; Qiao, Q.; Di, R.M.; Li, X.M.; Xu, Y.J.; Zhang, M.; Li, R.G.; Qiu, X.B.; et al. ISL1 loss-of-function mutation contributes to congenital heart defects. Heart Vessels 2019, 34, 658–668. [Google Scholar]
- Qiao, X.H.; Wang, F.; Zhang, X.L.; Huang, R.T.; Xue, S.; Wang, J.; Qiu, X.B.; Liu, X.Y.; Yang, Y.Q. MEF2C loss-of-function mutation contributes to congenital heart defects. Int. J. Med. Sci. 2017, 14, 1143–1153. [Google Scholar]
- Chung, I.M.; Rajakumar, G. Genetics of Congenital Heart Defects: The NKX2-5 Gene, a Key Player. Genes 2016, 7, 6. [Google Scholar]
- Zang, H.; Mathew, R.O.; Cui, T. The Dark Side of Nrf2 in the Heart. Front. Physiol. 2020, 11, 722. [Google Scholar]
- Oka, S.I.; Sabry, A.D.; Cawley, K.M.; Warren, J.S. Multiple Levels of PGC-1alpha Dysregulation in Heart Failure. Front. Cardiovasc. Med. 2020, 7, 2. [Google Scholar]
- Zhou, L.; Wang, Z.Z.; Xiao, Z.C.; Tu, L. Effects of PPAR-gamma in the Myocardium on the Development of Ventricular Septation. Curr. Med. Sci. 2020, 40, 313–319. [Google Scholar]
- Mori, A.D.; Bruneau, B.G. TBX5 mutations and congenital heart disease: Holt-Oram syndrome revealed. Curr. Opin. Cardiol. 2004, 19, 211–215. [Google Scholar]
- Ye, L.; Yin, M.; Xia, Y.; Jiang, C.; Hong, H.; Liu, J. Decreased Yes-Associated Protein-1 (YAP1) Expression in Pediatric Hearts with Ventricular Septal Defects. PLoS ONE 2015, 10, e0139712. [Google Scholar]
- Casini, S.; Verkerk, A.O.; Remme, C.A. Human iPSC-Derived Cardiomyocytes for Investigation of Disease Mechanisms and Therapeutic Strategies in Inherited Arrhythmia Syndromes: Strengths and Limitations. Cardiovasc. Drugs Ther. 2017, 31, 325–344. [Google Scholar]



{kind=link}
{kind=link}
| Gene Name | Transcriptional Targets in Cardiomyocytes | Role in Cardiomyocyte Development | Role in Postnatal Cardiomyocyte Maturation | Associated Human Heart Defects |
|---|---|---|---|---|
| Btg2 [65] | Unknown | Unknown | Contributes to cell cycle exit | Unknown |
| E2f2/4 [70,71,72,146] | repressor of p53; retinoblastoma protein; activator of cyclins A, E, and D3 (unknown if direct or indirect) | Promotes proliferation | Downregulation contributes to cell cycle exit | Unknown |
| ErbB2/4 [53,54,55,56,147] | activates MAPK and AKT signaling cascades | Promotes proliferation and ventricular trabeculation | Downregulation contributes to cell cycle exit | Abnormalities associated with left ventricular outflow tract defects |
| ERRs [118,119,120,121,122,148,149,150] | activator of Gata4, succinate dehydrogenase genes, electron-transferring flavoproteins, and components of oxidative phosphorylation and the electron transport chain (including Atp5g3, Coq7, Cox6c, Ndufa8, Ckmt2, and Slc25a4) | Not expressed | Promotes mitochondrial oxidative metabolism | Downregulated in human heart failure; alterations are predictive for heart failure |
| FoxM1 [7,69] | Activator of Igf1; repressor of p21, p27 | Promotes proliferation downstream of AKT | Downregulation contributes to cell cycle exit | Unknown |
| FoxO1/3 [69,77,131] | Repressor of Igf1; activator of p21, p27 | Not activated | Promotes postnatal cell cycle exit; promotes survival | Unknown |
| Gata4 [19,56,68,74,87,88,90,91,102,109,135,140,151] | Activator of Cdk2, Cdk4, Hand2, BNP, Myh6; repressor of Cdkn1c | Promotes early differentiation and proliferation | Promotes hypertrophic growth, promotes expression of mature sarcomeric protein isoforms | Mutations associated with instances of congenital heart defects |
| Hand2 [74,75,152] | Unknown | Promotes proliferation in the developing outflow tract and left ventricle | Not expressed | Mutations associated with familial congenital heart defects |
| HIF-1α [123,153] | Repressor of Mfn1, Mfn2, Opa1 | Maintains immature mitochondrial function in hypoxic environment | Downregulation promotes mitochondrial biogenesis, growth, and maturation | Elevated levels of protein in acyanotic congenital heart disease with hypoxemia |
| Isl1 [73,74,75,154] | Activator of Fgfs, Bmps, Hand2 | Promotes proliferation and heart field specification | Not expressed | Mutations associated with congenital heart defects |
| Maf [126,127,128] | Activators of ARE enhancers; Gsta1, HO-1 | Not expressed | Antioxidant effects to handle increased ROS production | Unknown |
| Mef2 [101,102,103,104,105,106,107,108,155] | Activators of Myh6 | Promotes myofibril stability and sarcomere organization | Promotes myofibril stability and sarcomere organization; promotes expression of mature sarcomeric protein isoforms | Mutations associated with familiar congenital heart defects |
| Meis1 [77,99] | Activator of p15, p16, p21 | Not expressed | Promotes cell cycle exit and hypertrophic growth in combination with Hoxb13 | Unknown |
| Nkx2.5 [90,91,110,156] | Activator of BNP; miR-1 | Promotes early differentiation and proliferation | Promotes hypertrophic growth and sarcomere organization | Mutations frequently associated with congenital heart defects |
| Nrf2 [128,129,157] | Activator of Nrf-1, ARE enhancers; Gsta1, HO-1 | Promotes mitochondrial biogenesis | Antioxidant effects to handle increased ROS production; rapidly degraded in non-stressed conditions | Abnormalities associated with heart failure progression |
| PGC1α [111,112,113,114,115,116,117,118,119,120,121,122,158] | Activator of ERRs, activator of succinate dehydrogenase genes, electron-transferring flavoproteins, and components of oxidative phosphorylation and the electron transport chain (including Atp5g3, Coq7, Cox6c, Ndufa8, Ckmt2, and Slc25a4) | Promotes mitochondrial biogenesis | Promotes fatty acid oxidation while inhibiting glycolysis, promotes antioxidant properties in stressed conditions | Mutations associated with congestive heart failure |
| PPARs [111,112,113,114,115,116,117,122,159] | Activator of ERRs, activator of succinate dehydrogenase genes, electron-transferring flavoproteins, and components of oxidative phosphorylation and the electron transport chain (including Atp5g3, Coq7, Cox6c, Ndufa8, Ckmt2, and Slc25a4) | Promotes mitochondrial biogenesis | Promotes fatty acid oxidation while inhibiting glycolysis, promotes antioxidant properties in stressed conditions | Mutations associated with ventricular septal defects |
| Tbx20 [60,61,62,63,64,65,66] | Activator of Ccna2, Cdde, Mycn, Erbb2; repressor of Cdkn1a, Meis1, Btg2 | Promotes cell specification and proliferation | Downregulation promotes cell cycle exit; promotes sarcomere and myofibrillar organization | Mutations associated with common congenital heart defects |
| Tbx5 [57,58,59,103,160] | Activator of Cdk2, Cdk4, Nppa, Gja5, Scn5a, Myh6 | Promotes heart chamber growth and proliferation | Promotes conduction and ion channel homeostasis | Mutations associated with multiple congenital heart defects, including Holt-Oram Syndrome |
| Yap1 [49,50,51,52,130,131,132,161] | Activator of Smads, Tcf4, Parkin; Repressor of Wnt signaling | Promotes proliferation | Downregulation promotes cell cycle arrest; promotes oxidative phosphorylation and mitochondrial homeostasis; promotes antioxidant properties in stressed conditions | Reduced levels associated with ventricular septal defects |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Padula, S.L.; Velayutham, N.; Yutzey, K.E. Transcriptional Regulation of Postnatal Cardiomyocyte Maturation and Regeneration. Int. J. Mol. Sci. 2021, 22, 3288. https://doi.org/10.3390/ijms22063288
Padula SL, Velayutham N, Yutzey KE. Transcriptional Regulation of Postnatal Cardiomyocyte Maturation and Regeneration. International Journal of Molecular Sciences. 2021; 22(6):3288. https://doi.org/10.3390/ijms22063288
Chicago/Turabian StylePadula, Stephanie L., Nivedhitha Velayutham, and Katherine E. Yutzey. 2021. "Transcriptional Regulation of Postnatal Cardiomyocyte Maturation and Regeneration" International Journal of Molecular Sciences 22, no. 6: 3288. https://doi.org/10.3390/ijms22063288
APA StylePadula, S. L., Velayutham, N., & Yutzey, K. E. (2021). Transcriptional Regulation of Postnatal Cardiomyocyte Maturation and Regeneration. International Journal of Molecular Sciences, 22(6), 3288. https://doi.org/10.3390/ijms22063288