
Obesogenic Diet-Induced Neuroinflammation: A Pathological Link between Hedonic and Homeostatic Control of Food Intake

 ,
,  , ,
, ,  and
and 
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
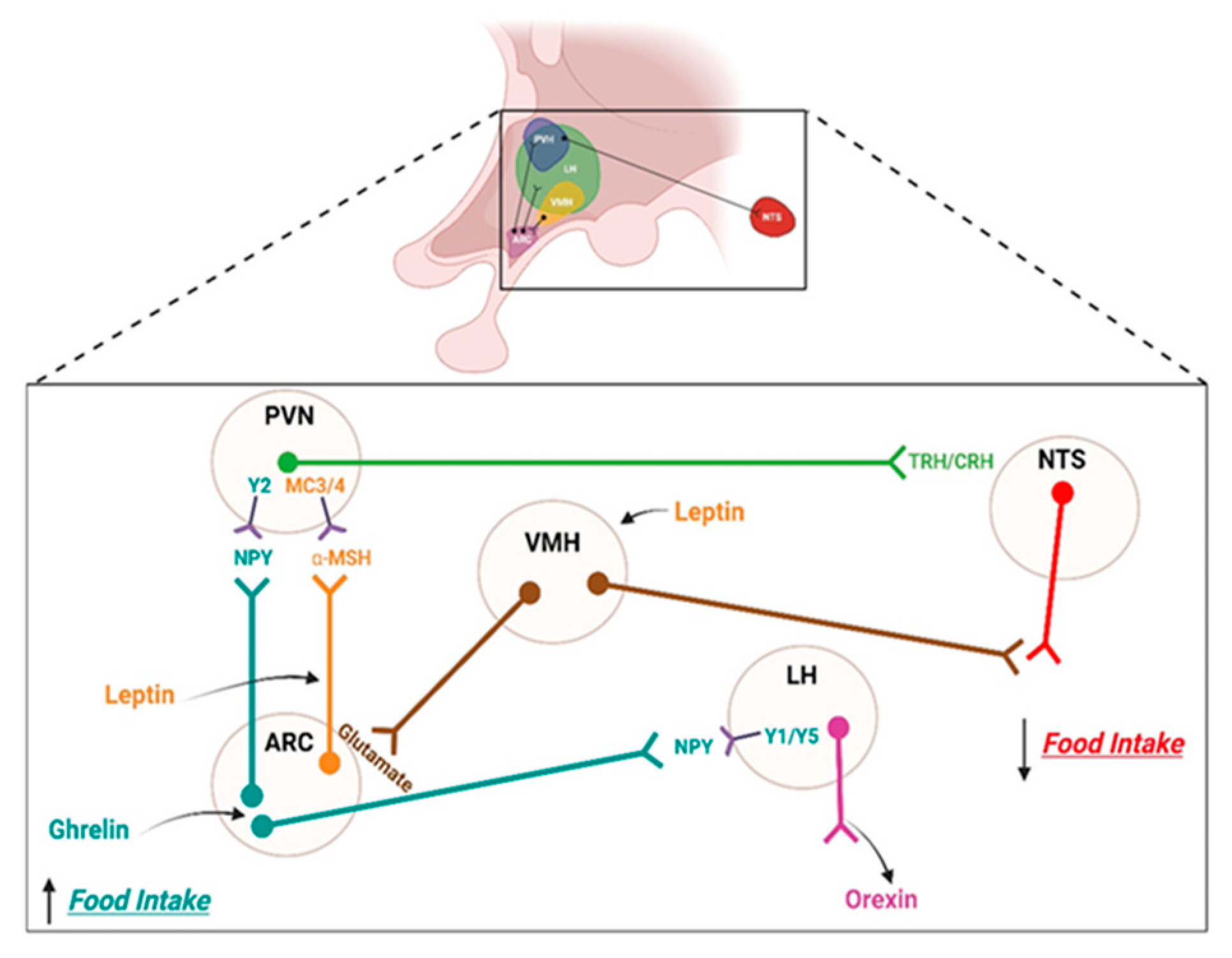
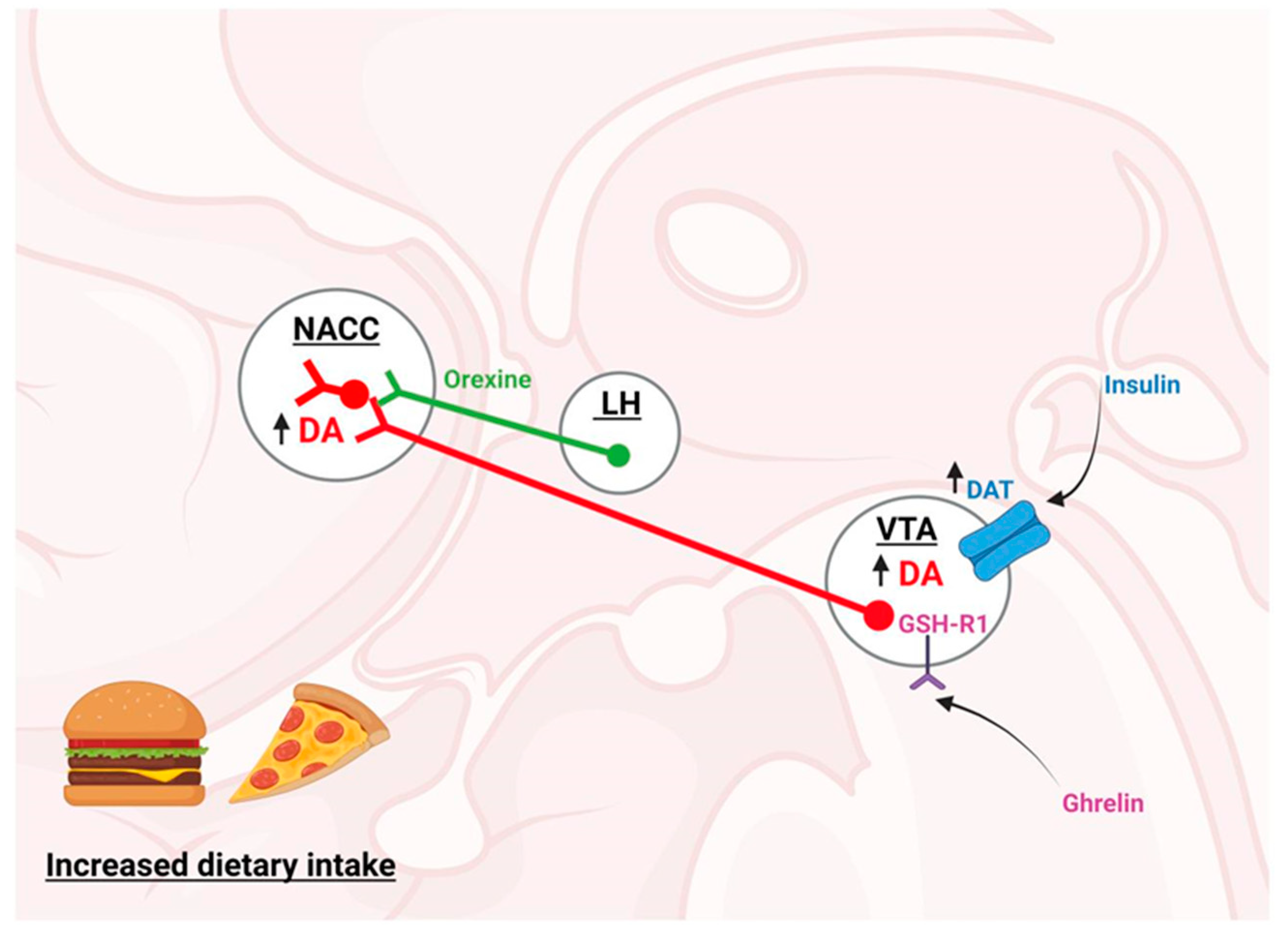
2. Brain Regulation of Feeding
2.1. Homeostatic Control System
2.2. Hedonic Control System
2.3. Other Changes Induced by a High-Fat Diet
3. Inflammation
3.1. Peripheral Inflammation
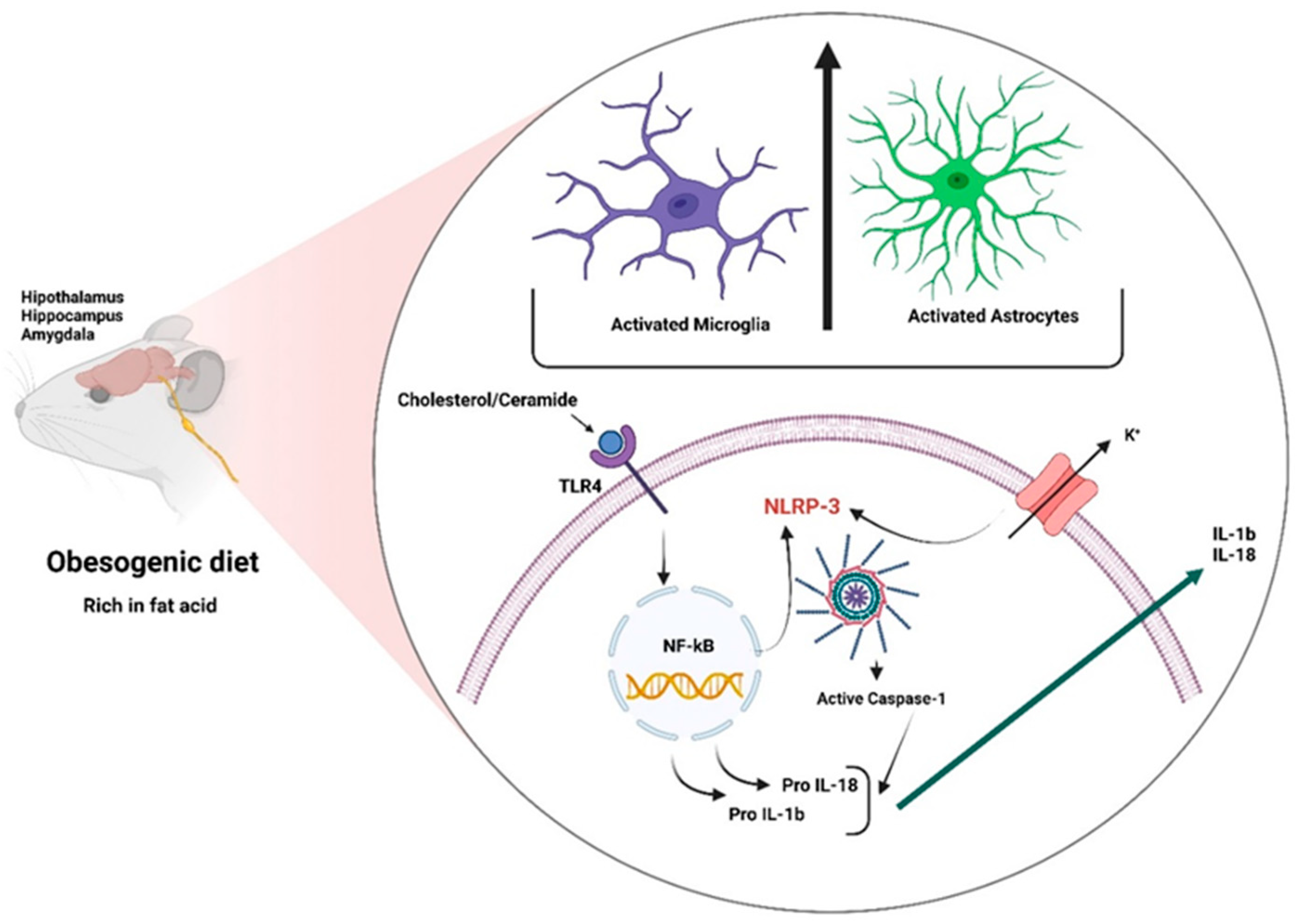
3.2. Central Inflammation or Neuroinflammation
3.2.1. Neuroinflammatory Mechanisms
Blood-Brain Barrier
Fatty Acids
3.2.2. Role of Glial Cells in Obesity
3.2.3. Inflammasomes
4. Neuroinflammation and Its Consequences
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- De Souza, C.T.; Araujo, E.P.; Bordin, S.; Ashimine, R.; Zollner, R.L.; Boschero, A.C.; Saad, M.J.; Velloso, L.A. Consumption of a fat-rich diet activates a proinflammatory response and induces insulin resistance in the hypothalamus. Endocrinology 2005, 146, 4192–4199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thaler, J.P.; Guyenet, S.J.; Dorfman, M.D.; Wisse, B.E.; Schwartz, M.W. Hypothalamic inflammation: Marker or mechanism of obesity pathogenesis? Diabetes 2013, 62, 2629–2634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buckman, L.B.; Thompson, M.M.; Moreno, H.N.; Ellacott, K.L. Regional astrogliosis in the mouse hypothalamus in response to obesity. J. Comp. Neurol. 2013, 521, 1322–1333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waise, T.M.Z.; Toshinai, K.; Naznin, F.; NamKoong, C.; Md Moin, A.S.; Sakoda, H.; Nakazato, M. One-day high-fat diet induces inflammation in the nodose ganglion and hypothalamus of mice. Biochem. Biophys. Res. Commun. 2015, 464, 1157–1162. [Google Scholar] [CrossRef] [Green Version]
- Lee, B.C.; Lee, J. Cellular and molecular players in adipose tissue inflammation in the development of obesity-induced insulin resistance. Biochim. Biophys. Acta 2014, 1842, 446–462. [Google Scholar] [CrossRef] [Green Version]
- Lee, T.J.; Hargrave, S.L.; Kinzig, K.P. Dual functions of CNS inflammation in food intake and metabolic regulation. Brain Res. 2020, 1740, 146859. [Google Scholar] [CrossRef]
- Pan, W.; Allison, M.B.; Sabatini, P.; Rupp, A.; Adams, J.; Patterson, C.; Jones, J.C.; Olson, D.P.; Myers, M.G., Jr. Transcriptional and physiological roles for STAT proteins in leptin action. Mol. Metab. 2019, 22, 121–131. [Google Scholar] [CrossRef]
- Dorfman, M.D.; Thaler, J.P. Hypothalamic inflammation and gliosis in obesity. Curr. Opin. Endocrinol. Diabetes Obes. 2015, 22, 325–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, M.; Wang, H.; Li, J.J.; Zhang, Y.L.; Xin, L.; Li, F.; Lou, S.J. The signaling mechanisms of hippocampal endoplasmic reticulum stress affecting neuronal plasticity-related protein levels in high fat diet-induced obese rats and the regulation of aerobic exercise. Brain Behav. Immun. 2016, 57, 347–359. [Google Scholar] [CrossRef] [PubMed]
- Seong, J.; Kang, J.Y.; Sun, J.S.; Kim, K.W. Hypothalamic inflammation and obesity: A mechanistic review. Arch. Pharm. Res. 2019, 42, 383–392. [Google Scholar] [CrossRef]
- Shabab, T.; Khanabdali, R.; Moghadamtousi, S.Z.; Kadir, H.A.; Mohan, G. Neuroinflammation pathways: A general review. Int. J. Neurosci. 2017, 127, 624–633. [Google Scholar] [CrossRef] [PubMed]
- Berthoud, H.R.; Munzberg, H. The lateral hypothalamus as integrator of metabolic and environmental needs: From electrical self-stimulation to opto-genetics. Physiol. Behav. 2011, 104, 29–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stuber, G.D.; Wise, R.A. Lateral hypothalamic circuits for feeding and reward. Nat. Neurosci. 2016, 19, 198–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klok, M.D.; Jakobsdottir, S.; Drent, M.L. The role of leptin and ghrelin in the regulation of food intake and body weight in humans: A review. Obes. Rev. 2007, 8, 21–34. [Google Scholar] [CrossRef]
- Won, J.C.; Jang, P.G.; Namkoong, C.; Koh, E.H.; Kim, S.K.; Park, J.Y.; Lee, K.U.; Kim, M.S. Central administration of an endoplasmic reticulum stress inducer inhibits the anorexigenic effects of leptin and insulin. Obesity 2009, 17, 1861–1865. [Google Scholar] [CrossRef]
- Kenny, P.J. Reward mechanisms in obesity: New insights and future directions. Neuron 2011, 69, 664–679. [Google Scholar] [CrossRef] [Green Version]
- Gysling, K.; Wang, R.Y. Morphine-induced activation of A10 dopamine neurons in the rat. Brain Res. 1983, 277, 119–127. [Google Scholar] [CrossRef]
- Johnson, S.W.; North, R.A. Opioids excite dopamine neurons by hyperpolarization of local interneurons. J. Neurosci. 1992, 12, 483–488. [Google Scholar] [CrossRef] [Green Version]
- Al Massadi, O.; Nogueiras, R.; Dieguez, C.; Girault, J.A. Ghrelin and food reward. Neuropharmacology 2019, 148, 131–138. [Google Scholar] [CrossRef]
- Floresco, S.B.; West, A.R.; Ash, B.; Moore, H.; Grace, A.A. Afferent modulation of dopamine neuron firing differentially regulates tonic and phasic dopamine transmission. Nat. Neurosci. 2003, 6, 968–973. [Google Scholar] [CrossRef]
- Kibaly, C.; Xu, C.; Cahill, C.M.; Evans, C.J.; Law, P.Y. Non-nociceptive roles of opioids in the CNS: Opioids’ effects on neurogenesis, learning, memory and affect. Nat. Rev. Neurosci. 2019, 20, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Di Chiara, G.; Imperato, A. Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proc. Natl. Acad. Sci. USA 1988, 85, 5274–5278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfaus, J.G.; Damsma, G.; Nomikos, G.G.; Wenkstern, D.G.; Blaha, C.D.; Phillips, A.G.; Fibiger, H.C. Sexual behavior enhances central dopamine transmission in the male rat. Brain Res. 1990, 530, 345–348. [Google Scholar] [CrossRef] [PubMed]
- Bassareo, V.; Di Chiara, G. Differential influence of associative and nonassociative learning mechanisms on the responsiveness of prefrontal and accumbal dopamine transmission to food stimuli in rats fed ad libitum. J. Neurosci. 1997, 17, 851–861. [Google Scholar] [CrossRef] [Green Version]
- Einhorn, L.C.; Johansen, P.A.; White, F.J. Electrophysiological effects of cocaine in the mesoaccumbens dopamine system: Studies in the ventral tegmental area. J. Neurosci. 1988, 8, 100–112. [Google Scholar] [CrossRef] [Green Version]
- Sagheddu, C.; Pintori, N.; Kalaba, P.; Dragacevic, V.; Piras, G.; Lubec, J.; Simola, N.; De Luca, M.A.; Lubec, G.; Pistis, M. Neurophysiological and Neurochemical Effects of the Putative Cognitive Enhancer (S)-CE-123 on Mesocorticolimbic Dopamine System. Biomolecules 2020, 10, 779. [Google Scholar] [CrossRef]
- Avelar, A.J.; Cao, J.; Newman, A.H.; Beckstead, M.J. Atypical dopamine transporter inhibitors R-modafinil and JHW 007 differentially affect D2 autoreceptor neurotransmission and the firing rate of midbrain dopamine neurons. Neuropharmacology 2017, 123, 410–419. [Google Scholar] [CrossRef]
- Ford, C.P. The role of D2-autoreceptors in regulating dopamine neuron activity and transmission. Neuroscience 2014, 282, 13–22. [Google Scholar] [CrossRef] [Green Version]
- Ikemoto, S. Dopamine reward circuitry: Two projection systems from the ventral midbrain to the nucleus accumbens-olfactory tubercle complex. Brain Res. Rev. 2007, 56, 27–78. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Liu, Q.; Wen, P.; Zhang, J.; Rao, X.; Zhou, Z.; Zhang, H.; He, X.; Li, J.; Zhou, Z.; et al. Activation of the dopaminergic pathway from VTA to the medial olfactory tubercle generates odor-preference and reward. Elife 2017, 6, e25423. [Google Scholar] [CrossRef]
- Wakabayashi, K.T.; Bruno, M.J.; Bass, C.E.; Park, J. Application of fast-scan cyclic voltammetry for the in vivo characterization of optically evoked dopamine in the olfactory tubercle of the rat brain. Analyst 2016, 141, 3746–3755. [Google Scholar] [CrossRef]
- Mebel, D.M.; Wong, J.C.; Dong, Y.J.; Borgland, S.L. Insulin in the ventral tegmental area reduces hedonic feeding and suppresses dopamine concentration via increased reuptake. Eur. J. Neurosci. 2012, 36, 2336–2346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aston-Jones, G.; Smith, R.J.; Sartor, G.C.; Moorman, D.E.; Massi, L.; Tahsili-Fahadan, P.; Richardson, K.A. Lateral hypothalamic orexin/hypocretin neurons: A role in reward-seeking and addiction. Brain Res. 2010, 1314, 74–90. [Google Scholar] [CrossRef] [Green Version]
- Espana, R.A.; Oleson, E.B.; Locke, J.L.; Brookshire, B.R.; Roberts, D.C.; Jones, S.R. The hypocretin-orexin system regulates cocaine self-administration via actions on the mesolimbic dopamine system. Eur. J. Neurosci. 2010, 31, 336–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Plasse, G.; van Zessen, R.; Luijendijk, M.C.; Erkan, H.; Stuber, G.D.; Ramakers, G.M.; Adan, R.A. Modulation of cue-induced firing of ventral tegmental area dopamine neurons by leptin and ghrelin. Int. J. Obes. 2015, 39, 1742–1749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thaler, J.P.; Yi, C.X.; Schur, E.A.; Guyenet, S.J.; Hwang, B.H.; Dietrich, M.O.; Zhao, X.; Sarruf, D.A.; Izgur, V.; Maravilla, K.R.; et al. Obesity is associated with hypothalamic injury in rodents and humans. J. Clin. Investig. 2012, 122, 153–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Zhang, G.; Zhang, H.; Karin, M.; Bai, H.; Cai, D. Hypothalamic IKKbeta/NF-kappaB and ER stress link overnutrition to energy imbalance and obesity. Cell 2008, 135, 61–73. [Google Scholar] [CrossRef] [Green Version]
- Naznin, F.; Toshinai, K.; Waise, T.M.; NamKoong, C.; Md Moin, A.S.; Sakoda, H.; Nakazato, M. Diet-induced obesity causes peripheral and central ghrelin resistance by promoting inflammation. J. Endocrinol. 2015, 226, 81–92. [Google Scholar] [CrossRef] [Green Version]
- Milanski, M.; Arruda, A.P.; Coope, A.; Ignacio-Souza, L.M.; Nunez, C.E.; Roman, E.A.; Romanatto, T.; Pascoal, L.B.; Caricilli, A.M.; Torsoni, M.A.; et al. Inhibition of hypothalamic inflammation reverses diet-induced insulin resistance in the liver. Diabetes 2012, 61, 1455–1462. [Google Scholar] [CrossRef] [Green Version]
- Guillemot-Legris, O.; Masquelier, J.; Everard, A.; Cani, P.D.; Alhouayek, M.; Muccioli, G.G. High-fat diet feeding differentially affects the development of inflammation in the central nervous system. J. Neuroinflammation 2016, 13, 206. [Google Scholar] [CrossRef]
- Wang, S.; Huang, X.F.; Zhang, P.; Wang, H.; Zhang, Q.; Yu, S.; Yu, Y. Chronic rhein treatment improves recognition memory in high-fat diet-induced obese male mice. J. Nutr. Biochem. 2016, 36, 42–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, J.; Wu, D.M.; Zheng, Y.L.; Hu, B.; Cheng, W.; Zhang, Z.F.; Shan, Q. Ursolic acid improves high fat diet-induced cognitive impairments by blocking endoplasmic reticulum stress and IkappaB kinase beta/nuclear factor-kappaB-mediated inflammatory pathways in mice. Brain Behav. Immun. 2011, 25, 1658–1667. [Google Scholar] [CrossRef] [PubMed]
- Dutheil, S.; Ota, K.T.; Wohleb, E.S.; Rasmussen, K.; Duman, R.S. High-Fat Diet Induced Anxiety and Anhedonia: Impact on Brain Homeostasis and Inflammation. Neuropsychopharmacology 2016, 41, 1874–1887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeon, B.T.; Jeong, E.A.; Shin, H.J.; Lee, Y.; Lee, D.H.; Kim, H.J.; Kang, S.S.; Cho, G.J.; Choi, W.S.; Roh, G.S. Resveratrol attenuates obesity-associated peripheral and central inflammation and improves memory deficit in mice fed a high-fat diet. Diabetes 2012, 61, 1444–1454. [Google Scholar] [CrossRef] [Green Version]
- Miao, Y.; Ren, J.; Jiang, L.; Liu, J.; Jiang, B.; Zhang, X. alpha-Lipoic acid attenuates obesity-associated hippocampal neuroinflammation and increases the levels of brain-derived neurotrophic factor in ovariectomized rats fed a high-fat diet. Int. J. Mol. Med. 2013, 32, 1179–1186. [Google Scholar] [CrossRef] [Green Version]
- Tucsek, Z.; Toth, P.; Sosnowska, D.; Gautam, T.; Mitschelen, M.; Koller, A.; Szalai, G.; Sonntag, W.E.; Ungvari, Z.; Csiszar, A. Obesity in aging exacerbates blood-brain barrier disruption, neuroinflammation, and oxidative stress in the mouse hippocampus: Effects on expression of genes involved in beta-amyloid generation and Alzheimer’s disease. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69, 1212–1226. [Google Scholar] [CrossRef]
- Castro, G.; MF, C.A.; Weissmann, L.; Quaresma, P.G.; Katashima, C.K.; Saad, M.J.; Prada, P.O. Diet-induced obesity induces endoplasmic reticulum stress and insulin resistance in the amygdala of rats. FEBS Open Bio. 2013, 3, 443–449. [Google Scholar] [CrossRef] [Green Version]
- Almeida-Suhett, C.P.; Graham, A.; Chen, Y.; Deuster, P. Behavioral changes in male mice fed a high-fat diet are associated with IL-1beta expression in specific brain regions. Physiol. Behav. 2017, 169, 130–140. [Google Scholar] [CrossRef]
- Unamuno, X.; Gomez-Ambrosi, J.; Rodriguez, A.; Becerril, S.; Fruhbeck, G.; Catalan, V. Adipokine dysregulation and adipose tissue inflammation in human obesity. Eur. J. Clin. Invest. 2018, 48, e12997. [Google Scholar] [CrossRef] [Green Version]
- Trim, W.V.; Lynch, L. Immune and non-immune functions of adipose tissue leukocytes. Nat Rev Immunol 2022, 22, 371–386. [Google Scholar] [CrossRef]
- Fritsche, K.L. The science of fatty acids and inflammation. Adv. Nutr. 2015, 6, 293S–301S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Liu, D.; Wang, F.; Liu, S.; Zhao, S.; Ling, E.A.; Hao, A. Saturated fatty acids activate microglia via Toll-like receptor 4/NF-kappaB signalling. Br. J. Nutr. 2012, 107, 229–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hotamisligil, G.S.; Peraldi, P.; Budavari, A.; Ellis, R.; White, M.F.; Spiegelman, B.M. IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-alpha- and obesity-induced insulin resistance. Science 1996, 271, 665–668. [Google Scholar] [CrossRef] [PubMed]
- Uysal, K.T.; Wiesbrock, S.M.; Marino, M.W.; Hotamisligil, G.S. Protection from obesity-induced insulin resistance in mice lacking TNF-alpha function. Nature 1997, 389, 610–614. [Google Scholar] [CrossRef] [PubMed]
- Camandola, S.; Mattson, M.P. Toll-like receptor 4 mediates fat, sugar, and umami taste preference and food intake and body weight regulation. Obesity 2017, 25, 1237–1245. [Google Scholar] [CrossRef] [Green Version]
- Reynolds, C.M.; McGillicuddy, F.C.; Harford, K.A.; Finucane, O.M.; Mills, K.H.; Roche, H.M. Dietary saturated fatty acids prime the NLRP3 inflammasome via TLR4 in dendritic cells-implications for diet-induced insulin resistance. Mol. Nutr. Food Res. 2012, 56, 1212–1222. [Google Scholar] [CrossRef]
- Genser, L.; Poitou, C.; Brot-Laroche, E.; Rousset, M.; Vaillant, J.C.; Clement, K.; Thenet, S.; Leturque, A. Alteration of intestinal permeability: The missing link between gut microbiota modifications and inflammation in obesity? Med. Sci. (Paris) 2016, 32, 461–469. [Google Scholar] [CrossRef] [Green Version]
- Cox, A.J.; West, N.P.; Cripps, A.W. Obesity, inflammation, and the gut microbiota. Lancet Diabetes Endocrinol 2015, 3, 207–215. [Google Scholar] [CrossRef]
- Kang, C.; Wang, B.; Kaliannan, K.; Wang, X.; Lang, H.; Hui, S.; Huang, L.; Zhang, Y.; Zhou, M.; Chen, M.; et al. Gut Microbiota Mediates the Protective Effects of Dietary Capsaicin against Chronic Low-Grade Inflammation and Associated Obesity Induced by High-Fat Diet. MBio 2017, 8, e00470-17. [Google Scholar] [CrossRef] [Green Version]
- Bauer, K.C.; Huus, K.E.; Finlay, B.B. Microbes and the mind: Emerging hallmarks of the gut microbiota-brain axis. Cell Microbiol. 2016, 18, 632–644. [Google Scholar] [CrossRef]
- Cai, Z.; Hussain, M.D.; Yan, L.J. Microglia, neuroinflammation, and beta-amyloid protein in Alzheimer’s disease. Int. J. Neurosci. 2014, 124, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Ganong, W.F. Circumventricular organs: Definition and role in the regulation of endocrine and autonomic function. Clin. Exp. Pharm. Physiol. 2000, 27, 422–427. [Google Scholar] [CrossRef]
- Mullier, A.; Bouret, S.G.; Prevot, V.; Dehouck, B. Differential distribution of tight junction proteins suggests a role for tanycytes in blood-hypothalamus barrier regulation in the adult mouse brain. J. Comp. Neurol. 2010, 518, 943–962. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, E.M.; Blazquez, J.L.; Guerra, M. The design of barriers in the hypothalamus allows the median eminence and the arcuate nucleus to enjoy private milieus: The former opens to the portal blood and the latter to the cerebrospinal fluid. Peptides 2010, 31, 757–776. [Google Scholar] [CrossRef] [PubMed]
- Buckman, L.B.; Hasty, A.H.; Flaherty, D.K.; Buckman, C.T.; Thompson, M.M.; Matlock, B.K.; Weller, K.; Ellacott, K.L. Obesity induced by a high-fat diet is associated with increased immune cell entry into the central nervous system. Brain Behav. Immun. 2014, 35, 33–42. [Google Scholar] [CrossRef] [Green Version]
- Hsuchou, H.; Kastin, A.J.; Mishra, P.K.; Pan, W. C-reactive protein increases BBB permeability: Implications for obesity and neuroinflammation. Cell Physiol. Biochem. 2012, 30, 1109–1119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stranahan, A.M.; Hao, S.; Dey, A.; Yu, X.; Baban, B. Blood-brain barrier breakdown promotes macrophage infiltration and cognitive impairment in leptin receptor-deficient mice. J. Cereb. Blood Flow Metab. 2016, 36, 2108–2121. [Google Scholar] [CrossRef] [Green Version]
- Nerurkar, P.V.; Johns, L.M.; Buesa, L.M.; Kipyakwai, G.; Volper, E.; Sato, R.; Shah, P.; Feher, D.; Williams, P.G.; Nerurkar, V.R. Momordica charantia (bitter melon) attenuates high-fat diet-associated oxidative stress and neuroinflammation. J. Neuroinflammation 2011, 8, 64. [Google Scholar] [CrossRef]
- Chang, H.C.; Tai, Y.T.; Cherng, Y.G.; Lin, J.W.; Liu, S.H.; Chen, T.L.; Chen, R.M. Resveratrol attenuates high-fat diet-induced disruption of the blood-brain barrier and protects brain neurons from apoptotic insults. J. Agric. Food Chem. 2014, 62, 3466–3475. [Google Scholar] [CrossRef]
- Elahy, M.; Lam, V.; Pallebage-Gamarallage, M.M.; Giles, C.; Mamo, J.C.; Takechi, R. Nicotine Attenuates Disruption of Blood-Brain Barrier Induced by Saturated-Fat Feeding in Wild-Type Mice. Nicotine Tob. Res. 2015, 17, 1436–1441. [Google Scholar] [CrossRef]
- Kanoski, S.E.; Zhang, Y.; Zheng, W.; Davidson, T.L. The effects of a high-energy diet on hippocampal function and blood-brain barrier integrity in the rat. J. Alzheimers Dis. 2010, 21, 207–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Douglass, J.D.; Dorfman, M.D.; Fasnacht, R.; Shaffer, L.D.; Thaler, J.P. Astrocyte IKKbeta/NF-kappaB signaling is required for diet-induced obesity and hypothalamic inflammation. Mol. Metab. 2017, 6, 366–373. [Google Scholar] [CrossRef]
- Valdearcos, M.; Robblee, M.M.; Benjamin, D.I.; Nomura, D.K.; Xu, A.W.; Koliwad, S.K. Microglia dictate the impact of saturated fat consumption on hypothalamic inflammation and neuronal function. Cell Rep. 2014, 9, 2124–2138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borg, M.L.; Omran, S.F.; Weir, J.; Meikle, P.J.; Watt, M.J. Consumption of a high-fat diet, but not regular endurance exercise training, regulates hypothalamic lipid accumulation in mice. J. Physiol. 2012, 590, 4377–4389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thaler, J.P.; Choi, S.J.; Schwartz, M.W.; Wisse, B.E. Hypothalamic inflammation and energy homeostasis: Resolving the paradox. Front. Neuroendocr. 2010, 31, 79–84. [Google Scholar] [CrossRef]
- Andre, C.; Guzman-Quevedo, O.; Rey, C.; Remus-Borel, J.; Clark, S.; Castellanos-Jankiewicz, A.; Ladeveze, E.; Leste-Lasserre, T.; Nadjar, A.; Abrous, D.N.; et al. Inhibiting Microglia Expansion Prevents Diet-Induced Hypothalamic and Peripheral Inflammation. Diabetes 2017, 66, 908–919. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Le, W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 1181–1194. [Google Scholar] [CrossRef]
- Kigerl, K.A.; de Rivero Vaccari, J.P.; Dietrich, W.D.; Popovich, P.G.; Keane, R.W. Pattern recognition receptors and central nervous system repair. Exp. Neurol. 2014, 258, 5–16. [Google Scholar] [CrossRef] [Green Version]
- Valdearcos, M.; Xu, A.W.; Koliwad, S.K. Hypothalamic inflammation in the control of metabolic function. Annu. Rev. Physiol. 2015, 77, 131–160. [Google Scholar] [CrossRef]
- Buttini, M.; Limonta, S.; Boddeke, H.W. Peripheral administration of lipopolysaccharide induces activation of microglial cells in rat brain. Neurochem. Int. 1996, 29, 25–35. [Google Scholar] [CrossRef]
- Garcia-Bueno, B.; Serrats, J.; Sawchenko, P.E. Cerebrovascular cyclooxygenase-1 expression, regulation, and role in hypothalamic-pituitary-adrenal axis activation by inflammatory stimuli. J. Neurosci. 2009, 29, 12970–12981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shih, R.H.; Wang, C.Y.; Yang, C.M. NF-kappaB Signaling Pathways in Neurological Inflammation: A Mini Review. Front. Mol. Neurosci. 2015, 8, 77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cintra, D.E.; Ropelle, E.R.; Moraes, J.C.; Pauli, J.R.; Morari, J.; Souza, C.T.; Grimaldi, R.; Stahl, M.; Carvalheira, J.B.; Saad, M.J.; et al. Unsaturated fatty acids revert diet-induced hypothalamic inflammation in obesity. PLoS ONE 2012, 7, e30571. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Qian, Y.; Fang, Q.; Zhong, P.; Li, W.; Wang, L.; Fu, W.; Zhang, Y.; Xu, Z.; Li, X.; et al. Saturated palmitic acid induces myocardial inflammatory injuries through direct binding to TLR4 accessory protein MD2. Nat. Commun. 2017, 8, 13997. [Google Scholar] [CrossRef] [Green Version]
- Valdearcos, M.; Douglass, J.D.; Robblee, M.M.; Dorfman, M.D.; Stifler, D.R.; Bennett, M.L.; Gerritse, I.; Fasnacht, R.; Barres, B.A.; Thaler, J.P.; et al. Microglial Inflammatory Signaling Orchestrates the Hypothalamic Immune Response to Dietary Excess and Mediates Obesity Susceptibility. Cell Metab. 2017, 26, 185–197 e3. [Google Scholar] [CrossRef] [Green Version]
- Cope, E.C.; LaMarca, E.A.; Monari, P.K.; Olson, L.B.; Martinez, S.; Zych, A.D.; Katchur, N.J.; Gould, E. Microglia Play an Active Role in Obesity-Associated Cognitive Decline. J. Neurosci. 2018, 38, 8889–8904. [Google Scholar] [CrossRef] [Green Version]
- Butler, M.J.; Cole, R.M.; Deems, N.P.; Belury, M.A.; Barrientos, R.M. Fatty food, fatty acids, and microglial priming in the adult and aged hippocampus and amygdala. Brain Behav. Immun. 2020, 89, 145–158. [Google Scholar] [CrossRef]
- Hahm, J.R.; Jo, M.H.; Ullah, R.; Kim, M.W.; Kim, M.O. Metabolic Stress Alters Antioxidant Systems, Suppresses the Adiponectin Receptor 1 and Induces Alzheimer’s Like Pathology in Mice Brain. Cells 2020, 9, 249. [Google Scholar] [CrossRef] [Green Version]
- Jin, S.; Kim, K.K.; Park, B.S.; Kim, D.H.; Jeong, B.; Kang, D.; Lee, T.H.; Park, J.W.; Kim, J.G.; Lee, B.J. Function of astrocyte MyD88 in high-fat-diet-induced hypothalamic inflammation. J. Neuroinflammation 2020, 17, 195. [Google Scholar] [CrossRef]
- Shi, H.; Wang, Q.; Zheng, M.; Hao, S.; Lum, J.S.; Chen, X.; Huang, X.F.; Yu, Y.; Zheng, K. Supplement of microbiota-accessible carbohydrates prevents neuroinflammation and cognitive decline by improving the gut microbiota-brain axis in diet-induced obese mice. J. Neuroinflammation 2020, 17, 77. [Google Scholar] [CrossRef]
- Kim, J.D.; Yoon, N.A.; Jin, S.; Diano, S. Microglial UCP2 Mediates Inflammation and Obesity Induced by High-Fat Feeding. Cell Metab. 2019, 30, 952–962 e5. [Google Scholar] [CrossRef]
- Robison, L.S.; Gannon, O.J.; Thomas, M.A.; Salinero, A.E.; Abi-Ghanem, C.; Poitelon, Y.; Belin, S.; Zuloaga, K.L. Role of sex and high-fat diet in metabolic and hypothalamic disturbances in the 3xTg-AD mouse model of Alzheimer’s disease. J. Neuroinflammation 2020, 17, 285. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Yu, Y.; Lin, D.; Zheng, P.; Zhang, P.; Hu, M.; Wang, Q.; Pan, W.; Yang, X.; Hu, T.; et al. beta-glucan attenuates cognitive impairment via the gut-brain axis in diet-induced obese mice. Microbiome 2020, 8, 143. [Google Scholar] [CrossRef]
- Park, J.H.; Ahn, J.H.; Song, M.; Kim, H.; Park, C.W.; Park, Y.E.; Lee, T.K.; Lee, J.C.; Kim, D.W.; Lee, C.H.; et al. A 2-Min Transient Ischemia Confers Cerebral Ischemic Tolerance in Non-Obese Gerbils, but Results in Neuronal Death in Obese Gerbils by Increasing Abnormal mTOR Activation-Mediated Oxidative Stress and Neuroinflammation. Cells 2019, 8, 1126. [Google Scholar] [CrossRef] [Green Version]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [Green Version]
- He, W.T.; Wan, H.; Hu, L.; Chen, P.; Wang, X.; Huang, Z.; Yang, Z.H.; Zhong, C.Q.; Han, J. Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. Cell Res. 2015, 25, 1285–1298. [Google Scholar] [CrossRef]
- Lamkanfi, M.; Dixit, V.M. Inflammasomes and their roles in health and disease. Annu. Rev. Cell Dev. Biol. 2012, 28, 137–161. [Google Scholar] [CrossRef] [Green Version]
- de Rivero Vaccari, J.P.; Dietrich, W.D.; Keane, R.W. Activation and regulation of cellular inflammasomes: Gaps in our knowledge for central nervous system injury. J. Cereb. Blood Flow Metab. 2014, 34, 369–375. [Google Scholar] [CrossRef] [Green Version]
- Jorquera, G.; Russell, J.; Monsalves-Alvarez, M.; Cruz, G.; Valladares-Ide, D.; Basualto-Alarcon, C.; Barrientos, G.; Estrada, M.; Llanos, P. NLRP3 Inflammasome: Potential Role in Obesity Related Low-Grade Inflammation and Insulin Resistance in Skeletal Muscle. Int. J. Mol. Sci. 2021, 22, 3254. [Google Scholar] [CrossRef]
- Man, S.M.; Kanneganti, T.D. Regulation of inflammasome activation. Immunol. Rev. 2015, 265, 6–21. [Google Scholar] [CrossRef]
- Rajamaki, K.; Lappalainen, J.; Oorni, K.; Valimaki, E.; Matikainen, S.; Kovanen, P.T.; Eklund, K.K. Cholesterol crystals activate the NLRP3 inflammasome in human macrophages: A novel link between cholesterol metabolism and inflammation. PLoS ONE 2010, 5, e11765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giordano, A.; Murano, I.; Mondini, E.; Perugini, J.; Smorlesi, A.; Severi, I.; Barazzoni, R.; Scherer, P.E.; Cinti, S. Obese adipocytes show ultrastructural features of stressed cells and die of pyroptosis. J. Lipid Res. 2013, 54, 2423–2436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rheinheimer, J.; de Souza, B.M.; Cardoso, N.S.; Bauer, A.C.; Crispim, D. Current role of the NLRP3 inflammasome on obesity and insulin resistance: A systematic review. Metabolism 2017, 74, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Barra, N.G.; Henriksbo, B.D.; Anhe, F.F.; Schertzer, J.D. The NLRP3 inflammasome regulates adipose tissue metabolism. Biochem. J. 2020, 477, 1089–1107. [Google Scholar] [CrossRef] [Green Version]
- Schroder, K.; Zhou, R.; Tschopp, J. The NLRP3 inflammasome: A sensor for metabolic danger? Science 2010, 327, 296–300. [Google Scholar] [CrossRef]
- Guillemot-Legris, O.; Muccioli, G.G. Obesity-Induced Neuroinflammation: Beyond the Hypothalamus. Trends Neurosci. 2017, 40, 237–253. [Google Scholar] [CrossRef]
- Gomes, J.A.S.; Silva, J.F.; Marcal, A.P.; Silva, G.C.; Gomes, G.F.; de Oliveira, A.C.P.; Soares, V.L.; Oliveira, M.C.; Ferreira, A.V.M.; Aguiar, D.C. High-refined carbohydrate diet consumption induces neuroinflammation and anxiety-like behavior in mice. J. Nutr. Biochem. 2020, 77, 108317. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.T.; Chen, P.S.; Kuo, Y.M.; Tzeng, S.F. Intermittent peripheral exposure to lipopolysaccharide induces exploratory behavior in mice and regulates brain glial activity in obese mice. J. Neuroinflammation 2020, 17, 163. [Google Scholar] [CrossRef] [PubMed]
- Jones, N.S.; Watson, K.Q.; Rebeck, G.W. High-fat diet increases gliosis and immediate early gene expression in APOE3 mice, but not APOE4 mice. J. Neuroinflammation 2021, 18, 214. [Google Scholar] [CrossRef]
- Morelli, S.; Piscioneri, A.; Guarnieri, G.; Morelli, A.; Drioli, E.; De Bartolo, L. Anti-neuroinflammatory effect of daidzein in human hypothalamic GnRH neurons in an in vitro membrane-based model. Biofactors 2021, 47, 93–111. [Google Scholar] [CrossRef]
- Bartolome, F.; Antequera, D.; de la Cueva, M.; Rubio-Fernandez, M.; Castro, N.; Pascual, C.; Camins, A.; Carro, E. Endothelial-specific deficiency of megalin in the brain protects mice against high-fat diet challenge. J. Neuroinflammation 2020, 17, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivanova, N.; Liu, Q.; Agca, C.; Agca, Y.; Noble, E.G.; Whitehead, S.N.; Cechetto, D.F. White matter inflammation and cognitive function in a co-morbid metabolic syndrome and prodromal Alzheimer’s disease rat model. J. Neuroinflammation 2020, 17, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Correia, S.S.; Iyengar, R.R.; Germano, P.; Tang, K.; Bernier, S.G.; Schwartzkopf, C.D.; Tobin, J.; Lee, T.W.; Liu, G.; Jacobson, S.; et al. The CNS-Penetrant Soluble Guanylate Cyclase Stimulator CY6463 Reveals its Therapeutic Potential in Neurodegenerative Diseases. Front. Pharm. 2021, 12, 656561. [Google Scholar] [CrossRef] [PubMed]
- Kacirova, M.; Zelezna, B.; Blazkova, M.; Holubova, M.; Popelova, A.; Kunes, J.; Sediva, B.; Maletinska, L. Aging and high-fat diet feeding lead to peripheral insulin resistance and sex-dependent changes in brain of mouse model of tau pathology THY-Tau22. J. Neuroinflammation 2021, 18, 141. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marcos, J.L.; Olivares-Barraza, R.; Ceballo, K.; Wastavino, M.; Ortiz, V.; Riquelme, J.; Martínez-Pinto, J.; Muñoz, P.; Cruz, G.; Sotomayor-Zárate, R. Obesogenic Diet-Induced Neuroinflammation: A Pathological Link between Hedonic and Homeostatic Control of Food Intake. Int. J. Mol. Sci. 2023, 24, 1468. https://doi.org/10.3390/ijms24021468
Marcos JL, Olivares-Barraza R, Ceballo K, Wastavino M, Ortiz V, Riquelme J, Martínez-Pinto J, Muñoz P, Cruz G, Sotomayor-Zárate R. Obesogenic Diet-Induced Neuroinflammation: A Pathological Link between Hedonic and Homeostatic Control of Food Intake. International Journal of Molecular Sciences. 2023; 24(2):1468. https://doi.org/10.3390/ijms24021468
Chicago/Turabian StyleMarcos, José Luis, Rossy Olivares-Barraza, Karina Ceballo, Melisa Wastavino, Víctor Ortiz, Julio Riquelme, Jonathan Martínez-Pinto, Pablo Muñoz, Gonzalo Cruz, and Ramón Sotomayor-Zárate. 2023. "Obesogenic Diet-Induced Neuroinflammation: A Pathological Link between Hedonic and Homeostatic Control of Food Intake" International Journal of Molecular Sciences 24, no. 2: 1468. https://doi.org/10.3390/ijms24021468
APA StyleMarcos, J. L., Olivares-Barraza, R., Ceballo, K., Wastavino, M., Ortiz, V., Riquelme, J., Martínez-Pinto, J., Muñoz, P., Cruz, G., & Sotomayor-Zárate, R. (2023). Obesogenic Diet-Induced Neuroinflammation: A Pathological Link between Hedonic and Homeostatic Control of Food Intake. International Journal of Molecular Sciences, 24(2), 1468. https://doi.org/10.3390/ijms24021468