
Discovering Biomarkers for Non-Alcoholic Steatohepatitis Patients with and without Hepatocellular Carcinoma Using Fecal Metaproteomics

 , , , and
, , , and
Abstract
:1. Introduction
2. Results
2.1. Characteristics of the Study Cohort
2.2. Characterization of Fecal Metaproteomics
2.3. Identification of Disease-Specific Metaprotein Patterns
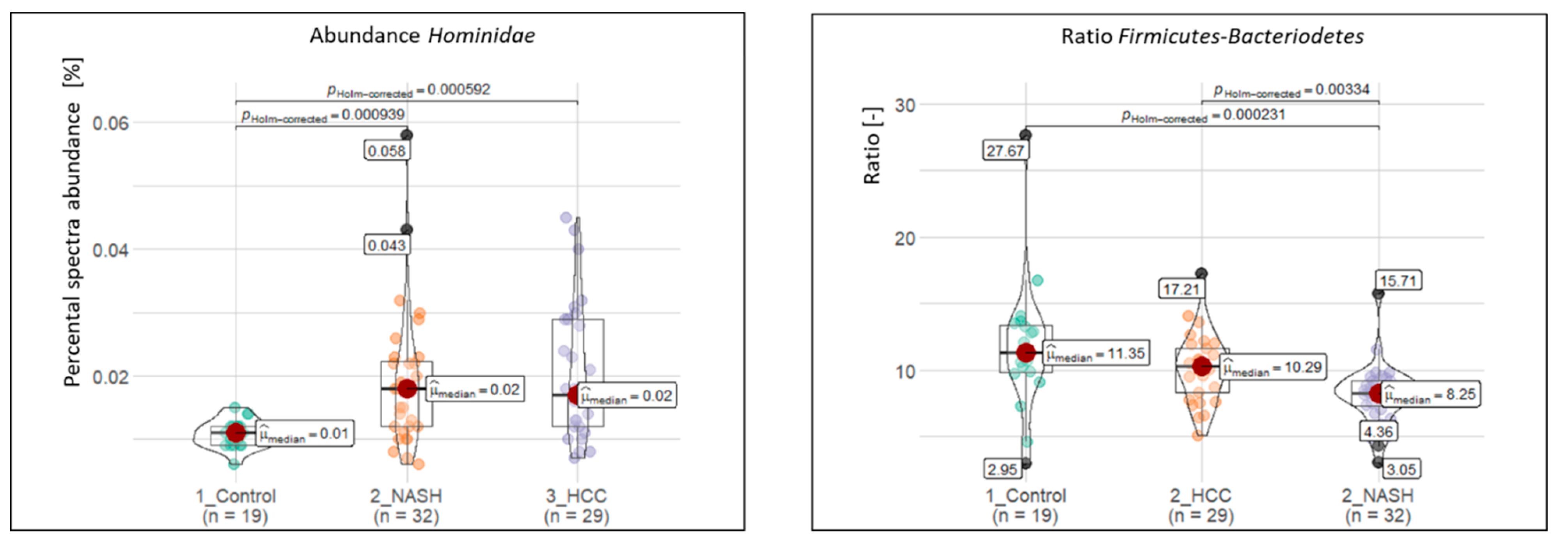
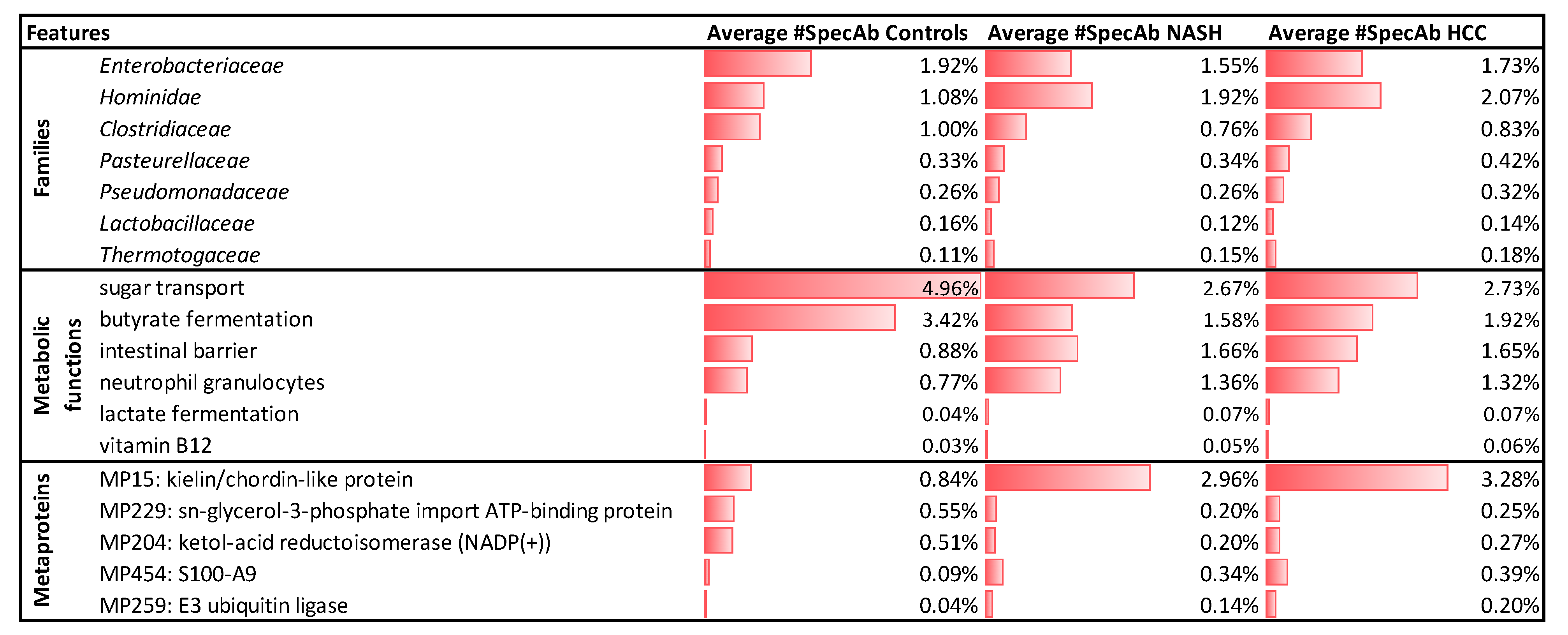
2.4. Significantly Altered Metaproteins, Taxonomies, and Functions
2.5. Potential Biomarkers to Distinguish NASH and HCC from Controls
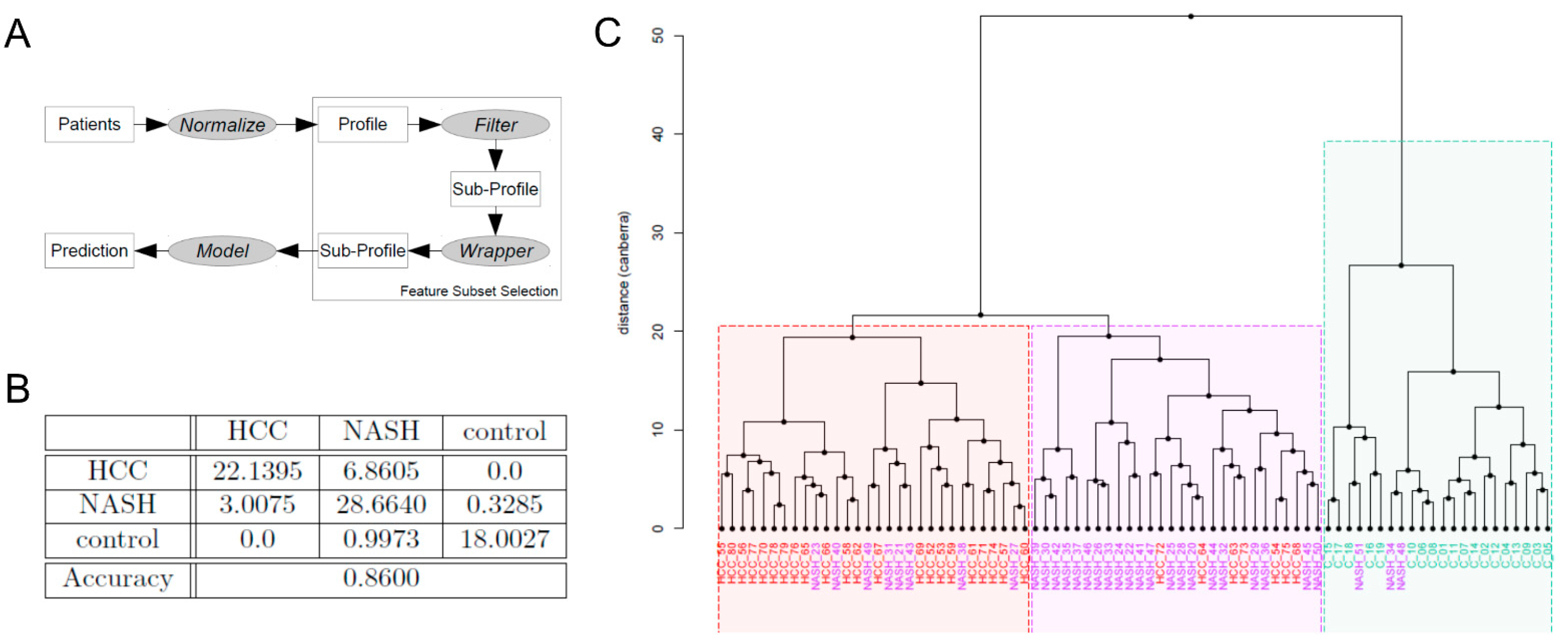
2.6. Machine Learning-Based Biomarker Panels to Separate NASH from HCC and Controls
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Patient Recruitment and Sample Collection
5.2. Transient Elastography and Controlled Attenuation Parameter
5.3. ELISA
5.4. Fecal Sample Preparation for Metaproteomics
5.5. Data Handling
5.6. Statistical Analysis
5.7. Development of a Biomarker Panel
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AFP/AFP-L3 | alpha-fetoprotein/lectin-3-reactive alpha-fetoprotein |
| ALT | alanine aminotransferase |
| ANOSIM | analysis of similarities |
| AP | alkaline phosphatase |
| AST | aspartate aminotransferase |
| BMI | body mass index |
| CAP | controlled attenuation parameter |
| DCP | des-gamma-carboxyprothrombin |
| ER | endoplasmic reticulum |
| FGF19/21 | fibroblast growth factor 19/21 |
| γGT | gamma-glutamyltransferase |
| GLDH | glutamate dehydrogenase |
| GLP1 | glucagon-like peptide |
| HCC | hepatocellular carcinoma |
| HDL | high-density lipoprotein |
| IL6 | interleukin 6 |
| LBP1 | lipoprotein-binding protein 1 |
| LDH | lactate dehydrogenase |
| LDL | low-density lipoprotein |
| NAFLD | non-alcoholic fatty liver disease |
| NASH | non-alcoholic steatohepatitis |
| PCA | principal component analysis |
| ROC | receiver operating characteristic |
| #SpecAb | spectral abundance |
| TAG | triglyceride |
| TE | transient elastography |
| TGFβ | tumor growth factor beta |
| TNFα | tumor necrosis factor alpha |
References
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global Burden of NAFLD and NASH: Trends, Predictions, Risk Factors and Prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Boyle, M.; Masson, S.; Anstee, Q.M. The Bidirectional Impacts of Alcohol Consumption and the Metabolic Syndrome: Cofactors for Progressive Fatty Liver Disease. J. Hepatol. 2018, 68, 251–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeh, M.M.; Brunt, E.M. Pathology of Nonalcoholic Fatty Liver Disease. Am. J. Clin. Pathol. 2007, 128, 837–847. [Google Scholar] [CrossRef] [PubMed]
- Powell, E.E.; Wong, V.W.-S.; Rinella, M. Non-Alcoholic Fatty Liver Disease. Lancet 2021, 397, 2212–2224. [Google Scholar] [CrossRef]
- Bugianesi, E. Non-Alcoholic Steatohepatitis and Cancer. Clin. Liver Dis. 2007, 11, 191–207. [Google Scholar] [CrossRef] [PubMed]
- Bechmann, L.P.; Hannivoort, R.A.; Gerken, G.; Hotamisligil, G.S.; Trauner, M.; Canbay, A. The Interaction of Hepatic Lipid and Glucose Metabolism in Liver Diseases. J. Hepatol. 2012, 56, 952–964. [Google Scholar] [CrossRef] [Green Version]
- El-Serag, H.B. Hepatocellular Carcinoma. N. Engl. J. Med. 2011, 365, 1118–1127. [Google Scholar] [CrossRef]
- Ertle, J.; Dechêne, A.; Sowa, J.-P.; Penndorf, V.; Herzer, K.; Kaiser, G.; Schlaak, J.F.; Gerken, G.; Syn, W.-K.; Canbay, A. Non-Alcoholic Fatty Liver Disease Progresses to Hepatocellular Carcinoma in the Absence of Apparent Cirrhosis. Int. J. Cancer 2011, 128, 2436–2443. [Google Scholar] [CrossRef]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD Development and Therapeutic Strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef]
- Yoshimoto, S.; Loo, T.M.; Atarashi, K.; Kanda, H.; Sato, S.; Oyadomari, S.; Iwakura, Y.; Oshima, K.; Morita, H.; Hattori, M.; et al. Obesity-Induced Gut Microbial Metabolite Promotes Liver Cancer through Senescence Secretome. Nature 2013, 499, 97–101. [Google Scholar] [CrossRef]
- Mouzaki, M.; Wang, A.Y.; Bandsma, R.; Comelli, E.M.; Arendt, B.M.; Zhang, L.; Fung, S.; Fischer, S.E.; McGilvray, I.G.; Allard, J.P. Bile Acids and Dysbiosis in Non-Alcoholic Fatty Liver Disease. PLoS ONE 2016, 11, e0151829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konturek, P.C.; Harsch, I.A.; Konturek, K.; Schink, M.; Konturek, T.; Neurath, M.F.; Zopf, Y. Gut-Liver Axis: How Do Gut Bacteria Influence the Liver? Med. Sci. 2018, 6, 79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, J.-W.; Chen, X.-H.; Ren, Z.; Zheng, S.-S. Gut Microbial Dysbiosis Associates Hepatocellular Carcinoma via the Gut-Liver Axis. Hepatobiliary Pancreat. Dis. Int. 2019, 18, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Sydor, S.; Best, J.; Messerschmidt, I.; Manka, P.; Vilchez-Vargas, R.; Brodesser, S.; Lucas, C.; Wegehaupt, A.; Wenning, C.; Aßmuth, S.; et al. Altered Microbiota Diversity and Bile Acid Signaling in Cirrhotic and Noncirrhotic NASH-HCC. Clin. Transl. Gastroenterol. 2020, 11, e00131. [Google Scholar] [CrossRef]
- Lehmann, T.; Schallert, K.; Vilchez-Vargas, R.; Benndorf, D.; Püttker, S.; Sydor, S.; Schulz, C.; Bechmann, L.; Canbay, A.; Heidrich, B.; et al. Metaproteomics of Fecal Samples of Crohn’s Disease and Ulcerative Colitis. J. Proteom. 2019, 201, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Biemann, R.; Buß, E.; Benndorf, D.; Lehmann, T.; Schallert, K.; Püttker, S.; Reichl, U.; Isermann, B.; Schneider, J.G.; Saake, G.; et al. Fecal Metaproteomics Reveals Reduced Gut Inflammation and Changed Microbial Metabolism Following Lifestyle-Induced Weight Loss. Biomolecules 2021, 11, 726. [Google Scholar] [CrossRef]
- Kupčová, V.; Fedelešová, M.; Bulas, J.; Kozmonová, P.; Turecký, L. Overview of the Pathogenesis, Genetic, and Non-Invasive Clinical, Biochemical, and Scoring Methods in the Assessment of NAFLD. Int. J. Environ. Res. Public Health 2019, 16, 3570. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Liu, J.; Luo, F. Serum Tumor Markers for Detection of Hepatocellular Carcinoma. World J. Gastroenterol. 2006, 12, 1175–1181. [Google Scholar] [CrossRef] [Green Version]
- Negro, F. Natural History of NASH and HCC. Liver Int. 2020, 40 (Suppl. S1), 72–76. [Google Scholar] [CrossRef] [Green Version]
- Bashiardes, S.; Shapiro, H.; Rozin, S.; Shibolet, O.; Elinav, E. Non-Alcoholic Fatty Liver and the Gut Microbiota. Mol. Metab. 2016, 5, 782–794. [Google Scholar] [CrossRef]
- Michelotti, G.A.; Machado, M.V.; Diehl, A.M. NAFLD, NASH and Liver Cancer. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 656–665. [Google Scholar] [CrossRef] [PubMed]
- Anstee, Q.M.; Reeves, H.L.; Kotsiliti, E.; Govaere, O.; Heikenwalder, M. From NASH to HCC: Current Concepts and Future Challenges. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 411–428. [Google Scholar] [CrossRef] [PubMed]
- Biernacka, A.; Dobaczewski, M.; Frangogiannis, N.G. TGF-β Signaling in Fibrosis. Growth Factors 2011, 29, 196–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kandeil, M.A.; Hashem, R.M.; Mahmoud, M.O.; Hetta, M.H.; Tohamy, M.A. Zingiber Officinale Extract and Omega-3 Fatty Acids Ameliorate Endoplasmic Reticulum Stress in a Nonalcoholic Fatty Liver Rat Model. J. Food Biochem. 2019, 43, e13076. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Li, C.; Kang, N.; Malhi, H.; Shah, V.H.; Maiers, J.L. Transforming Growth Factor β (TGFβ) Cross-Talk with the Unfolded Protein Response Is Critical for Hepatic Stellate Cell Activation. J. Biol. Chem. 2019, 294, 3137–3151. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y. E3 Ubiquitin Ligases as Cancer Targets and Biomarkers. Neoplasia 2006, 8, 645–654. [Google Scholar] [CrossRef] [Green Version]
- Soofi, A.; Wolf, K.I.; Emont, M.P.; Qi, N.; Martinez-Santibanez, G.; Grimley, E.; Ostwani, W.; Dressler, G.R. The Kielin/Chordin-like Protein (KCP) Attenuates High-Fat Diet-Induced Obesity and Metabolic Syndrome in Mice. J. Biol. Chem. 2017, 292, 9051–9062. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y.; Gui, W.; Koo, I.; Smith, P.B.; Allman, E.L.; Nichols, R.G.; Rimal, B.; Cai, J.; Liu, Q.; Patterson, A.D. The Microbiome Modulating Activity of Bile Acids. Gut Microbes 2020, 11, 979–996. [Google Scholar] [CrossRef]
- Voland, L.; Le Roy, T.; Debédat, J.; Clément, K. Gut Microbiota and Vitamin Status in Persons with Obesity: A Key Interplay. Obes. Rev. 2022, 23, e13377. [Google Scholar] [CrossRef]
- Xu, Y.; Xiang, S.; Ye, K.; Zheng, Y.; Feng, X.; Zhu, X.; Chen, J.; Chen, Y. Cobalamin (Vitamin B12) Induced a Shift in Microbial Composition and Metabolic Activity in an in Vitro Colon Simulation. Front. Microbiol. 2018, 9, 2780. [Google Scholar] [CrossRef] [Green Version]
- Xie, G.; Wang, X.; Huang, F.; Zhao, A.; Chen, W.; Yan, J.; Zhang, Y.; Lei, S.; Ge, K.; Zheng, X.; et al. Dysregulated Hepatic Bile Acids Collaboratively Promote Liver Carcinogenesis. Int. J. Cancer 2016, 139, 1764–1775. [Google Scholar] [CrossRef] [PubMed]
- Miura, S.; Mitsuhashi, N.; Shimizu, H.; Kimura, F.; Yoshidome, H.; Otsuka, M.; Kato, A.; Shida, T.; Okamura, D.; Miyazaki, M. Fibroblast Growth Factor 19 Expression Correlates with Tumor Progression and Poorer Prognosis of Hepatocellular Carcinoma. BMC Cancer 2012, 12, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Zhang, W.; Doughtie, A.; Cui, G.; Li, X.; Pandit, H.; Yang, Y.; Li, S.; Martin, R. Up-Regulation of Fibroblast Growth Factor 19 and Its Receptor Associates with Progression from Fatty Liver to Hepatocellular Carcinoma. Oncotarget 2016, 7, 52329–52339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canova, M.J.; Molle, V. Bacterial Serine/Threonine Protein Kinases in Host-Pathogen Interactions. J. Biol. Chem. 2014, 289, 9473–9479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, L.; Baker, S.S.; Gill, C.; Liu, W.; Alkhouri, R.; Baker, R.D.; Gill, S.R. Characterization of Gut Microbiomes in Nonalcoholic Steatohepatitis (NASH) Patients: A Connection between Endogenous Alcohol and NASH. Hepatology 2013, 57, 601–609. [Google Scholar] [CrossRef]
- Yue, X.; Ai, J.; Xu, Y.; Chen, Y.; Huang, M.; Yang, X.; Hu, B.; Zhang, H.; He, C.; Yang, X.; et al. Polymeric Immunoglobulin Receptor Promotes Tumor Growth in Hepatocellular Carcinoma. Hepatology 2017, 65, 1948–1962. [Google Scholar] [CrossRef]
- Zhang, Y.; Lu, W.; Chen, X.; Cao, Y.; Yang, Z. A Bioinformatic Analysis of Correlations between Polymeric Immunoglobulin Receptor (PIGR) and Liver Fibrosis Progression. BioMed Res. Int. 2021, 2021, 5541780. [Google Scholar] [CrossRef]
- Samraj, A.N.; Pearce, O.M.T.; Läubli, H.; Crittenden, A.N.; Bergfeld, A.K.; Banda, K.; Gregg, C.J.; Bingman, A.E.; Secrest, P.; Diaz, S.L.; et al. A Red Meat-Derived Glycan Promotes Inflammation and Cancer Progression. Proc. Natl. Acad. Sci. USA 2015, 112, 542–547. [Google Scholar] [CrossRef] [Green Version]
- Dawson, D.A.; Li, T.-K.; Grant, B.F. A Prospective Study of Risk Drinking: At Risk for What? Drug Alcohol Depend. 2008, 95, 62–72. [Google Scholar] [CrossRef] [Green Version]
- Heyer, R.; Schallert, K.; Büdel, A.; Zoun, R.; Dorl, S.; Behne, A.; Kohrs, F.; Püttker, S.; Siewert, C.; Muth, T.; et al. A Robust and Universal Metaproteomics Workflow for Research Studies and Routine Diagnostics Within 24 h Using Phenol Extraction, FASP Digest, and the MetaProteomeAnalyzer. Front. Microbiol. 2019, 10, 1883. [Google Scholar] [CrossRef] [Green Version]
- Human Microbiome Jumpstart Reference Strains Consortium; Nelson, K.E.; Weinstock, G.M.; Highlander, S.K.; Worley, K.C.; Creasy, H.H.; Wortman, J.R.; Rusch, D.B.; Mitreva, M.; Sodergren, E.; et al. A Catalog of Reference Genomes from the Human Microbiome. Science 2010, 328, 994–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanai, M.; Maeda, Y.; Okada, Y. Grimon: Graphical Interface to Visualize Multi-Omics Networks. Bioinformatics 2018, 34, 3934–3936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ondov, B.D.; Bergman, N.H.; Phillippy, A.M. Interactive Metagenomic Visualization in a Web Browser. BMC Bioinform. 2011, 12, 385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ROC Analysis: Online ROC Curve Calculator. Available online: http://www.rad.jhmi.edu/jeng/javarad/roc/JROCFITi.html (accessed on 23 May 2022).
- Fisher, R.A. The Use of Multiple Measurements in Taxonomic Problems. Ann. Eugen. 1936, 7, 179–188. [Google Scholar] [CrossRef]
- Dudoit, S.; Fridlyand, J.; Speed, T.P. Comparison of Discrimination Methods for the Classification of Tumors Using Gene Expression Data. J. Am. Stat. Assoc. 2002, 97, 77–87. [Google Scholar] [CrossRef] [Green Version]
- Berkson, J. Application of the Logistic Function to Bio-Assay. J. Am. Stat. Assoc. 1944, 39, 357–365. [Google Scholar] [CrossRef]
- Vapnik, V. The Nature of Statistical Learning Theory; Springer: New York, NY, USA, 2000. [Google Scholar]
- Breiman, L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef] [Green Version]
- Geurts, P.; Ernst, D.; Wehenkel, L. Extremely Randomized Trees. Mach. Learn. 2006, 63, 3–42. [Google Scholar] [CrossRef] [Green Version]
- Fix, E.; Hodges, J.L. Discriminatory Analysis—Nonparametric Discrimination: Consistency Properties. Int. Stat. Rev. 1989, 57, 238–247. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metaproteins | #SpecAb | Area under Curve |
|---|---|---|
| Kielin/chordin-like protein | 2.568% | 0.893 |
| Sn-glycerol-3-phosphate import ATP-binding protein | 0.303% | 0.868 |
| Ketol-acid reductoisomerase (NADP(+)) | 0.297% | 0.862 |
| Protein S100-A9 | 0.296% | 0.815 |
| Probable E3 ubiquitin ligase complex SCF | 0.135% | 0.839 |
| 30S ribosomal protein S3 | 0.120% | 0.879 |
| Formate-tetrahydrofolate ligase 2 | 0.073% | 0.913 |
| 30S ribosomal protein S2 | 0.066% | 0.842 |
| Acyl-CoA dehydrogenase, short-chain specific | 0.066% | 0.883 |
| Glyceraldehyde-3-phosphate dehydrogenate | 0.063% | 0.905 |
| Comparison | Accuracy | Number of Features |
|---|---|---|
| NASH vs. Control | 0.9998 | 7 features |
| HCC vs. Control: | 1 | 5 features |
| HCC vs. NASH | 0.8640 | 10 features |
| HCC vs. NASH vs. Control | 0.86 | 11 features |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sydor, S.; Dandyk, C.; Schwerdt, J.; Manka, P.; Benndorf, D.; Lehmann, T.; Schallert, K.; Wolf, M.; Reichl, U.; Canbay, A.; et al. Discovering Biomarkers for Non-Alcoholic Steatohepatitis Patients with and without Hepatocellular Carcinoma Using Fecal Metaproteomics. Int. J. Mol. Sci. 2022, 23, 8841. https://doi.org/10.3390/ijms23168841
Sydor S, Dandyk C, Schwerdt J, Manka P, Benndorf D, Lehmann T, Schallert K, Wolf M, Reichl U, Canbay A, et al. Discovering Biomarkers for Non-Alcoholic Steatohepatitis Patients with and without Hepatocellular Carcinoma Using Fecal Metaproteomics. International Journal of Molecular Sciences. 2022; 23(16):8841. https://doi.org/10.3390/ijms23168841
Chicago/Turabian StyleSydor, Svenja, Christian Dandyk, Johannes Schwerdt, Paul Manka, Dirk Benndorf, Theresa Lehmann, Kay Schallert, Maximilian Wolf, Udo Reichl, Ali Canbay, and et al. 2022. "Discovering Biomarkers for Non-Alcoholic Steatohepatitis Patients with and without Hepatocellular Carcinoma Using Fecal Metaproteomics" International Journal of Molecular Sciences 23, no. 16: 8841. https://doi.org/10.3390/ijms23168841
APA StyleSydor, S., Dandyk, C., Schwerdt, J., Manka, P., Benndorf, D., Lehmann, T., Schallert, K., Wolf, M., Reichl, U., Canbay, A., Bechmann, L. P., & Heyer, R. (2022). Discovering Biomarkers for Non-Alcoholic Steatohepatitis Patients with and without Hepatocellular Carcinoma Using Fecal Metaproteomics. International Journal of Molecular Sciences, 23(16), 8841. https://doi.org/10.3390/ijms23168841