
The Yin/Yang Balance of Communication between Sensory Neurons and Macrophages in Traumatic Peripheral Neuropathic Pain


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction: Neuropathic Pain Associated with Traumatic Peripheral Nerve Injuries
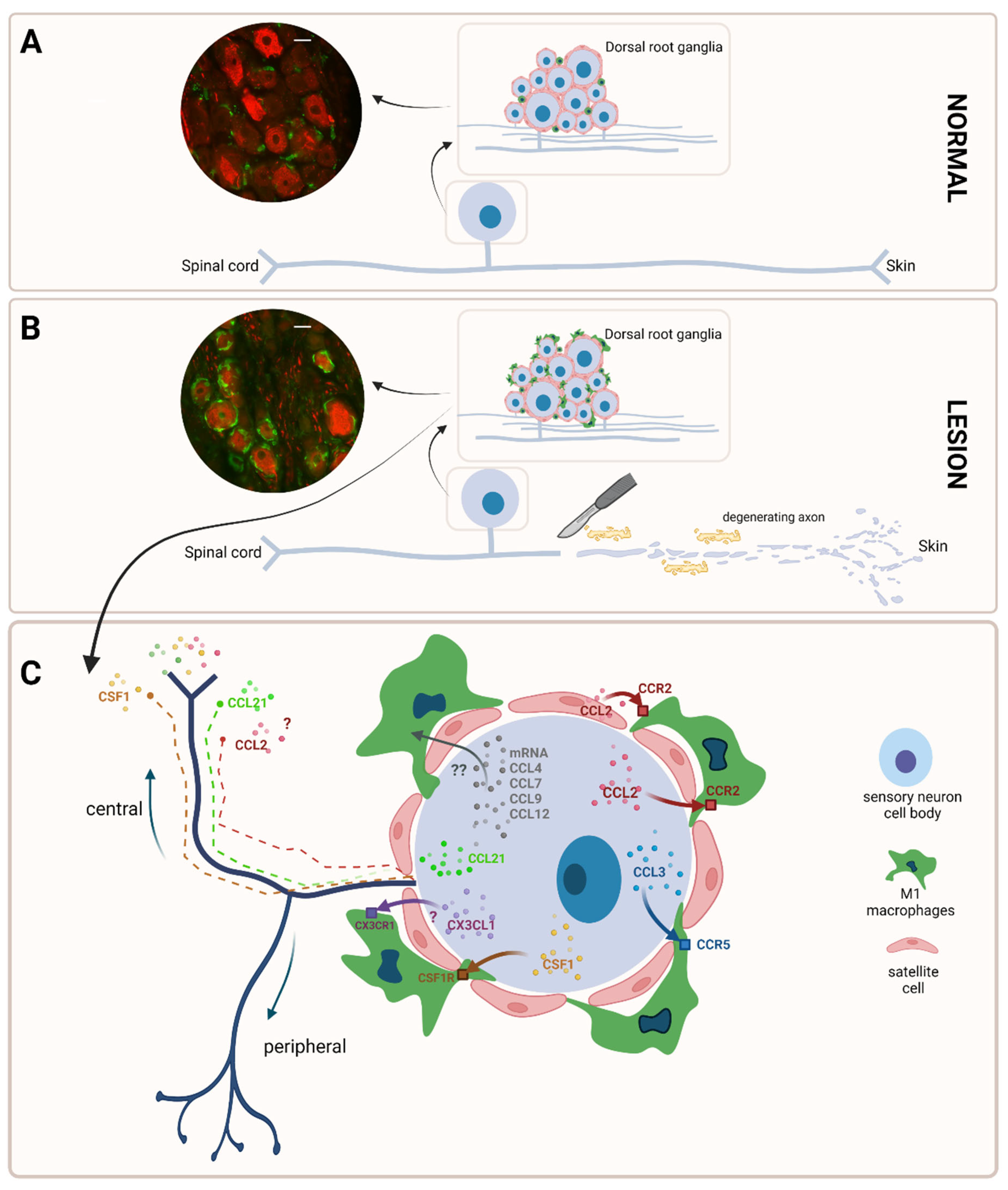
2. Macrophages in the Peripheral Nervous System: A Distinct Population
3. Macrophages Are Quickly Activated after a Traumatic Peripheral Nerve Lesion
4. There Is an Intense Cross-Talk between Macrophages and Sensory Neurons Facilitated by Pro-Inflammatory Mediators, in Which Ion Channels Are Important Players
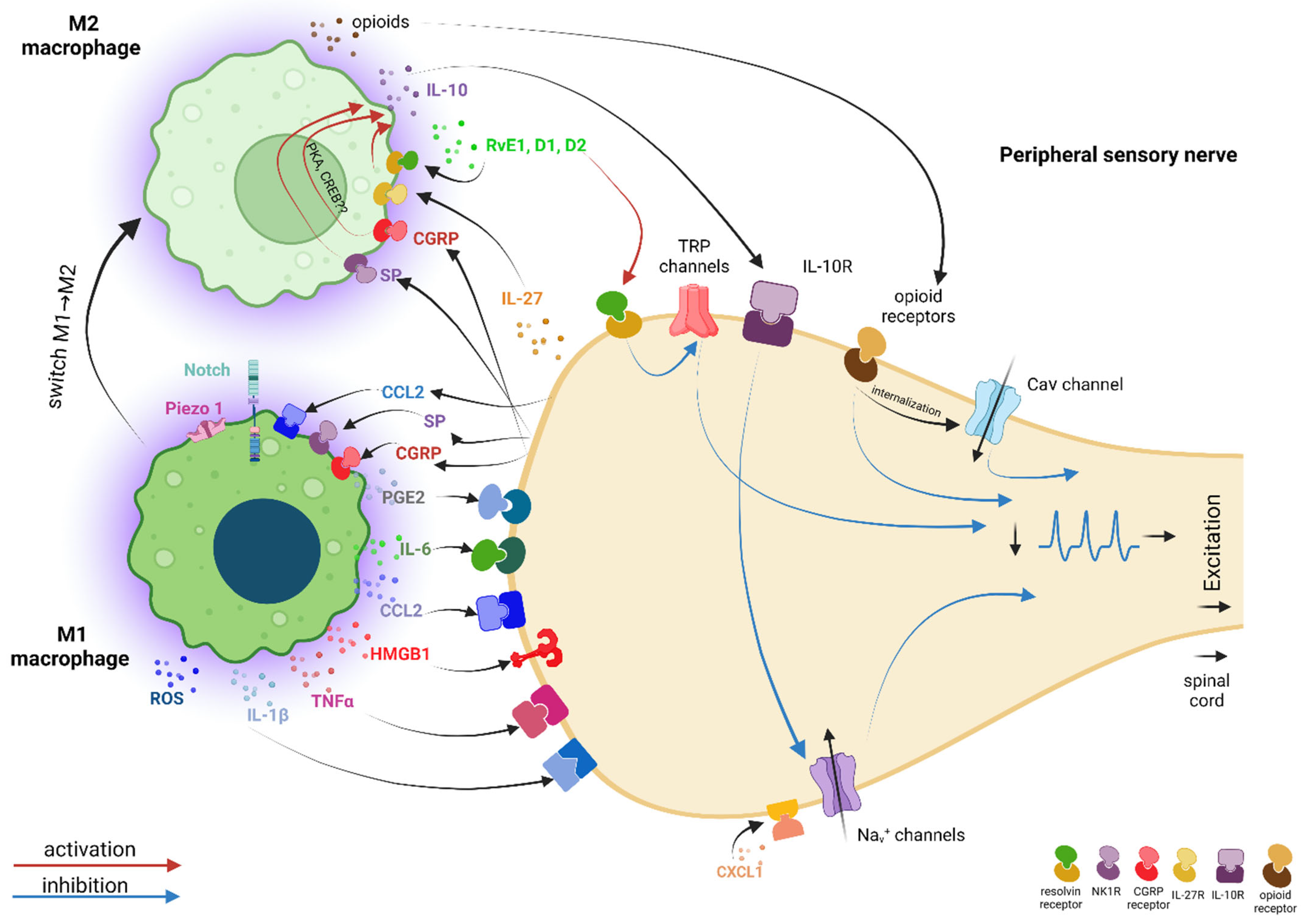
5. In Parallel with the “Pro-nociceptive” Dialogue between Primary Sensory Neurons and Macrophages, There Is Also an “Anti-nociceptive” Dialogue with M2 Macrophages That Helps to Reduce Pain
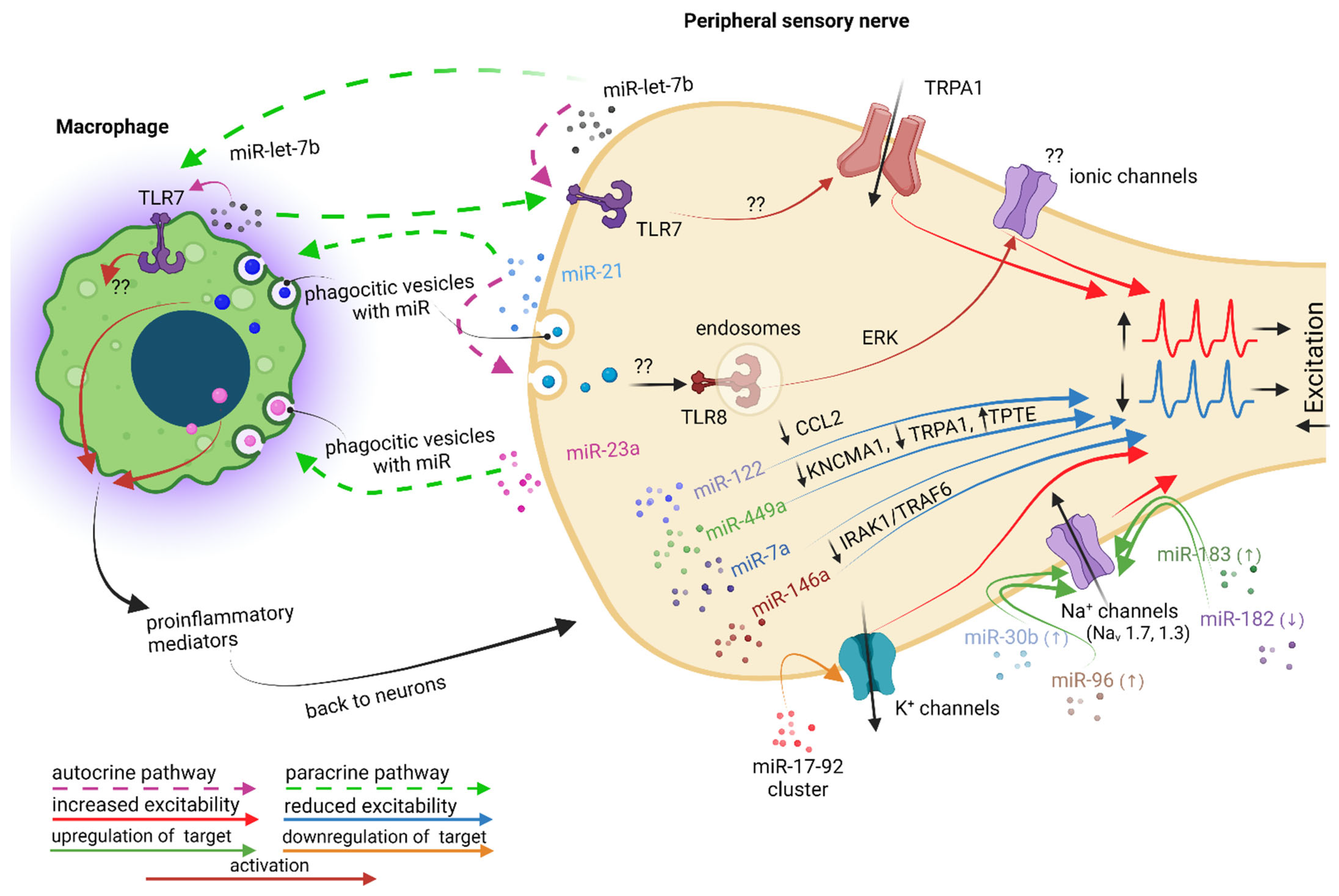
6. MicroRNAs May Also Facilitate Pro- and Anti-nociceptive Effects on Primary Sensory Neurons, with Macrophage Involvement More or Less Well Documented
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AAV | adeno-associated virus |
| Atf3 | activating transcription factor 3 |
| BDNF | brain-derived neurotrophic factor |
| BMDMs | bone marrow-derived macrophages |
| Cav3.2 T-type | voltage-gated calcium channels isoform 3.2 |
| CCD | chronic compression of the DRG |
| CCI | chronic constriction injury |
| CCL12 | chemokine (C-C motif) ligand 12 |
| CCL2/MCP1 | chemokine (C-C motif) ligand 2/monocyte chemoattractant protein-1 |
| CCL21 | chemokine (C-C motif) ligand 21 |
| CCL3 | chemokine (C-C motif) ligand 3 |
| CCL4 | chemokine (C-C motif) ligand 4 |
| CCL6 | chemokine (C-C motif) ligand 6 |
| CCL7 | chemokine (C-C motif) ligand 7 |
| CCL8 | chemokine (C-C motif) ligand 8 |
| CCL9 | chemokine (C-C motif) ligand 9 |
| CCR2 | C-C chemokine receptor type 2 |
| CD11b | cluster of differentiation molecule 11B |
| CD64 | cluster of differentiation 64 |
| CGRP | calcitonin gene-related peptide |
| CNS | central nervous system |
| CREB | cyclic adenosine monophosphate (cAMP) response element-binding protein |
| CSF1/M-CSF | colony-stimulating factor 1/macrophage colony-stimulating factor |
| CSF1R | colony-stimulating factor 1 receptor |
| CX3CL1 | chemokine (C-X3-C motif) ligand 1 |
| CX3CR1 | C-X3-C motif chemokine receptor 1 |
| CXCR2 | C-X-C chemokine receptor 2 |
| DRG | dorsal root ganglia |
| Egr-1 | early growth response factor 1 |
| EP1 | E-prostanoid 1 |
| EP3 | E-prostanoid 3 |
| EP4 | E-prostanoid 4 |
| ERK | extracellular signal-regulated protein kinase |
| Fos | fos proto-oncogene, AP-1 transcription factor subunit |
| GFP | green fluorescent protein |
| GM-CSF | granulocyte-macrophage colony-stimulating factor |
| GPCR | G-protein-coupled receptors |
| HMGB1 | high-mobility group box 1 |
| IASP | International Association for the Study of Pain |
| IB4 | isolectin B4 |
| Iba1 | ionized calcium-binding adapter molecule 1 |
| IFNγ | interferon gamma |
| IL-10 | interleukin 10 |
| IL-1α | interleukin 1 α |
| IL-1β | interleukin 1 β |
| IL-27 | interleukin 27 |
| IL-4 | interleukin 4 |
| IL-6 | interleukin 6 |
| iNOS | inducible nitric oxide synthase |
| IRAK | IL-1R-associated kinase |
| JAK-STAT-Janus-kinase (JAK) | signal transducers and activators of transcription (STAT) |
| JNK | c-Jun N-terminal kinases |
| Jun | Jun proto-oncogene, AP-1 transcription factor subunit |
| KCNMA1 | calcium-activated K+ channel subunit alpha-1 |
| KV1.4 | voltage-gated K+ channels isoform 1.4 |
| KV3.4 | voltage-gated K+ channels isoform 3.4 |
| KV4.3 | voltage-gated K+ channels isoform 4.3 |
| L4/L5/L6 | lumbar 4/5/6 |
| LPS | lipopolysaccharide |
| Ly6C | lymphocyte antigen 6 complex, locus C |
| Ly6G | lymphocyte antigen 6 complex locus G6 |
| MAFIA | macrophage Fas-induced apoptosis |
| Mgl1 | macrophage galactose-type lectin 1 |
| MHCII | major histocompatibility complex class II molecules |
| MIP-2 | macrophage inflammatory protein-2 |
| miRNAs or miRs | microRNAs |
| MMP-9 | matrix metalloproteinase-9 |
| mRNA | messenger RNA |
| NaV1.1 | voltage-gated Na+ channels isoform 1.1 |
| NaV1.3 | voltage-gated Na+ channels isoform 1.3 |
| NaV1.7 | voltage-gated Na+ channels isoform 1.7 |
| NaV1.8 | voltage-gated Na+ channels isoform 1.8 |
| NF-κB | nuclear factor κB |
| NMDA | N-methyl-d-aspartate |
| NOX1 | nicotinamide adenine dinucleotide phosphate (NADPH) oxidase isoform 1 |
| NOX2 | nicotinamide adenine dinucleotide phosphate (NADPH) oxidase isoform 2 |
| p38 MAPK | p38 mitogen-activated protein kinase |
| p-ERK | phospho-ERK |
| PGE2 | prostaglandin E2 |
| PKA | cyclic adenosine monophosphate (cAMP)-dependent protein kinase |
| PKC | protein kinase C |
| PNS | peripheral nervous system |
| PSNL | partial sciatic nerve ligation |
| RAGE | receptor for advanced glycation end-products |
| Relmα | resistin-like molecule alpha |
| RFP | red fluorescent protein |
| RNA | ribonucleic acid |
| RNAseq | RNA sequencing |
| ROS | reactive oxygen species |
| Rv E1, D1, D2 | resolvins E1, D1, and D2 |
| sIL-6R | soluble IL-6 receptor |
| SNI | spared nerve injury |
| SNL | spinal nerve ligation |
| SNT | sciatic nerve transection |
| SP | substance P |
| TLR2, 4, 7, 8 | toll-like receptor isoform 2, 4, 7, or 8 |
| TNF-α | tumor necrosis factor alpha |
| TPTE | transmembrane phosphatase with tension homology |
| TRAF6 | tumor necrosis factor receptor (TNFR)-associated factor 6 |
| Trem2 | triggering receptor expressed on myeloid cells 2 |
| TRP | transient receptor potential |
| TRPA1 | transient receptor potential ankyrin 1 |
| TRPV1 | transient receptor potential vanilloid type-1 |
| TTX | tetrodotoxin |
| WD | Wallerian degeneration |
| WSX-1 | receptor for IL-27 |
References
- Kouyoumdjian, J.A.; Graca, C.R.; Ferreira, V.F.M. Peripheral nerve injuries: A retrospective survey of 1124 cases. Neurol. India 2017, 65, 551–555. [Google Scholar] [CrossRef]
- Seddon, H. Three types of nerve injury. Brain J. Neurol. 1943, 66, 237–288. [Google Scholar] [CrossRef]
- Sunderland, S. A classification of peripheral nerve injuries producing loss of function. Brain J. Neurol. 1951, 74, 491–516. [Google Scholar] [CrossRef]
- Hussain, G.; Wang, J.; Rasul, A.; Anwar, H.; Qasim, M.; Zafar, S.; Aziz, N.; Razzaq, A.; Hussain, R.; de Aguilar, J.G.; et al. Current Status of Therapeutic Approaches against Peripheral Nerve Injuries: A Detailed Story from Injury to Recovery. Int. J. Biol. Sci. 2020, 16, 116–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menorca, R.M.; Fussell, T.S.; Elfar, J.C. Nerve physiology: Mechanisms of injury and recovery. Hand Clin. 2013, 29, 317–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rotshenker, S. Wallerian degeneration: The innate-immune response to traumatic nerve injury. J. Neuroinflamm. 2011, 8, 109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, P.; Piao, X.; Bonaldo, P. Role of macrophages in Wallerian degeneration and axonal regeneration after peripheral nerve injury. Acta Neuropathol. 2015, 130, 605–618. [Google Scholar] [CrossRef] [PubMed]
- Treede, R.D.; Rief, W.; Barke, A.; Aziz, Q.; Bennett, M.I.; Benoliel, R.; Cohen, M.; Evers, S.; Finnerup, N.B.; First, M.B.; et al. A classification of chronic pain for ICD-11. Pain 2015, 156, 1003–1007. [Google Scholar] [CrossRef] [Green Version]
- Breivik, H.; Collett, B.; Ventafridda, V.; Cohen, R.; Gallacher, D. Survey of chronic pain in Europe: Prevalence, impact on daily life, and treatment. Eur. J. Pain 2006, 10, 287–333. [Google Scholar] [CrossRef] [PubMed]
- Yong, R.J.; Mullins, P.M.; Bhattacharyya, N. Prevalence of chronic pain among adults in the United States. Pain 2022, 163, e328–e332. [Google Scholar] [CrossRef] [PubMed]
- Chronic Pain in Australia; Australian Institute of Health and Welfare: Canberra, Australia, 2020.
- Fayaz, A.; Croft, P.; Langford, R.M.; Donaldson, L.J.; Jones, G.T. Prevalence of chronic pain in the UK: A systematic review and meta-analysis of population studies. BMJ Open 2016, 6, e010364. [Google Scholar] [CrossRef] [PubMed]
- Benn, S.C.; Woolf, C.J. Adult neuron survival strategies--slamming on the brakes. Nat. Rev. Neurosci. 2004, 5, 686–700. [Google Scholar] [CrossRef]
- Costigan, M.; Scholz, J.; Woolf, C.J. Neuropathic pain: A maladaptive response of the nervous system to damage. Annu. Rev. Neurosci. 2009, 32, 1–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- von Hehn, C.A.; Baron, R.; Woolf, C.J. Deconstructing the neuropathic pain phenotype to reveal neural mechanisms. Neuron 2012, 73, 638–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gangadharan, V.; Kuner, R. Pain hypersensitivity mechanisms at a glance. Dis. Model. Mech. 2013, 6, 889–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuner, R.; Flor, H. Structural plasticity and reorganisation in chronic pain. Nat. Rev. Neurosci. 2017, 18, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- IASP, International Association for the Study of Pain (IASP) Terminology. International Association for the Study of Pain. 2017. Available online: https://www.iasp-pain.org/terminology?navItemNumber=576 (accessed on 10 October 2022).
- Dowdall, T.; Robinson, I.; Meert, T.F. Comparison of five different rat models of peripheral nerve injury. Pharmacol. Biochem. Behav. 2005, 80, 93–108. [Google Scholar] [CrossRef]
- Sorkin, L.S.; Yaksh, T.L. Behavioral models of pain states evoked by physical injury to the peripheral nerve. Neurother. J. Am. Soc. Exp. Neurother. 2009, 6, 609–619. [Google Scholar] [CrossRef] [Green Version]
- Gangadharan, V.; Zheng, H.; Taberner, F.J.; Landry, J.; Nees, T.A.; Pistolic, J.; Agarwal, N.; Mannich, D.; Benes, V.; Helmstaedter, M.; et al. Neuropathic pain caused by miswiring and abnormal end organ targeting. Nature 2022, 606, 137–145. [Google Scholar] [CrossRef]
- Raoof, R.; Willemen, H.; Eijkelkamp, N. Divergent roles of immune cells and their mediators in pain. Rheumatology 2018, 57, 429–440. [Google Scholar] [CrossRef]
- Lim, J.S.; Kam, P.C. Neuroimmune mechanisms of pain: Basic science and potential therapeutic modulators. Anaesth. Intensive Care 2020, 48, 167–178. [Google Scholar] [CrossRef]
- Calvo, M.; Dawes, J.M.; Bennett, D.L. The role of the immune system in the generation of neuropathic pain. Lancet Neurol. 2012, 11, 629–642. [Google Scholar] [CrossRef]
- Grace, P.M.; Hutchinson, M.R.; Maier, S.F.; Watkins, L.R. Pathological pain and the neuroimmune interface. Nat. Rev. Immunol. 2014, 14, 217–231. [Google Scholar] [CrossRef] [Green Version]
- Wynn, T.A.; Chawla, A.; Pollard, J.W. Macrophage biology in development, homeostasis and disease. Nature 2013, 496, 445–455. [Google Scholar] [CrossRef] [Green Version]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef]
- Italiani, P.; Boraschi, D. From Monocytes to M1/M2 Macrophages: Phenotypical vs. Functional Differentiation. Front. Immunol. 2014, 5, 514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.L.; Yim, A.K.Y.; Kim, K.W.; Avey, D.; Czepielewski, R.S.; Colonna, M.; Milbrandt, J.; Randolph, G.J. Peripheral nerve resident macrophages share tissue-specific programming and features of activated microglia. Nat. Commun. 2020, 11, 2552. [Google Scholar] [CrossRef] [PubMed]
- Kolter, J.; Kierdorf, K.; Henneke, P. Origin and Differentiation of Nerve-Associated Macrophages. J. Immunol. 2020, 204, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Ydens, E.; Amann, L.; Asselbergh, B.; Scott, C.L.; Martens, L.; Sichien, D.; Mossad, O.; Blank, T.; De Prijck, S.; Low, D.; et al. Profiling peripheral nerve macrophages reveals two macrophage subsets with distinct localization, transcriptome and response to injury. Nat. Neurosci. 2020, 23, 676–689. [Google Scholar] [CrossRef] [PubMed]
- Pirzgalska, R.M.; Seixas, E.; Seidman, J.S.; Link, V.M.; Sanchez, N.M.; Mahu, I.; Mendes, R.; Gres, V.; Kubasova, N.; Morris, I.; et al. Sympathetic neuron-associated macrophages contribute to obesity by importing and metabolizing norepinephrine. Nat. Med. 2017, 23, 1309–1318. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.; Hore, Z.; Harley, P.; Uchenna Stanley, F.; Michrowska, A.; Dahiya, M.; La Russa, F.; Jager, S.E.; Villa-Hernandez, S.; Denk, F. A transcriptional toolbox for exploring peripheral neuroimmune interactions. Pain 2020, 161, 2089–2106. [Google Scholar] [CrossRef] [PubMed]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 2017, 47, 566–581.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavin, Y.; Winter, D.; Blecher-Gonen, R.; David, E.; Keren-Shaul, H.; Merad, M.; Jung, S.; Amit, I. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell 2014, 159, 1312–1326. [Google Scholar] [CrossRef] [Green Version]
- Gosselin, D.; Link, V.M.; Romanoski, C.E.; Fonseca, G.J.; Eichenfield, D.Z.; Spann, N.J.; Stender, J.D.; Chun, H.B.; Garner, H.; Geissmann, F.; et al. Environment drives selection and function of enhancers controlling tissue-specific macrophage identities. Cell 2014, 159, 1327–1340. [Google Scholar] [CrossRef] [PubMed]
- Ton, B.H.; Chen, Q.; Gaina, G.; Tucureanu, C.; Georgescu, A.; Strungaru, C.; Flonta, M.L.; Sah, D.; Ristoiu, V. Activation profile of dorsal root ganglia Iba-1 (+) macrophages varies with the type of lesion in rats. Acta Histochem. 2013, 115, 840–850. [Google Scholar] [CrossRef]
- Vega-Avelaira, D.; Geranton, S.M.; Fitzgerald, M. Differential regulation of immune responses and macrophage/neuron interactions in the dorsal root ganglion in young and adult rats following nerve injury. Mol. Pain 2009, 5, 70. [Google Scholar] [CrossRef] [Green Version]
- Kwon, M.J.; Kim, J.; Shin, H.; Jeong, S.R.; Kang, Y.M.; Choi, J.Y.; Hwang, D.H.; Kim, B.G. Contribution of macrophages to enhanced regenerative capacity of dorsal root ganglia sensory neurons by conditioning injury. J. Neurosci. 2013, 33, 15095–15108. [Google Scholar] [CrossRef] [Green Version]
- Kwon, M.J.; Shin, H.Y.; Cui, Y.; Kim, H.; Thi, A.H.; Choi, J.Y.; Kim, E.Y.; Hwang, D.H.; Kim, B.G. CCL2 Mediates Neuron-Macrophage Interactions to Drive Proregenerative Macrophage Activation Following Preconditioning Injury. J. Neurosci. 2015, 35, 15934–15947. [Google Scholar] [CrossRef] [Green Version]
- Ristoiu, V. Contribution of macrophages to peripheral neuropathic pain pathogenesis. Life Sci. 2013, 93, 870–881. [Google Scholar] [CrossRef]
- Yu, X.; Liu, H.; Hamel, K.A.; Morvan, M.G.; Yu, S.; Leff, J.; Guan, Z.; Braz, J.M.; Basbaum, A.I. Dorsal root ganglion macrophages contribute to both the initiation and persistence of neuropathic pain. Nat. Commun. 2020, 11, 264. [Google Scholar] [CrossRef]
- De Logu, F.; Nassini, R.; Materazzi, S.; Carvalho Goncalves, M.; Nosi, D.; Rossi Degl’Innocenti, D.; Marone, I.M.; Ferreira, J.; Li Puma, S.; Benemei, S.; et al. Schwann cell TRPA1 mediates neuroinflammation that sustains macrophage-dependent neuropathic pain in mice. Nat. Commun. 2017, 8, 1887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martini, R.; Fischer, S.; Lopez-Vales, R.; David, S. Interactions between Schwann cells and macrophages in injury and inherited demyelinating disease. Glia 2008, 56, 1566–1577. [Google Scholar] [CrossRef] [PubMed]
- Perrin, F.E.; Lacroix, S.; Aviles-Trigueros, M.; David, S. Involvement of monocyte chemoattractant protein-1, macrophage inflammatory protein-1alpha and interleukin-1beta in Wallerian degeneration. Brain J. Neurol. 2005, 128, 854–866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shubayev, V.I.; Angert, M.; Dolkas, J.; Campana, W.M.; Palenscar, K.; Myers, R.R. TNFalpha-induced MMP-9 promotes macrophage recruitment into injured peripheral nerve. Mol. Cell. Neurosci. 2006, 31, 407–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dailey, A.T.; Avellino, A.M.; Benthem, L.; Silver, J.; Kliot, M. Complement depletion reduces macrophage infiltration and activation during Wallerian degeneration and axonal regeneration. J. Neurosci. 1998, 18, 6713–6722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeon, S.M.; Lee, K.M.; Cho, H.J. Expression of monocyte chemoattractant protein-1 in rat dorsal root ganglia and spinal cord in experimental models of neuropathic pain. Brain Res. 2009, 1251, 103–111. [Google Scholar] [CrossRef]
- Bravo-Caparros, I.; Ruiz-Cantero, M.C.; Perazzoli, G.; Cronin, S.J.F.; Vela, J.M.; Hamed, M.F.; Penninger, J.M.; Baeyens, J.M.; Cobos, E.J.; Nieto, F.R. Sigma-1 receptors control neuropathic pain and macrophage infiltration into the dorsal root ganglion after peripheral nerve injury. FASEB J. 2020, 34, 5951–5966. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, T.; Minami, M.; Nakagawa, T.; Satoh, M. Enhanced production of monocyte chemoattractant protein-1 in the dorsal root ganglia in a rat model of neuropathic pain: Possible involvement in the development of neuropathic pain. Neurosci. Res. 2004, 48, 463–469. [Google Scholar] [CrossRef]
- Zhang, J.; De Koninck, Y. Spatial and temporal relationship between monocyte chemoattractant protein-1 expression and spinal glial activation following peripheral nerve injury. J. Neurochem. 2006, 97, 772–783. [Google Scholar] [CrossRef]
- Niemi, J.P.; DeFrancesco-Lisowitz, A.; Cregg, J.M.; Howarth, M.; Zigmond, R.E. Overexpression of the monocyte chemokine CCL2 in dorsal root ganglion neurons causes a conditioning-like increase in neurite outgrowth and does so via a STAT3 dependent mechanism. Exp. Neurol. 2016, 275 Pt 1, 25–37. [Google Scholar] [CrossRef]
- Okubo, M.; Yamanaka, H.; Kobayashi, K.; Dai, Y.; Kanda, H.; Yagi, H.; Noguchi, K. Macrophage-Colony Stimulating Factor Derived from Injured Primary Afferent Induces Proliferation of Spinal Microglia and Neuropathic Pain in Rats. PLoS ONE 2016, 11, e0153375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnan, A.; Bhavanam, S.; Zochodne, D. An Intimate Role for Adult Dorsal Root Ganglia Resident Cycling Cells in the Generation of Local Macrophages and Satellite Glial Cells. J. Neuropathol. Exp. Neurol. 2018, 77, 929–941. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; You, B.; Lim, H.; Lee, S.J. Toll-like receptor 2 contributes to chemokine gene expression and macrophage infiltration in the dorsal root ganglia after peripheral nerve injury. Mol. Pain 2011, 7, 74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verge, G.M.; Milligan, E.D.; Maier, S.F.; Watkins, L.R.; Naeve, G.S.; Foster, A.C. Fractalkine (CX3CL1) and fractalkine receptor (CX3CR1) distribution in spinal cord and dorsal root ganglia under basal and neuropathic pain conditions. Eur. J. Neurosci. 2004, 20, 1150–1160. [Google Scholar] [CrossRef]
- Lindborg, J.A.; Mack, M.; Zigmond, R.E. Neutrophils Are Critical for Myelin Removal in a Peripheral Nerve Injury Model of Wallerian Degeneration. J. Neurosci. 2017, 37, 10258–10277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boakye, P.A.; Tang, S.J.; Smith, P.A. Mediators of Neuropathic Pain; Focus on Spinal Microglia, CSF-1, BDNF, CCL21, TNF-alpha, Wnt Ligands, and Interleukin 1beta. Front. Pain Res. 2021, 2, 698157. [Google Scholar] [CrossRef] [PubMed]
- Cobos, E.J.; Nickerson, C.A.; Gao, F.; Chandran, V.; Bravo-Caparros, I.; Gonzalez-Cano, R.; Riva, P.; Andrews, N.A.; Latremoliere, A.; Seehus, C.R.; et al. Mechanistic Differences in Neuropathic Pain Modalities Revealed by Correlating Behavior with Global Expression Profiling. Cell Rep. 2018, 22, 1301–1312. [Google Scholar] [CrossRef] [Green Version]
- de Jong, E.K.; Vinet, J.; Stanulovic, V.S.; Meijer, M.; Wesseling, E.; Sjollema, K.; Boddeke, H.W.; Biber, K. Expression, transport, and axonal sorting of neuronal CCL21 in large dense-core vesicles. FASEB J. 2008, 22, 4136–4145. [Google Scholar] [CrossRef] [Green Version]
- de Jong, E.K.; Dijkstra, I.M.; Hensens, M.; Brouwer, N.; van Amerongen, M.; Liem, R.S.; Boddeke, H.W.; Biber, K. Vesicle-mediated transport and release of CCL21 in endangered neurons: A possible explanation for microglia activation remote from a primary lesion. J. Neurosci. 2005, 25, 7548–7557. [Google Scholar] [CrossRef] [Green Version]
- Honjoh, K.; Nakajima, H.; Hirai, T.; Watanabe, S.; Matsumine, A. Relationship of Inflammatory Cytokines From M1-Type Microglia/Macrophages at the Injured Site and Lumbar Enlargement With Neuropathic Pain After Spinal Cord Injury in the CCL21 Knockout (plt) Mouse. Front. Cell. Neurosci. 2019, 13, 525. [Google Scholar] [CrossRef]
- Jung, H.; Toth, P.T.; White, F.A.; Miller, R.J. Monocyte chemoattractant protein-1 functions as a neuromodulator in dorsal root ganglia neurons. J. Neurochem. 2008, 104, 254–263. [Google Scholar] [CrossRef] [Green Version]
- Jung, H.; Bhangoo, S.; Banisadr, G.; Freitag, C.; Ren, D.; White, F.A.; Miller, R.J. Visualization of chemokine receptor activation in transgenic mice reveals peripheral activation of CCR2 receptors in states of neuropathic pain. J. Neurosci. 2009, 29, 8051–8062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Ren, J.; Morgan, S.; Liu, Z.; Dou, C.; Liu, B. Monocyte chemoattractant protein-1 (MCP-1) regulates macrophage cytotoxicity in abdominal aortic aneurysm. PLoS ONE 2014, 9, e92053. [Google Scholar] [CrossRef]
- Sodhi, A.; Biswas, S.K. Monocyte chemoattractant protein-1-induced activation of p42/44 MAPK and c-Jun in murine peritoneal macrophages: A potential pathway for macrophage activation. J. Interferon Cytokine Res. 2002, 22, 517–526. [Google Scholar] [CrossRef]
- Mu, J. RhoA signaling in CCL2-induced macrophage polarization. J. Allergy Clin. Immunol. 2018, 141, AB114. [Google Scholar] [CrossRef]
- Sierra-Filardi, E.; Nieto, C.; Dominguez-Soto, A.; Barroso, R.; Sanchez-Mateos, P.; Puig-Kroger, A.; Lopez-Bravo, M.; Joven, J.; Ardavin, C.; Rodriguez-Fernandez, J.L.; et al. CCL2 shapes macrophage polarization by GM-CSF and M-CSF: Identification of CCL2/CCR2-dependent gene expression profile. J. Immunol. 2014, 192, 3858–3867. [Google Scholar] [CrossRef] [Green Version]
- Fleetwood, A.J.; Lawrence, T.; Hamilton, J.A.; Cook, A.D. Granulocyte-macrophage colony-stimulating factor (CSF) and macrophage CSF-dependent macrophage phenotypes display differences in cytokine profiles and transcription factor activities: Implications for CSF blockade in inflammation. J. Immunol. 2007, 178, 5245–5252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, C.V.; Ricardo, S.D. Macrophages and CSF-1: Implications for development and beyond. Organogenesis 2013, 9, 249–260. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Peng, J.; Han, G.H.; Ding, X.; Wei, S.; Gao, G.; Huang, K.; Chang, F.; Wang, Y. Role of macrophages in peripheral nerve injury and repair. Neural Regen. Res. 2019, 14, 1335–1342. [Google Scholar] [CrossRef] [PubMed]
- Kroner, A.; Greenhalgh, A.D.; Zarruk, J.G.; Passos Dos Santos, R.; Gaestel, M.; David, S. TNF and increased intracellular iron alter macrophage polarization to a detrimental M1 phenotype in the injured spinal cord. Neuron 2014, 83, 1098–1116. [Google Scholar] [CrossRef]
- Silva, C.E.A.; Guimaraes, R.M.; Cunha, T.M. Sensory neuron-associated macrophages as novel modulators of neuropathic pain. Pain Rep. 2021, 6, e873. [Google Scholar] [CrossRef]
- Domoto, R.; Sekiguchi, F.; Tsubota, M.; Kawabata, A. Macrophage as a Peripheral Pain Regulator. Cells 2021, 10, 1881. [Google Scholar] [CrossRef] [PubMed]
- Verri, W.A., Jr.; Cunha, T.M.; Parada, C.A.; Poole, S.; Cunha, F.Q.; Ferreira, S.H. Hypernociceptive role of cytokines and chemokines: Targets for analgesic drug development? Pharmacol. Ther. 2006, 112, 116–138. [Google Scholar] [CrossRef]
- Binshtok, A.M.; Wang, H.; Zimmermann, K.; Amaya, F.; Vardeh, D.; Shi, L.; Brenner, G.J.; Ji, R.R.; Bean, B.P.; Woolf, C.J.; et al. Nociceptors are interleukin-1beta sensors. J. Neurosci. 2008, 28, 14062–14073. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.Q.; Liu, Z.; Liu, Z.H.; Chen, S.P.; Li, M.; Shahveranov, A.; Ye, D.W.; Tian, Y.K. Interleukin-6: An emerging regulator of pathological pain. J. Neuroinflamm. 2016, 13, 141. [Google Scholar] [CrossRef] [Green Version]
- Ma, W.; Quirion, R. Up-regulation of interleukin-6 induced by prostaglandin E from invading macrophages following nerve injury: An in vivo and in vitro study. J. Neurochem. 2005, 93, 664–673. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Chen, W.; Fan, X.; Wang, J.; Fu, S.; Cui, S.; Liao, F.; Cai, J.; Wang, X.; Huang, Y.; et al. Upregulation of interleukin-6 on Cav3.2 T-type calcium channels in dorsal root ganglion neurons contributes to neuropathic pain in rats with spinal nerve ligation. Exp. Neurol. 2019, 317, 226–243. [Google Scholar] [CrossRef]
- Ma, W.; St-Jacques, B.; Duarte, P.C. Targeting pain mediators induced by injured nerve-derived COX2 and PGE2 to treat neuropathic pain. Expert Opin. Ther. Targets 2012, 16, 527–540. [Google Scholar] [CrossRef]
- Ma, W.; Eisenach, J.C. Morphological and pharmacological evidence for the role of peripheral prostaglandins in the pathogenesis of neuropathic pain. Eur. J. Neurosci. 2002, 15, 1037–1047. [Google Scholar] [CrossRef] [PubMed]
- Moriyama, T.; Higashi, T.; Togashi, K.; Iida, T.; Segi, E.; Sugimoto, Y.; Tominaga, T.; Narumiya, S.; Tominaga, M. Sensitization of TRPV1 by EP1 and IP reveals peripheral nociceptive mechanism of prostaglandins. Mol. Pain 2005, 1, 3. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.A.; Davis, C.L.; Burgess, G.M. Prostaglandin E2-induced sensitization of bradykinin-evoked responses in rat dorsal root ganglion neurons is mediated by cAMP-dependent protein kinase A. Eur. J. Neurosci. 2000, 12, 3250–3258. [Google Scholar] [CrossRef] [PubMed]
- Ma, W. Chronic prostaglandin E2 treatment induces the synthesis of the pain-related peptide substance P and calcitonin gene-related peptide in cultured sensory ganglion explants. J. Neurochem. 2010, 115, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Treutlein, E.M.; Kern, K.; Weigert, A.; Tarighi, N.; Schuh, C.D.; Nusing, R.M.; Schreiber, Y.; Ferreiros, N.; Brune, B.; Geisslinger, G.; et al. The prostaglandin E2 receptor EP3 controls CC-chemokine ligand 2-mediated neuropathic pain induced by mechanical nerve damage. J. Biol. Chem. 2018, 293, 9685–9695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gschwandtner, M.; Derler, R.; Midwood, K.S. More Than Just Attractive: How CCL2 Influences Myeloid Cell Behavior Beyond Chemotaxis. Front. Immunol. 2019, 10, 2759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, F.A.; Sun, J.; Waters, S.M.; Ma, C.; Ren, D.; Ripsch, M.; Steflik, J.; Cortright, D.N.; Lamotte, R.H.; Miller, R.J. Excitatory monocyte chemoattractant protein-1 signaling is up-regulated in sensory neurons after chronic compression of the dorsal root ganglion. Proc. Natl. Acad. Sci. USA 2005, 102, 14092–14097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J.H.; Yang, B.; Donnelly, D.F.; Ma, C.; LaMotte, R.H. MCP-1 enhances excitability of nociceptive neurons in chronically compressed dorsal root ganglia. J. Neurophysiol. 2006, 96, 2189–2199. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Xie, W.; Strong, J.A.; Zhang, J.M. Systemic anti-inflammatory corticosteroid reduces mechanical pain behavior, sympathetic sprouting, and elevation of pro-inflammatory cytokines in a rat model of neuropathic pain. Anesthesiology 2007, 107, 469–477. [Google Scholar] [CrossRef] [Green Version]
- Silva, R.L.; Lopes, A.H.; Guimaraes, R.M.; Cunha, T.M. CXCL1/CXCR2 signaling in pathological pain: Role in peripheral and central sensitization. Neurobiol. Dis. 2017, 105, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.G.; Strong, J.A.; Xie, W.; Yang, R.H.; Coyle, D.E.; Wick, D.M.; Dorsey, E.D.; Zhang, J.M. The chemokine CXCL1/growth related oncogene increases sodium currents and neuronal excitability in small diameter sensory neurons. Mol. Pain 2008, 4, 38. [Google Scholar] [CrossRef] [Green Version]
- Yang, R.H.; Strong, J.A.; Zhang, J.M. NF-kappaB mediated enhancement of potassium currents by the chemokine CXCL1/growth related oncogene in small diameter rat sensory neurons. Mol. Pain 2009, 5, 26. [Google Scholar] [CrossRef]
- Shibasaki, M.; Sasaki, M.; Miura, M.; Mizukoshi, K.; Ueno, H.; Hashimoto, S.; Tanaka, Y.; Amaya, F. Induction of high mobility group box-1 in dorsal root ganglion contributes to pain hypersensitivity after peripheral nerve injury. Pain 2010, 149, 514–521. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.F.; Morioka, N.; Harano, S.; Nakamura, Y.; Liu, K.; Nishibori, M.; Hisaoka-Nakashima, K.; Nakata, Y. Perineural expression of high-mobility group box-1 contributes to long-lasting mechanical hypersensitivity via matrix metalloprotease-9 up-regulation in mice with painful peripheral neuropathy. J. Neurochem. 2016, 136, 837–850. [Google Scholar] [CrossRef] [Green Version]
- Yamasoba, D.; Tsubota, M.; Domoto, R.; Sekiguchi, F.; Nishikawa, H.; Liu, K.; Nishibori, M.; Ishikura, H.; Yamamoto, T.; Taga, A.; et al. Peripheral HMGB1-induced hyperalgesia in mice: Redox state-dependent distinct roles of RAGE and TLR4. J. Pharmacol. Sci. 2016, 130, 139–142. [Google Scholar] [CrossRef] [Green Version]
- Tomita, S.; Sekiguchi, F.; Kasanami, Y.; Naoe, K.; Tsubota, M.; Wake, H.; Nishibori, M.; Kawabata, A. Cav3.2 overexpression in L4 dorsal root ganglion neurons after L5 spinal nerve cutting involves Egr-1, USP5 and HMGB1 in rats: An emerging signaling pathway for neuropathic pain. Eur. J. Pharmacol. 2020, 888, 173587. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, A.J.; Copits, B.A.; Mickle, A.D.; Karlsson, P.; Kadunganattil, S.; Haroutounian, S.; Tadinada, S.M.; de Kloet, A.D.; Valtcheva, M.V.; McIlvried, L.A.; et al. Angiotensin II Triggers Peripheral Macrophage-to-Sensory Neuron Redox Crosstalk to Elicit Pain. J. Neurosci. 2018, 38, 7032–7057. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Deng, B.; Liu, S.; Luo, S.; Ning, Y.; Pan, X.; Wan, R.; Chen, Y.; Zhang, Z.; Jiang, J.; et al. Myeloid Piezo1 Deletion Protects Renal Fibrosis by Restraining Macrophage Infiltration and Activation. Hypertension 2022, 79, 918–931. [Google Scholar] [CrossRef]
- Jain, N.; Moeller, J.; Vogel, V. Mechanobiology of Macrophages: How Physical Factors Coregulate Macrophage Plasticity and Phagocytosis. Annu. Rev. Biomed. Eng. 2019, 21, 267–297. [Google Scholar] [CrossRef]
- Atcha, H.; Jairaman, A.; Holt, J.R.; Meli, V.S.; Nagalla, R.R.; Veerasubramanian, P.K.; Brumm, K.T.; Lim, H.E.; Othy, S.; Cahalan, M.D.; et al. Mechanically activated ion channel Piezo1 modulates macrophage polarization and stiffness sensing. Nat. Commun. 2021, 12, 3256. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Ramnath, R.D.; Zhi, L.; Tamizhselvi, R.; Bhatia, M. Substance P enhances NF-kappaB transactivation and chemokine response in murine macrophages via ERK1/2 and p38 MAPK signaling pathways. Am. J. Physiol. Cell Physiol. 2008, 294, C1586–C1596. [Google Scholar] [CrossRef]
- Lucrezi, J.D.; Burns, T.J.; Matesic, D.F.; Oldham, C.D.; May, S.W. Inhibition of JNK and p38 MAPK phosphorylation by 5-(acetylamino)-4-oxo-6-phenyl-2-hexenoic acid methyl ester and 4-phenyl-butenoic acid decreases substance P-induced TNF-alpha upregulation in macrophages. Int. Immunopharmacol. 2014, 21, 44–50. [Google Scholar] [CrossRef]
- Lim, J.E.; Chung, E.; Son, Y. A neuropeptide, Substance-P, directly induces tissue-repairing M2 like macrophages by activating the PI3K/Akt/mTOR pathway even in the presence of IFNgamma. Sci. Rep. 2017, 7, 9417. [Google Scholar] [CrossRef] [Green Version]
- Iyengar, S.; Ossipov, M.H.; Johnson, K.W. The role of calcitonin gene-related peptide in peripheral and central pain mechanisms including migraine. Pain 2017, 158, 543–559. [Google Scholar] [CrossRef] [Green Version]
- Natura, G.; von Banchet, G.S.; Schaible, H.G. Calcitonin gene-related peptide enhances TTX-resistant sodium currents in cultured dorsal root ganglion neurons from adult rats. Pain 2005, 116, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Benarroch, E.E. CGRP: Sensory neuropeptide with multiple neurologic implications. Neurology 2011, 77, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Baliu-Pique, M.; Jusek, G.; Holzmann, B. Neuroimmunological communication via CGRP promotes the development of a regulatory phenotype in TLR4-stimulated macrophages. Eur. J. Immunol. 2014, 44, 3708–3716. [Google Scholar] [CrossRef]
- Ichinose, M.; Sawada, M. Enhancement of phagocytosis by calcitonin gene-related peptide (CGRP) in cultured mouse peritoneal macrophages. Peptides 1996, 17, 1405–1414. [Google Scholar] [CrossRef]
- Ma, W.; Quirion, R. Increased calcitonin gene-related peptide in neuroma and invading macrophages is involved in the up-regulation of interleukin-6 and thermal hyperalgesia in a rat model of mononeuropathy. J. Neurochem. 2006, 98, 180–192. [Google Scholar] [CrossRef] [PubMed]
- Recasens, M.; Almolda, B.; Perez-Clausell, J.; Campbell, I.L.; Gonzalez, B.; Castellano, B. Chronic exposure to IL-6 induces a desensitized phenotype of the microglia. J. Neuroinflamm. 2021, 18, 31. [Google Scholar] [CrossRef] [PubMed]
- Sikandar, S.; Minett, M.S.; Millet, Q.; Santana-Varela, S.; Lau, J.; Wood, J.N.; Zhao, J. Brain-derived neurotrophic factor derived from sensory neurons plays a critical role in chronic pain. Brain J. Neurol. 2018, 141, 1028–1039. [Google Scholar] [CrossRef] [Green Version]
- Lever, I.J.; Bradbury, E.J.; Cunningham, J.R.; Adelson, D.W.; Jones, M.G.; McMahon, S.B.; Marvizon, J.C.; Malcangio, M. Brain-derived neurotrophic factor is released in the dorsal horn by distinctive patterns of afferent fiber stimulation. J. Neurosci. 2001, 21, 4469–4477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fonseca, M.M.; Davoli-Ferreira, M.; Santa-Cecilia, F.; Guimaraes, R.M.; Oliveira, F.F.B.; Kusuda, R.; Ferreira, D.W.; Alves-Filho, J.C.; Cunha, F.Q.; Cunha, T.M. IL-27 Counteracts Neuropathic Pain Development Through Induction of IL-10. Front. Immunol. 2019, 10, 3059. [Google Scholar] [CrossRef]
- Shen, K.F.; Zhu, H.Q.; Wei, X.H.; Wang, J.; Li, Y.Y.; Pang, R.P.; Liu, X.G. Interleukin-10 down-regulates voltage gated sodium channels in rat dorsal root ganglion neurons. Exp. Neurol. 2013, 247, 466–475. [Google Scholar] [CrossRef]
- Pannell, M.; Labuz, D.; Celik, M.O.; Keye, J.; Batra, A.; Siegmund, B.; Machelska, H. Adoptive transfer of M2 macrophages reduces neuropathic pain via opioid peptides. J. Neuroinflamm. 2016, 13, 262. [Google Scholar] [CrossRef] [Green Version]
- Malafoglia, V.; Ilari, S.; Vitiello, L.; Tenti, M.; Balzani, E.; Muscoli, C.; Raffaeli, W.; Bonci, A. The Interplay between Chronic Pain, Opioids, and the Immune System. Neurosci. Rev. J. Bringing Neurobiol. Neurol. Psychiatry 2021, 10738584211030493. [Google Scholar] [CrossRef] [PubMed]
- Corder, G.; Castro, D.C.; Bruchas, M.R.; Scherrer, G. Endogenous and Exogenous Opioids in Pain. Annu. Rev. Neurosci. 2018, 41, 453–473. [Google Scholar] [CrossRef]
- Altier, C.; Khosravani, H.; Evans, R.M.; Hameed, S.; Peloquin, J.B.; Vartian, B.A.; Chen, L.; Beedle, A.M.; Ferguson, S.S.; Mezghrani, A.; et al. ORL1 receptor-mediated internalization of N-type calcium channels. Nat. Neurosci. 2006, 9, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Chavez-Castillo, M.; Ortega, A.; Cudris-Torres, L.; Duran, P.; Rojas, M.; Manzano, A.; Garrido, B.; Salazar, J.; Silva, A.; Rojas-Gomez, D.M.; et al. Specialized Pro-Resolving Lipid Mediators: The Future of Chronic Pain Therapy? Int. J. Mol. Sci. 2021, 22, 10370. [Google Scholar] [CrossRef]
- Ohira, T.; Arita, M.; Omori, K.; Recchiuti, A.; Van Dyke, T.E.; Serhan, C.N. Resolvin E1 receptor activation signals phosphorylation and phagocytosis. J. Biol. Chem. 2010, 285, 3451–3461. [Google Scholar] [CrossRef] [Green Version]
- Schmid, M.; Gemperle, C.; Rimann, N.; Hersberger, M. Resolvin D1 Polarizes Primary Human Macrophages toward a Proresolution Phenotype through GPR32. J. Immunol. 2016, 196, 3429–3437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.F.; Li, Q.; Liu, S.B.; Mi, W.L.; Hu, S.; Zhao, J.; Tian, Y.; Mao-Ying, Q.L.; Jiang, J.W.; Ma, H.J.; et al. Aspirin-triggered Lipoxin A4 attenuates mechanical allodynia in association with inhibiting spinal JAK2/STAT3 signaling in neuropathic pain in rats. Neuroscience 2014, 273, 65–78. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.Z.; Berta, T.; Ji, R.R. Resolvin E1 inhibits neuropathic pain and spinal cord microglial activation following peripheral nerve injury. J. Neuroimmune Pharmacol. 2013, 8, 37–41. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.Z.; Liu, X.J.; Berta, T.; Park, C.K.; Lu, N.; Serhan, C.N.; Ji, R.R. Neuroprotectin/protectin D1 protects against neuropathic pain in mice after nerve trauma. Ann. Neurol. 2013, 74, 490–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jo, Y.Y.; Lee, J.Y.; Park, C.K. Resolvin E1 Inhibits Substance P-Induced Potentiation of TRPV1 in Primary Sensory Neurons. Mediat. Inflamm. 2016, 2016, 5259321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perna, E.; Aguilera-Lizarraga, J.; Florens, M.V.; Jain, P.; Theofanous, S.A.; Hanning, N.; De Man, J.G.; Berg, M.; De Winter, B.; Alpizar, Y.A.; et al. Effect of resolvins on sensitisation of TRPV1 and visceral hypersensitivity in IBS. Gut 2021, 70, 1275–1286. [Google Scholar] [CrossRef] [PubMed]
- Park, C.K.; Xu, Z.Z.; Liu, T.; Lu, N.; Serhan, C.N.; Ji, R.R. Resolvin D2 is a potent endogenous inhibitor for transient receptor potential subtype V1/A1, inflammatory pain, and spinal cord synaptic plasticity in mice: Distinct roles of resolvin D1, D2, and E1. J. Neurosci. 2011, 31, 18433–18438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalpachidou, T.; Kummer, K.K.; Kress, M. Non-coding RNAs in neuropathic pain. Neuronal Signal. 2020, 4, NS20190099. [Google Scholar] [CrossRef] [Green Version]
- Park, C.K.; Xu, Z.Z.; Berta, T.; Han, Q.; Chen, G.; Liu, X.J.; Ji, R.R. Extracellular microRNAs activate nociceptor neurons to elicit pain via TLR7 and TRPA1. Neuron 2014, 82, 47–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehmann, S.M.; Kruger, C.; Park, B.; Derkow, K.; Rosenberger, K.; Baumgart, J.; Trimbuch, T.; Eom, G.; Hinz, M.; Kaul, D.; et al. An unconventional role for miRNA: Let-7 activates Toll-like receptor 7 and causes neurodegeneration. Nat. Neurosci. 2012, 15, 827–835. [Google Scholar] [CrossRef]
- Cobos Jimenez, V.; Bradley, E.J.; Willemsen, A.M.; van Kampen, A.H.; Baas, F.; Kootstra, N.A. Next-generation sequencing of microRNAs uncovers expression signatures in polarized macrophages. Physiol. Genom. 2014, 46, 91–103. [Google Scholar] [CrossRef] [Green Version]
- Simeoli, R.; Montague, K.; Jones, H.R.; Castaldi, L.; Chambers, D.; Kelleher, J.H.; Vacca, V.; Pitcher, T.; Grist, J.; Al-Ahdal, H.; et al. Exosomal cargo including microRNA regulates sensory neuron to macrophage communication after nerve trauma. Nat. Commun. 2017, 8, 1778. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.J.; Guo, J.S.; Li, S.S.; Wu, X.B.; Cao, D.L.; Jiang, B.C.; Jing, P.B.; Bai, X.Q.; Li, C.H.; Wu, Z.H.; et al. TLR8 and its endogenous ligand miR-21 contribute to neuropathic pain in murine DRG. J. Exp. Med. 2018, 215, 3019–3037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Liu, J.; Wang, X.; Zhang, J.; Xie, C. Extracellular vesicle-encapsulated microRNA-23a from dorsal root ganglia neurons binds to A20 and promotes inflammatory macrophage polarization following peripheral nerve injury. Aging 2021, 13, 6752–6764. [Google Scholar] [CrossRef] [PubMed]
- Sakai, A.; Saitow, F.; Maruyama, M.; Miyake, N.; Miyake, K.; Shimada, T.; Okada, T.; Suzuki, H. MicroRNA cluster miR-17-92 regulates multiple functionally related voltage-gated potassium channels in chronic neuropathic pain. Nat. Commun. 2017, 8, 16079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, J.; Cao, J.; Wang, J.; Ren, X.; Su, S.; Li, M.; Li, Z.; Zhao, Q.; Zang, W. MicroRNA-30b regulates expression of the sodium channel Nav1.7 in nerve injury-induced neuropathic pain in the rat. Mol. Pain 2016, 12, 1744806916671523. [Google Scholar] [CrossRef]
- Chen, H.P.; Zhou, W.; Kang, L.M.; Yan, H.; Zhang, L.; Xu, B.H.; Cai, W.H. Intrathecal miR-96 inhibits Nav1.3 expression and alleviates neuropathic pain in rat following chronic construction injury. Neurochem. Res. 2014, 39, 76–83. [Google Scholar] [CrossRef]
- Lin, C.R.; Chen, K.H.; Yang, C.H.; Huang, H.W.; Sheen-Chen, S.M. Intrathecal miR-183 delivery suppresses mechanical allodynia in mononeuropathic rats. Eur. J. Neurosci. 2014, 39, 1682–1689. [Google Scholar] [CrossRef]
- Cai, W.; Zhao, Q.; Shao, J.; Zhang, J.; Li, L.; Ren, X.; Su, S.; Bai, Q.; Li, M.; Chen, X.; et al. MicroRNA-182 Alleviates Neuropathic Pain by Regulating Nav1.7 Following Spared Nerve Injury in Rats. Sci. Rep. 2018, 8, 16750. [Google Scholar] [CrossRef] [Green Version]
- Liao, M.F.; Hsu, J.L.; Lu, K.T.; Chao, P.K.; Cheng, M.Y.; Hsu, H.C.; Lo, A.L.; Lee, Y.L.; Hung, Y.H.; Lyu, R.K.; et al. Granulocyte Colony Stimulating Factor (GCSF) Can Attenuate Neuropathic Pain by Suppressing Monocyte Chemoattractant Protein-1 (MCP-1) Expression, through Upregulating the Early MicroRNA-122 Expression in the Dorsal Root Ganglia. Cells 2020, 9, 1669. [Google Scholar] [CrossRef]
- Lu, S.; Ma, S.; Wang, Y.; Huang, T.; Zhu, Z.; Zhao, G. Mus musculus-microRNA-449a ameliorates neuropathic pain by decreasing the level of KCNMA1 and TRPA1, and increasing the level of TPTE. Mol. Med. Rep. 2017, 16, 353–360. [Google Scholar] [CrossRef] [Green Version]
- Yang, F.R.; Chen, J.; Yi, H.; Peng, L.Y.; Hu, X.L.; Guo, Q.L. MicroRNA-7a ameliorates neuropathic pain in a rat model of spinal nerve ligation via the neurofilament light polypeptide-dependent signal transducer and activator of transcription signaling pathway. Mol. Pain 2019, 15, 1744806919842464. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, F.; Wei, M.; Qiu, Y.; Ma, C.; Shen, L.; Huang, Y. Chronic constriction injury-induced microRNA-146a-5p alleviates neuropathic pain through suppression of IRAK1/TRAF6 signaling pathway. J. Neuroinflamm. 2018, 15, 179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.; van Rooijen, N.; Tracey, D.J. Depletion of macrophages reduces axonal degeneration and hyperalgesia following nerve injury. Pain 2000, 86, 25–32. [Google Scholar] [CrossRef]
- Shepherd, A.J.; Mickle, A.D.; Golden, J.P.; Mack, M.R.; Halabi, C.M.; de Kloet, A.D.; Samineni, V.K.; Kim, B.S.; Krause, E.G.; Gereau, R.W.t.; et al. Macrophage angiotensin II type 2 receptor triggers neuropathic pain. Proc. Natl. Acad. Sci. USA 2018, 115, E8057–E8066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, J.; Gu, N.; Zhou, L.; BEyo, U.; Murugan, M.; Gan, W.B.; Wu, L.J. Microglia and monocytes synergistically promote the transition from acute to chronic pain after nerve injury. Nat. Commun. 2016, 7, 12029. [Google Scholar] [CrossRef] [PubMed]
- Rutkowski, M.D.; Pahl, J.L.; Sweitzer, S.; van Rooijen, N.; DeLeo, J.A. Limited role of macrophages in generation of nerve injury-induced mechanical allodynia. Physiol. Behav. 2000, 71, 225–235. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gheorghe, R.-O.; Grosu, A.V.; Bica-Popi, M.; Ristoiu, V. The Yin/Yang Balance of Communication between Sensory Neurons and Macrophages in Traumatic Peripheral Neuropathic Pain. Int. J. Mol. Sci. 2022, 23, 12389. https://doi.org/10.3390/ijms232012389
Gheorghe R-O, Grosu AV, Bica-Popi M, Ristoiu V. The Yin/Yang Balance of Communication between Sensory Neurons and Macrophages in Traumatic Peripheral Neuropathic Pain. International Journal of Molecular Sciences. 2022; 23(20):12389. https://doi.org/10.3390/ijms232012389
Chicago/Turabian StyleGheorghe, Roxana-Olimpia, Andreea Violeta Grosu, Melania Bica-Popi, and Violeta Ristoiu. 2022. "The Yin/Yang Balance of Communication between Sensory Neurons and Macrophages in Traumatic Peripheral Neuropathic Pain" International Journal of Molecular Sciences 23, no. 20: 12389. https://doi.org/10.3390/ijms232012389
APA StyleGheorghe, R. -O., Grosu, A. V., Bica-Popi, M., & Ristoiu, V. (2022). The Yin/Yang Balance of Communication between Sensory Neurons and Macrophages in Traumatic Peripheral Neuropathic Pain. International Journal of Molecular Sciences, 23(20), 12389. https://doi.org/10.3390/ijms232012389