
Dynamics of Inflammatory and Neurodegenerative Biomarkers after Autologous Hematopoietic Stem Cell Transplantation in Multiple Sclerosis
 , , ,
, , ,
Abstract
:1. Introduction
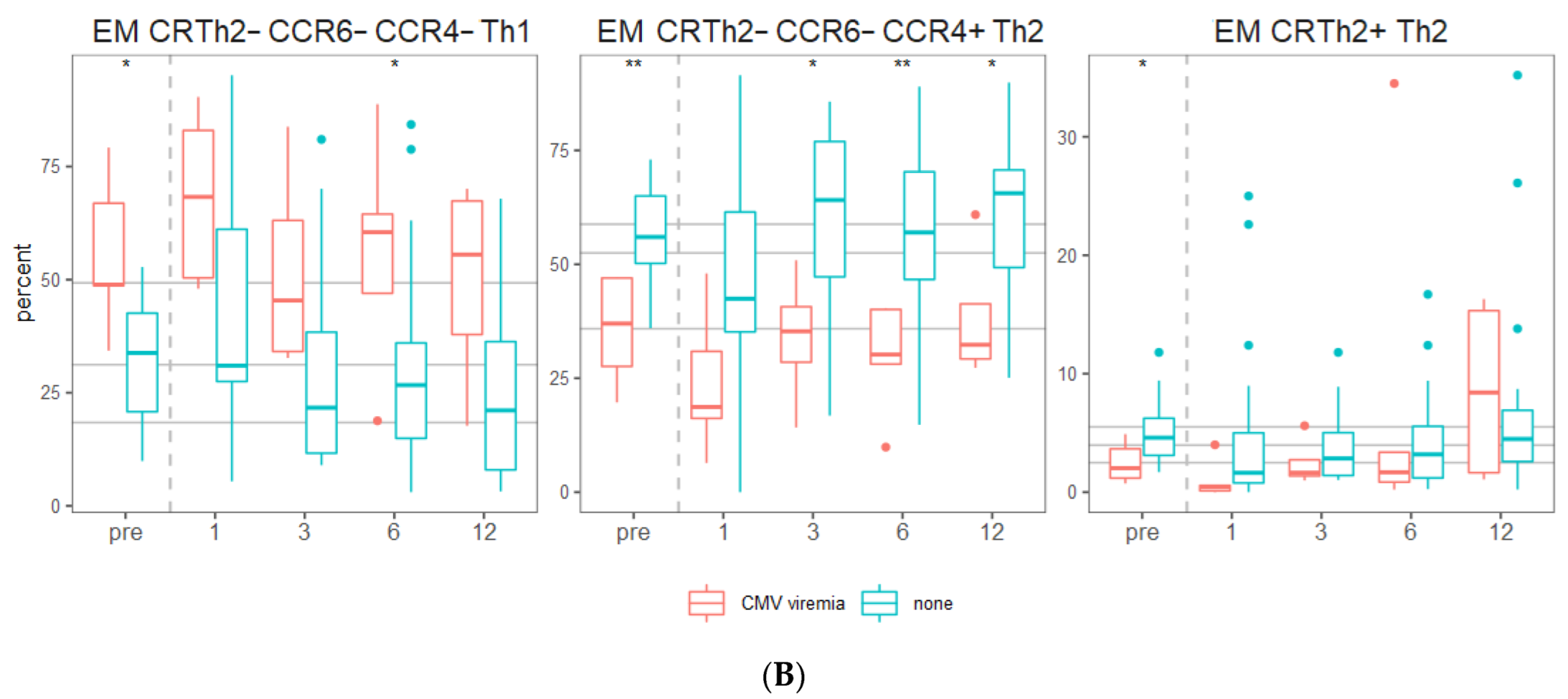
2. Results
3. Discussion
4. Material and Methods
4.1. Patients, Treatment Regimen and Samples
4.2. Detection of Biomarkers
4.3. Flow Cytometry Staining
4.4. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sospedra, M.; Martin, R. Immunology of Multiple Sclerosis. Semin. Neurol. 2016, 36, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Sawcer, S.; Hellenthal, G.; Pirinen, M.; Spencer, C.C.; Patsopoulos, N.A.; Moutsianas, L.; Dilthey, A.; Su, Z.; Freeman, C.; Hunt, S.E.; et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 2011, 476, 214–219. [Google Scholar] [PubMed]
- Belbasis, L.; Bellou, V.; Evangelou, E.; Ioannidis, J.P.A.; Tzoulaki, I. Environmental risk factors and multiple sclerosis: An umbrella review of systematic reviews and meta-analyses. Lancet Neurol. 2015, 14, 263–273. [Google Scholar] [CrossRef]
- Olsson, T.; Barcellos, L.F.; Alfredsson, L. Interactions between genetic, lifestyle and environmental risk factors for multiple sclerosis. Nat. Rev. Neurol. 2017, 13, 25–36. [Google Scholar] [CrossRef]
- Mirza, A.; Forbes, J.D.; Zhu, F.; Bernstein, C.N.; Van Domselaar, G.; Graham, M.; Waubant, E.; Tremlett, H. The multiple sclerosis gut microbiota: A systematic review. Mult. Scler. Relat. Disord. 2020, 37, 101427. [Google Scholar] [CrossRef]
- Jelcic, I.; Al Nimer, F.; Wang, J.; Lentsch, V.; Planas, R.; Jelcic, I.; Madjovski, A.; Ruhrmann, S.; Faigle, W.; Frauenknecht, K.; et al. Memory B Cells Activate Brain-Homing, Autoreactive CD4(+) T Cells in Multiple Sclerosis. Cell 2018, 175, 85–100.e23. [Google Scholar] [CrossRef]
- Wang, J.; Jelcic, I.; Mühlenbruch, L.; Haunerdinger, V.; Toussaint, N.C.; Zhao, Y.; Cruciani, C.; Faigle, W.; Naghavian, R.; Foege, M.; et al. HLA-DR15 Molecules Jointly Shape an Autoreactive T Cell Repertoire in Multiple Sclerosis. Cell 2020, 183, 1264–1281.e20. [Google Scholar] [CrossRef]
- Sospedra, M.; Martin, R. Immunology of multiple sclerosis. Annu. Rev. Immunol. 2005, 23, 683–747. [Google Scholar] [CrossRef]
- Lünemann, J.D.; Jelcic, I.; Roberts, S.; Lutterotti, A.; Tackenberg, B.; Martin, R.; Münz, C. EBNA1-specific T cells from patients with multiple sclerosis cross react with myelin antigens and co-produce IFN-gamma and IL-2. J. Exp. Med. 2008, 205, 1763–1773. [Google Scholar] [CrossRef]
- Planas, R.; Santos, R.; Tomas-Ojer, P.; Cruciani, C.; Lutterotti, A.; Faigle, W.; Scharen-Wiemers, N.; Espejo, C.; Eixarch, H.; Pinilla, C.; et al. GDP-l-fucose synthase is a CD4(+) T cell-specific autoantigen in DRB3*02, 02 patients with multiple sclerosis. Sci. Transl. Medicine. 2018, 10, eaat4301. [Google Scholar] [CrossRef] [Green Version]
- Becattini, S.; Latorre, D.; Mele, F.; Foglierini, M.; De Gregorio, C.; Cassotta, A.; Fernandez, B.; Kelderman, S.; Schumacher, T.N.; Corti, D.; et al. T cell immunity. Functional heterogeneity of human memory CD4⁺ T cell clones primed by pathogens or vaccines. Science 2015, 347, 400–406. [Google Scholar] [CrossRef]
- Cao, Y.; Goods, B.A.; Raddassi, K.; Nepom, G.T.; Kwok, W.W.; Love, J.C.; Hafler, D.A. Functional inflammatory profiles distinguish myelin-reactive T cells from patients with multiple sclerosis. Sci. Transl. Med. 2015, 7, 287ra74. [Google Scholar] [CrossRef]
- Kebir, H.; Kreymborg, K.; Ifergan, I.; Dodelet-Devillers, A.; Cayrol, R.; Bernard, M.; Giuliani, F.; Arbour, N.; Becher, B.; Prat, A. Human TH17 lymphocytes promote blood-brain barrier disruption and central nervous system inflammation. Nat. Med. 2007, 13, 1173–1175. [Google Scholar] [CrossRef]
- Lassmann, H.; Ransohoff, R.M. The CD4-Th1 model for multiple sclerosis: A critical [correction of crucial] re-appraisal. Trends Immunol. 2004, 25, 132–137. [Google Scholar] [CrossRef]
- Lock, C.; Hermans, G.; Pedotti, R.; Brendolan, A.; Schadt, E.; Garren, H.; Langer-Gould, A.; Strober, S.; Cannella, B.; Allard, J.; et al. Gene-microarray analysis of multiple sclerosis lesions yields new targets validated in autoimmune encephalomyelitis. Nat. Med. 2002, 8, 500–508. [Google Scholar] [CrossRef]
- Panitch, H.S.; Hirsch, R.L.; Schindler, J.; Johnson, K.P. Treatment of multiple sclerosis with gamma interferon: Exacerbations associated with activation of the immune system. Neurology 1987, 37, 1097–1102. [Google Scholar] [CrossRef]
- van Langelaar, J.; van der Vuurst de Vries, R.M.; Janssen, M.; Wierenga-Wolf, A.F.; Spilt, I.M.; Siepman, T.A.; Dankers, W.; Verjans, G.; de Vries, H.E.; Lubberts, E.; et al. T helper 17.1 cells associate with multiple sclerosis disease activity: Perspectives for early intervention. Brain J. Neurol. 2018, 141, 1334–1349. [Google Scholar] [CrossRef]
- Tzartos, J.S.; Friese, M.A.; Craner, M.J.; Palace, J.; Newcombe, J.; Esiri, M.M.; Fugger, L. Interleukin-17 production in central nervous system-infiltrating T cells and glial cells is associated with active disease in multiple sclerosis. Am. J. Pathol. 2008, 172, 146–155. [Google Scholar] [CrossRef]
- Greenfield, A.L.; Hauser, S.L. B-cell Therapy for Multiple Sclerosis: Entering an era. Ann. Neurol. 2018, 83, 13–26. [Google Scholar] [CrossRef]
- Thompson, A.J.; Banwell, B.L.; Barkhof, F.; Carroll, W.M.; Coetzee, T.; Comi, G.; Correale, J.; Fazekas, F.; Filippi, M.; Freedman, M.S.; et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. 2018, 17, 162–173. [Google Scholar] [CrossRef]
- Baecher-Allan, C.; Kaskow, B.J.; Weiner, H.L. Multiple Sclerosis: Mechanisms and Immunotherapy. Neuron 2018, 97, 742–768. [Google Scholar] [CrossRef] [PubMed]
- Comabella, M.; Montalban, X. Body fluid biomarkers in multiple sclerosis. Lancet Neurol. 2014, 13, 113–126. [Google Scholar] [CrossRef]
- El Ayoubi, N.K.; Khoury, S.J. Blood Biomarkers as Outcome Measures in Inflammatory Neurologic Diseases. Neurotherapeutics 2017, 14, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Khademi, M.; Fugger, L.; Lindhe, Ö.; Novakova, L.; Axelsson, M.; Malmeström, C.; Constantinescu, C.; Lycke, J.; Piehl, F.; et al. Inflammation-related plasma and CSF biomarkers for multiple sclerosis. Proc. Natl. Acad. Sci. USA 2020, 117, 12952–12960. [Google Scholar] [CrossRef]
- Muraro, P.A.; Martin, R.; Mancardi, G.L.; Nicholas, R.; Sormani, M.P.; Saccardi, R. Autologous haematopoietic stem cell transplantation for treatment of multiple sclerosis. Nat. Rev. Neurol. 2017, 13, 391–405. [Google Scholar] [CrossRef]
- Rush, C.A.; Atkins, H.L.; Freedman, M.S. Autologous Hematopoietic Stem Cell Transplantation in the Treatment of Multiple Sclerosis. Cold Spring Harb. Perspect. Med. 2019, 9, a029082. [Google Scholar] [CrossRef]
- Saccardi, R.; Kozak, T.; Bocelli-Tyndall, C.; Fassas, A.; Kazis, A.; Havrdova, E.; Carreras, E.; Saiz, A.; Löwenberg, B.; te Boekhorst, P.A.; et al. Autologous stem cell transplantation for progressive multiple sclerosis: Update of the European Group for Blood and Marrow Transplantation autoimmune diseases working party database. Mult. Scler. 2006, 12, 814–823. [Google Scholar] [CrossRef]
- Burt, R.K.; Balabanov, R.; Burman, J.; Sharrack, B.; Snowden, J.A.; Oliveira, M.C.; Fagius, J.; Rose, J.; Nelson, F.; Barreira, A.A.; et al. Effect of Nonmyeloablative Hematopoietic Stem Cell Transplantation vs Continued Disease-Modifying Therapy on Disease Progression in Patients with Relapsing-Remitting Multiple Sclerosis: A Randomized Clinical Trial. JAMA 2019, 321, 165–174. [Google Scholar] [CrossRef]
- Khalil, M.; Teunissen, C.E.; Otto, M.; Piehl, F.; Sormani, M.P.; Gattringer, T.; Barro, C.; Kappos, L.; Comabella, M.; Fazekas, F.; et al. Neurofilaments as biomarkers in neurological disorders. Nat. Rev. Neurol. 2018, 14, 577–589. [Google Scholar] [CrossRef]
- Benkert, P.; Meier, S.; Schaedelin, S.; Manouchehrinia, A.; Yaldizli, Ö.; Maceski, A.; Oechtering, J.; Achtnichts, L.; Conen, D.; Derfuss, T.; et al. Serum neurofilament light chain for individual prognostication of disease activity in people with multiple sclerosis: A retrospective modelling and validation study. Lancet Neurol. 2022, 21, 246–257. [Google Scholar] [CrossRef]
- Norgren, N.; Sundström, P.; Svenningsson, A.; Rosengren, L.; Stigbrand, T.; Gunnarsson, M. Neurofilament and glial fibrillary acidic protein in multiple sclerosis. Neurology 2004, 63, 1586–1590. [Google Scholar] [CrossRef]
- Petzold, A.; Eikelenboom, M.J.; Gveric, D.; Keir, G.; Chapman, M.; Lazeron, R.H.; Cuzner, M.L.; Polman, C.H.; Uitdehaag, B.M.; Thompson, E.J.; et al. Markers for different glial cell responses in multiple sclerosis: Clinical and pathological correlations. Brain 2002, 125 Pt 7, 1462–1473. [Google Scholar] [CrossRef]
- Axelsson, M.; Malmeström, C.; Nilsson, S.; Haghighi, S.; Rosengren, L.; Lycke, J. Glial fibrillary acidic protein: A potential biomarker for progression in multiple sclerosis. J. Neurol. 2011, 258, 882–888. [Google Scholar] [CrossRef]
- Martínez, M.A.; Olsson, B.; Bau, L.; Matas, E.; Cobo Calvo, Á.; Andreasson, U.; Blennow, K.; Romero-Pinel, L.; Martínez-Yélamos, S.; Zetterberg, H. Glial and neuronal markers in cerebrospinal fluid predict progression in multiple sclerosis. Mult. Scler. 2015, 21, 550–561. [Google Scholar] [CrossRef]
- Abdelhak, A.; Huss, A.; Kassubek, J.; Tumani, H.; Otto, M. Serum GFAP as a biomarker for disease severity in multiple sclerosis. Sci. Rep. 2018, 8, 14798. [Google Scholar] [CrossRef]
- Högel, H.; Rissanen, E.; Barro, C.; Matilainen, M.; Nylund, M.; Kuhle, J.; Airas, L. Serum glial fibrillary acidic protein correlates with multiple sclerosis disease severity. Mult. Scler. 2020, 26, 210–219. [Google Scholar] [CrossRef]
- Bonneh-Barkay, D.; Wang, G.; Starkey, A.; Hamilton, R.L.; Wiley, C.A. In vivo CHI3L1 (YKL-40) expression in astrocytes in acute and chronic neurological diseases. J. Neuroinflamm. 2010, 7, 34. [Google Scholar] [CrossRef]
- Hinsinger, G.; Galéotti, N.; Nabholz, N.; Urbach, S.; Rigau, V.; Demattei, C.; Lehmann, S.; Camu, W.; Labauge, P.; Castelnovo, G.; et al. Chitinase 3-like proteins as diagnostic and prognostic biomarkers of multiple sclerosis. Mult. Scler. 2015, 21, 1251–1261. [Google Scholar] [CrossRef]
- Paul, A.; Comabella, M.; Gandhi, R. Biomarkers in Multiple Sclerosis. Cold Spring Harb. Perspect. Med. 2019, 9, a029058. [Google Scholar] [CrossRef]
- Ziemssen, T.; Akgün, K.; Brück, W. Molecular biomarkers in multiple sclerosis. J. Neuroinflamm. 2019, 16, 272. [Google Scholar] [CrossRef] [Green Version]
- Cantó, E.; Tintoré, M.; Villar, L.M.; Costa, C.; Nurtdinov, R.; Álvarez-Cermeño, J.C.; Arrambide, G.; Reverter, F.; Deisenhammer, F.; Hegen, H.; et al. Chitinase 3-like 1: Prognostic biomarker in clinically isolated syndromes. Brain 2015, 138 Pt 4, 918–931. [Google Scholar] [CrossRef] [PubMed]
- Comabella, M.; Fernández, M.; Martin, R.; Rivera-Vallvé, S.; Borrás, E.; Chiva, C.; Julià, E.; Rovira, A.; Cantó, E.; Alvarez-Cermeño, J.C.; et al. Cerebrospinal fluid chitinase 3-like 1 levels are associated with conversion to multiple sclerosis. Brain 2010, 133 Pt 4, 1082–1093. [Google Scholar] [CrossRef] [PubMed]
- Cantó, E.; Reverter, F.; Morcillo-Suárez, C.; Matesanz, F.; Fernández, O.; Izquierdo, G.; Vandenbroeck, K.; Rodríguez-Antigüedad, A.; Urcelay, E.; Arroyo, R.; et al. Chitinase 3-like 1 plasma levels are increased in patients with progressive forms of multiple sclerosis. Mult. Scler. 2012, 18, 983–990. [Google Scholar] [CrossRef] [PubMed]
- Modvig, S.; Degn, M.; Roed, H.; Sørensen, T.L.; Larsson, H.B.; Langkilde, A.R.; Frederiksen, J.L.; Sellebjerg, F. Cerebrospinal fluid levels of chitinase 3-like 1 and neurofilament light chain predict multiple sclerosis development and disability after optic neuritis. Mult. Scler. 2015, 21, 1761–1770. [Google Scholar] [CrossRef]
- Ransohoff, R.M.; Kivisäkk, P.; Kidd, G. Three or more routes for leukocyte migration into the central nervous system. Nat. Rev. Immunol. 2003, 3, 569–581. [Google Scholar] [CrossRef]
- Sellebjerg, F.; Börnsen, L.; Khademi, M.; Krakauer, M.; Olsson, T.; Frederiksen, J.L.; Sørensen, P.S. Increased cerebrospinal fluid concentrations of the chemokine CXCL13 in active MS. Neurology 2009, 73, 2003–2010. [Google Scholar] [CrossRef]
- Khademi, M.; Kockum, I.; Andersson, M.L.; Iacobaeus, E.; Brundin, L.; Sellebjerg, F.; Hillert, J.; Piehl, F.; Olsson, T. Cerebrospinal fluid CXCL13 in multiple sclerosis: A suggestive prognostic marker for the disease course. Mult. Scler. 2011, 17, 335–343. [Google Scholar] [CrossRef]
- Ragheb, S.; Li, Y.; Simon, K.; VanHaerents, S.; Galimberti, D.; De Riz, M.; Fenoglio, C.; Scarpini, E.; Lisak, R. Multiple sclerosis: BAFF and CXCL13 in cerebrospinal fluid. Mult. Scler. 2011, 17, 819–829. [Google Scholar] [CrossRef]
- Magliozzi, R.; Scalfari, A.; Pisani, A.I.; Ziccardi, S.; Marastoni, D.; Pizzini, F.B.; Bajrami, A.; Tamanti, A.; Guandalini, M.; Bonomi, S.; et al. The CSF Profile Linked to Cortical Damage Predicts Multiple Sclerosis Activity. Ann. Neurol. 2020, 88, 562–573. [Google Scholar] [CrossRef]
- Brettschneider, J.; Czerwoniak, A.; Senel, M.; Fang, L.; Kassubek, J.; Pinkhardt, E.; Lauda, F.; Kapfer, T.; Jesse, S.; Lehmensiek, V.; et al. The chemokine CXCL13 is a prognostic marker in clinically isolated syndrome (CIS). PLoS ONE 2010, 5, e11986. [Google Scholar] [CrossRef]
- Festa, E.D.; Hankiewicz, K.; Kim, S.; Skurnick, J.; Wolansky, L.J.; Cook, S.D.; Cadavid, D. Serum levels of CXCL13 are elevated in active multiple sclerosis. Mult. Scler. 2009, 15, 1271–1279. [Google Scholar] [CrossRef]
- Cheng, W.; Chen, G. Chemokines and chemokine receptors in multiple sclerosis. Mediat. Inflamm. 2014, 2014, 659206. [Google Scholar] [CrossRef]
- Sørensen, T.L.; Trebst, C.; Kivisäkk, P.; Klaege, K.L.; Majmudar, A.; Ravid, R.; Lassmann, H.; Olsen, D.B.; Strieter, R.M.; Ransohoff, R.M.; et al. Multiple sclerosis: A study of CXCL10 and CXCR3 co-localization in the inflamed central nervous system. J. Neuroimmunol. 2002, 127, 59–68. [Google Scholar] [CrossRef]
- Bonecchi, R.; Bianchi, G.; Bordignon, P.P.; D’Ambrosio, D.; Lang, R.; Borsatti, A.; Sozzani, S.; Allavena, P.; Gray, P.A.; Mantovani, A.; et al. Differential expression of chemokine receptors and chemotactic responsiveness of type 1 T helper cells (Th1s) and Th2s. J. Exp. Med. 1998, 187, 129–134. [Google Scholar] [CrossRef]
- Vazirinejad, R.; Ahmadi, Z.; Kazemi Arababadi, M.; Hassanshahi, G.; Kennedy, D. The biological functions, structure and sources of CXCL10 and its outstanding part in the pathophysiology of multiple sclerosis. Neuroimmunomodulation 2014, 21, 322–330. [Google Scholar] [CrossRef]
- Rotondi, M.; Chiovato, L.; Romagnani, S.; Serio, M.; Romagnani, P. Role of chemokines in endocrine autoimmune diseases. Endocr. Rev. 2007, 28, 492–520. [Google Scholar] [CrossRef]
- Ruder, J.; Rex, J.; Obahor, S.; Docampo, M.J.; Müller, A.M.S.; Schanz, U.; Jelcic, I.; Martin, R. NK Cells and Innate-Like T Cells After Autologous Hematopoietic Stem Cell Transplantation in Multiple Sclerosis. Front. Immunol. 2021, 12, 794077. [Google Scholar] [CrossRef]
- Gangur, V.; Simons, F.E.; Hayglass, K.T. Human IP-10 selectively promotes dominance of polyclonally activated and environmental antigen-driven IFN-gamma over IL-4 responses. FASEB J. 1998, 12, 705–713. [Google Scholar] [CrossRef]
- Mehra, V.; Rhone, E.; Widya, S.; Zuckerman, M.; Potter, V.; Raj, K.; Kulasekararaj, A.; McLornan, D.; de Lavallade, H.; Benson-Quarm, N.; et al. Epstein-Barr Virus and Monoclonal Gammopathy of Clinical Significance in Autologous Stem Cell Transplantation for Multiple Sclerosis. Clin. Infect. Dis. 2019, 69, 1757–1763. [Google Scholar] [CrossRef]
- Momtazmanesh, S.; Shobeiri, P.; Saghazadeh, A.; Teunissen, C.E.; Burman, J.; Szalardy, L.; Klivenyi, P.; Bartos, A.; Fernandes, A.; Rezaei, N. Neuronal and glial CSF biomarkers in multiple sclerosis: A systematic review and meta-analysis. Rev. Neurosci. 2021, 32, 573–595. [Google Scholar] [CrossRef]
- Larsson, D.; Åkerfeldt, T.; Carlson, K.; Burman, J. Intrathecal immunoglobulins and neurofilament light after autologous haematopoietic stem cell transplantation for multiple sclerosis. Mult. Scler. 2020, 26, 1351–1359. [Google Scholar] [CrossRef]
- Mariottini, A.; Marchi, L.; Innocenti, C.; Di Cristinzi, M.; Pasca, M.; Filippini, S.; Barilaro, A.; Mechi, C.; Fani, A.; Mazzanti, B.; et al. Intermediate-Intensity Autologous Hematopoietic Stem Cell Transplantation Reduces Serum Neurofilament Light Chains and Brain Atrophy in Aggressive Multiple Sclerosis. Front. Neurol. 2022, 13, 820256. [Google Scholar] [CrossRef]
- Thebault, S.; Daniel, R.T.; Lee, H.; Bowman, M.; Bar-Or, A.; Arnold, D.L.; Tabard-Cossa, V.; Freedman, M.S. High serum neurofilament light chain normalizes after hematopoietic stem cell transplantation for MS. Neurol. Neuroimmunol. Neuroinflamm. 2019, 6, e598. [Google Scholar] [CrossRef]
- Petzold, A.; Mondria, T.; Kuhle, J.; Rocca, M.A.; Cornelissen, J.; te Boekhorst, P.; Lowenberg, B.; Giovannoni, G.; Filippi, M.; Kappos, L.; et al. Evidence for acute neurotoxicity after chemotherapy. Ann. Neurol. 2010, 68, 806–815. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Nakamura, K.; Narayanan, S.; Brown, R.; Chen, J.; Atkins, H.L.; Freedman, M.S.; Arnold, D.L. Impact of immunoablation and autologous hematopoietic stem cell transplantation on gray and white matter atrophy in multiple sclerosis. Mult. Scler. J. 2018, 24, 1055–1066. [Google Scholar] [CrossRef] [PubMed]
- Rocca, M.A.; Mondria, T.; Valsasina, P.; Sormani, M.P.; Flach, Z.H.; Te Boekhorst, P.A.; Comi, G.; Hintzen, R.Q.; Filippi, M. A three-year study of brain atrophy after autologous hematopoietic stem cell transplantation in rapidly evolving secondary progressive multiple sclerosis. Am. J. Neuroradiol. 2007, 28, 1659–1661. [Google Scholar] [CrossRef] [PubMed]
- Mariottini, A.; Filippini, S.; Innocenti, C.; Forci, B.; Mechi, C.; Barilaro, A.; Fani, A.; Carlucci, G.; Saccardi, R.; Massacesi, L.; et al. Impact of autologous haematopoietic stem cell transplantation on disability and brain atrophy in secondary progressive multiple sclerosis. Mult. Scler. 2021, 27, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Ruder Josefine, F.S. Immune Reconstitution after Autologous Hematopoietic Stem Cell Transplantation in Multiple Sclerosis. Ph.D. Thesis, Universität Zürich, Zürich, Switzerland, 2022. [Google Scholar]
- Wiberg, A.; Olsson-Strömberg, U.; Herman, S.; Kultima, K.; Burman, J. Profound but Transient Changes in the Inflammatory Milieu of the Blood during Autologous Hematopoietic Stem Cell Transplantation. Biol. Blood Marrow Transplant. 2020, 26, 50–57. [Google Scholar] [CrossRef]
- Weseslindtner, L.; Nachbagauer, R.; Kundi, M.; Jaksch, P.; Kerschner, H.; Simon, B.; Hatos-Agyi, L.; Scheed, A.; Aberle, J.H.; Klepetko, W.; et al. Human cytomegalovirus infection in lung transplant recipients triggers a CXCL-10 response. Am. J. Transplant 2011, 11, 542–552. [Google Scholar] [CrossRef]
- Uhlin, M.; Mattsson, J.; Maeurer, M. Update on viral infections in lung transplantation. Curr. Opin. Pulm. Med. 2012, 18, 264–270. [Google Scholar] [CrossRef]
- Noronha, B.P.; Mambrini, J.V.M.; Torres, K.C.L.; Martins-Filho, O.A.; Teixeira-Carvalho, A.; Lima-Costa, M.F.; Peixoto, S.V. Cytomegalovirus and herpes simplex type 1 infections and immunological profile of community-dwelling older adults. Exp. Gerontol. 2021, 149, 111337. [Google Scholar] [CrossRef]
- Miyazaki, D.; Uotani, R.; Inoue, M.; Haruki, T.; Shimizu, Y.; Yakura, K.; Yamagami, S.; Suzutani, T.; Hosogai, M.; Isomura, H.; et al. Corneal endothelial cells activate innate and acquired arm of anti-viral responses after cytomegalovirus infection. Exp. Eye Res. 2017, 161, 143–152. [Google Scholar] [CrossRef]
- van de Groep, K.; Nierkens, S.; Cremer, O.L.; Peelen, L.M.; Klein Klouwenberg, P.M.C.; Schultz, M.J.; Hack, C.E.; van der Poll, T.; Bonten, M.J.M.; Ong, D.S.Y. Effect of cytomegalovirus reactivation on the time course of systemic host response biomarkers in previously immunocompetent critically ill patients with sepsis: A matched cohort study. Crit. Care 2018, 22, 348. [Google Scholar] [CrossRef]
- Scott, G.M.; Chow, S.S.; Craig, M.E.; Pang, C.N.; Hall, B.; Wilkins, M.R.; Jones, C.A.; Lloyd, A.R.; Rawlinson, W.D. Cytomegalovirus infection during pregnancy with maternofetal transmission induces a proinflammatory cytokine bias in placenta and amniotic fluid. J. Infect. Dis. 2012, 205, 1305–1310. [Google Scholar] [CrossRef]
- Asadzadeh, R.; Ahmadpoor, P.; Nafar, M.; Samavat, S.; Nikoueinejad, H.; Hosseinzadeh, M.; Mamizadeh, N.; Hatami, S.; Masoumi, E.; Amirzargar, A. Association of IL-15 and IP-10 Serum Levels with Cytomegalovirus Infection, CMV Viral Load and Cyclosporine Level after Kidney Transplantation. Rep. Biochem. Mol. Biol. 2021, 10, 216–223. [Google Scholar] [CrossRef]
- Ho, J.; Schaub, S.; Wiebe, C.; Gao, A.; Wehmeierm, C.; Koller, M.T.; Hirsch, H.H.; Hopfer, H.; Nickerson, P.; Hirt-Minkowski, P. Urinary CXCL10 Chemokine Is Associated With Alloimmune and Virus Compartment-Specific Renal Allograft Inflammation. Transplantation 2018, 102, 521–529. [Google Scholar] [CrossRef]
- Reusser, P.; Riddell, S.R.; Meyers, J.D.; Greenberg, P.D. Cytotoxic T-lymphocyte response to cytomegalovirus after human allogeneic bone marrow transplantation: Pattern of recovery and correlation with cytomegalovirus infection and disease. Blood 1991, 78, 1373–1380. [Google Scholar] [CrossRef]
- Mackus, W.J.; Frakking, F.N.; Grummels, A.; Gamadia, L.E.; De Bree, G.J.; Hamann, D.; Van Lier, R.A.; Van Oers, M.H. Expansion of CMV-specific CD8+CD45RA+CD27- T cells in B-cell chronic lymphocytic leukemia. Blood 2003, 102, 1057–1063. [Google Scholar] [CrossRef]
- Harari, A.; Zimmerli, S.C.; Pantaleo, G. Cytomegalovirus (CMV)-specific cellular immune responses. Hum. Immunol. 2004, 65, 500–506. [Google Scholar] [CrossRef]
- Cheeran, M.C.; Hu, S.; Sheng, W.S.; Peterson, P.K.; Lokensgard, J.R. CXCL10 production from cytomegalovirus-stimulated microglia is regulated by both human and viral interleukin-10. J. Virol. 2003, 77, 4502–4515. [Google Scholar] [CrossRef] [Green Version]
- Van de Berg, P.J.; Yong, S.L.; Remmerswaal, E.B.; van Lier, R.A.; ten Berge, I.J. Cytomegalovirus-induced effector T cells cause endothelial cell damage. Clin. Vaccine Immunol. 2012, 19, 772–779. [Google Scholar] [CrossRef]
- Sundqvist, E.; Bergström, T.; Daialhosein, H.; Nyström, M.; Sundström, P.; Hillert, J.; Alfredsson, L.; Kockum, I.; Olsson, T. Cytomegalovirus seropositivity is negatively associated with multiple sclerosis. Mult. Scler. 2014, 20, 165–173. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Age | Sex | Diagnosis | Last Therapy before aHSCT | EDSS at aHSCT | CMV Viremia | EBV Viremia | |
|---|---|---|---|---|---|---|---|
| aHSCT_MS_01 | 38 | female | RRMS | Natalizumab | 4 | ||
| aHSCT_MS_02 | 36 | male | RRMS | Fingolimod | 5.5 | ||
| aHSCT_MS_03 | 53 | female | PPMS | Fingolimod | 6.5 | ||
| aHSCT_MS_04 | 32 | female | RRMS | Rituximab | 4 | ||
| aHSCT_MS_05 | 47 | male | PPMS | Rituximab | 6.5 | ||
| aHSCT_MS_06 | 49 | female | PPMS | Rituximab | 4 | ||
| aHSCT_MS_08 | 30 | male | PPMS | Ocrelizumab | 4.5 | ||
| aHSCT_MS_09 | 48 | female | SPMS | Rituximab | 6 | ||
| aHSCT_MS_10 | 36 | female | SPMS | Ocrelizumab | 4.5 | ||
| aHSCT_MS_11 | 47 | male | RRMS | Ocrelizumab | 3 | ||
| aHSCT_MS_12 | 45 | male | SPMS | Rituximab | 6.5 | ||
| aHSCT_MS_13 | 41 | male | PPMS | Ocrelizumab | 3 | ||
| aHSCT_MS_15 | 54 | female | SPMS | Rituximab | 5.5 | ||
| aHSCT_MS_16 | 33 | female | RRMS | Ocrelizumab | 4.5 | ||
| aHSCT_MS_19 | 43 | male | PPMS | Rituximab | 6 | ||
| aHSCT_MS_20 | 47 | male | PPMS | Ocrelizumab | 6 | ||
| aHSCT_MS_22 | 54 | female | SPMS | Rituximab | 4.5 | ||
| aHSCT_MS_24 | 44 | female | SPMS | Rituximab | 4 | ||
| aHSCT_MS_25 | 44 | male | SPMS | Ocrelizumab | 3.5 | ||
| aHSCT_MS_21 | 25 | male | RRMS | Ocrelizumab | 2 | x | |
| aHSCT_MS_23 | 43 | male | RRMS | Natalizumab | 3 | x | |
| aHSCT_MS_26 | 40 | female | RRMS | Natalizumab | 3.5 | x | |
| aHSCT_MS_28 | 32 | female | RRMS | Ocrelizumab | 2 | x | |
| aHSCT_MS_36 | 39 | female | RRMS | Ocrelizumab | 3 | x | |
| aHSCT_MS_07 | 33 | female | SPMS | Rituximab | 5 | x | |
| aHSCT_MS_14 | 25 | male | PPMS | Rituximab | 6.5 | x | |
| aHSCT_MS_27 | 51 | male | SPMS | Ocrelizumab | 4.5 | x | |
| aHSCT_MS_31 | 40 | female | RRMS | Ocrelizumab | 3.5 | x | |
| aHSCT_MS_17 | 39 | female | RRMS | Ocrelizumab | 2.5 | x | x |
| aHSCT_MS_18 | 47 | male | RRMS | Natalizumab | 4 | x | x |
| aHSCT_MS_32 | 35 | male | RRMS | Ocrelizumab | 3 | x | x |
| aHSCT_MS_34 | 29 | female | RRMS | Ocrelizumab | 2 | x | x |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruder, J.; Dinner, G.; Maceski, A.; Berenjeno-Correa, E.; Müller, A.M.; Jelcic, I.; Kuhle, J.; Martin, R. Dynamics of Inflammatory and Neurodegenerative Biomarkers after Autologous Hematopoietic Stem Cell Transplantation in Multiple Sclerosis. Int. J. Mol. Sci. 2022, 23, 10946. https://doi.org/10.3390/ijms231810946
Ruder J, Dinner G, Maceski A, Berenjeno-Correa E, Müller AM, Jelcic I, Kuhle J, Martin R. Dynamics of Inflammatory and Neurodegenerative Biomarkers after Autologous Hematopoietic Stem Cell Transplantation in Multiple Sclerosis. International Journal of Molecular Sciences. 2022; 23(18):10946. https://doi.org/10.3390/ijms231810946
Chicago/Turabian StyleRuder, Josefine, Gianna Dinner, Aleksandra Maceski, Ernesto Berenjeno-Correa, Antonia Maria Müller, Ilijas Jelcic, Jens Kuhle, and Roland Martin. 2022. "Dynamics of Inflammatory and Neurodegenerative Biomarkers after Autologous Hematopoietic Stem Cell Transplantation in Multiple Sclerosis" International Journal of Molecular Sciences 23, no. 18: 10946. https://doi.org/10.3390/ijms231810946
APA StyleRuder, J., Dinner, G., Maceski, A., Berenjeno-Correa, E., Müller, A. M., Jelcic, I., Kuhle, J., & Martin, R. (2022). Dynamics of Inflammatory and Neurodegenerative Biomarkers after Autologous Hematopoietic Stem Cell Transplantation in Multiple Sclerosis. International Journal of Molecular Sciences, 23(18), 10946. https://doi.org/10.3390/ijms231810946