
Integrative Proteomics and Metabolomics Analysis Reveals the Role of Small Signaling Peptide Rapid Alkalinization Factor 34 (RALF34) in Cucumber Roots


 , , , , , , ,
, , , , , , ,  ,
, 

Abstract
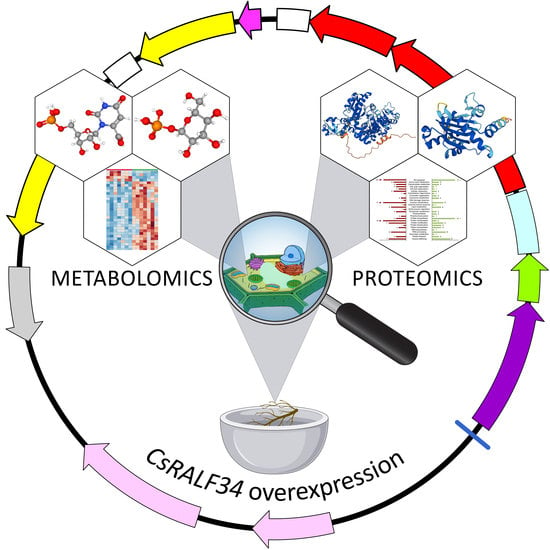
:
1. Introduction
2. Results
2.1. Overexpression of CsRALF34 Does Not Affect Lateral Root Initiation in Cucumber
2.2. Treatment of Cucumber Roots with Synthetic CsRALF34 Affects Root Growth but Not Lateral Root Initiation
2.3. Overexpression of CsRALF34 Does Not Affect the Expression of CsGATA14, CsGATA24, and CsE2F/DP Genes
2.4. Analysis of Biochemical Stress Response Markers in Cucumis Sativus Roots
2.5. Metabolomics Analysis
2.5.1. Analysis of Primary Metabolites
2.5.2. Analysis of Semi-Polar Secondary Metabolites
2.6. Protein Isolation and Tryptic Digestion
2.7. Annotation of Cucumis sativus Proteins
2.8. Label-Free Quantification
2.9. Functional Annotation of CsRALF34-Regulated Proteins
2.10. The Effects of CsRALF34 on Root Metabolic and Signaling Pathways
3. Discussion
3.1. CsRALF34 Is Not Involved in the Lateral Root Initiation
3.2. The Functions of CsRALF34
3.2.1. Activation of Protein Biosynthesis
3.2.2. Inhibition of Root Growth and Regulation of Cell Proliferation
3.2.3. Impact of CsRALF34 on ROS Signaling and Stress Adaptation
3.2.4. RALF-Related Dynamics of Cellular Metabolism
3.2.5. RALF34 as a Modulator of Phytohormone Responses
4. Materials and Methods
4.1. Plant Material and Bacterial Strains
4.2. Reagents
4.3. Phylogeny and Bioinformatics
4.4. RT-qPCR Assays
4.5. Lateral Root Initiation Index (ILRI) Estimation
4.6. Treatments with Synthetic CsRALF34 Peptide
4.7. Molecular Cloning and Vector Design
4.8. Plant Transformation and Handling for CsRALF34 Overexpression Assays
4.9. Determination of Hydrogen Peroxide Contents
4.10. Determination of Lipid Peroxidation Product Contents
4.11. Determination of Lipid Hydroperoxide Contents
4.12. Determination of Ascorbic Acid Contents
4.13. Analysis of Primary Metabolites
4.14. Analysis of Semi-Polar Secondary Metabolites
4.15. Processing of the Acquired Metabolomics Data
4.16. Protein Isolation and Determination
4.17. Tryptic Digestion and Sample Pre-Cleaning
4.18. Solid Phase Extraction
4.19. Nano LC-MS/MS
4.20. Data Post-Processing and Analysis
4.21. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Fletcher, J.C.; Brand, U.; Running, M.P.; Simon, R.; Meyerowitz, E.M. Signaling of cell fate decisions by CLAVATA3 in Arabidopsis shoot meristems. Science 1999, 283, 1911–1914. [Google Scholar] [CrossRef] [PubMed]
- Matsubayashi, Y.; Sakagami, Y. Phytosulfokine, sulfated peptides that induce the proliferation of single mesophyll cells of Asparagus officinalis L. Proc. Natl. Acad. Sci. USA 1996, 93, 7623–7627. [Google Scholar] [CrossRef] [PubMed]
- Pearce, G.; Strydom, D.; Johnson, S.; Ryan, C.A. A polypeptide from tomato leaves induces wound-inducible proteinase inhibitor proteins. Science 1991, 253, 895–897. [Google Scholar] [CrossRef] [PubMed]
- Schopfer, C.R.; Nasrallah, M.E.; Nasrallah, J.B. The male determinant of self-incompatibility in Brassica. Science 1999, 286, 1697–1700. [Google Scholar] [CrossRef]
- Araya, T.; Miyamoto, M.; Wibowo, J.; Suzuki, A.; Kojima, S.; Tsuchiya, Y.N.; Sawa, S.; Fukuda, H.; von Wirén, N.; Takahashi, H. CLE-CLAVATA1 peptide-receptor signaling module regulates the expansion of plant root systems in a nitrogen-dependent manner. Proc. Natl. Acad. Sci. USA 2014, 111, 2029–2034. [Google Scholar] [CrossRef]
- Chilley, P.M.; Casson, S.A.; Tarkowski, P.; Hawkins, N.; Wang, K.L.; Hussey, P.J.; Beale, M.; Ecker, J.R.; Sandberg, G.K.; Lindsey, K. The POLARIS peptide of Arabidopsis regulates auxin transport and root growth via effects on ethylene signaling. Plant Cell 2006, 18, 3058–3072. [Google Scholar] [CrossRef]
- Delay, C.; Imin, N.; Djordjevic, M.A. CEP genes regulate root and shoot development in response to environmental cues and are specific to seed plants. J. Exp. Bot. 2013, 64, 5383–5394. [Google Scholar] [CrossRef] [PubMed]
- Kumpf, R.P.; Shi, C.L.; Larrieu, A.; Sto, I.M.; Butenko, M.A.; Peret, B.; Riiser, E.S.; Bennett, M.J.; Aalen, R.B. Floral organ abscission peptide IDA and its HAE/HSL2 receptors control cell separation during lateral root emergence. Proc. Natl. Acad. Sci. USA 2013, 110, 5235–5240. [Google Scholar] [CrossRef]
- Yamaguchi, Y.L.; Ishida, T.; Sawa, S. CLE peptides and their signaling pathways in plant development. J. Exp. Bot. 2016, 67, 4813–4826. [Google Scholar] [CrossRef]
- Jourquin, J.; Fukaki, H.; Beeckman, T. Peptide-receptor signaling controls lateral root development. Plant Physiol. 2020, 182, 1645–1656. [Google Scholar] [CrossRef]
- Pearce, G.; Moura, D.S.; Stratmann, J.; Ryan, C.A. RALF, a 5-kDa ubiquitous polypeptide in plants, arrests root growth and development. Proc. Natl. Acad. Sci. USA 2001, 98, 12843–12847. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, M.R.; Haruta, M.; Moura, D.S. Twenty years of progress in physiological and biochemical investigation of RALF peptides. Plant Physiol. 2020, 182, 1657–1666. [Google Scholar] [CrossRef]
- Bergonci, T.; Ribeiro, B.; Ceciliato, P.H.O.; Guerrero-Abad, J.C.; Silva-Filho, M.C.; Moura, D.S. Arabidopsis thaliana RALF1 opposes brassinosteroid effects on root cell elongation and lateral root formation. J. Exp. Bot. 2014, 65, 2219–2230. [Google Scholar] [CrossRef]
- Murphy, E.; Vu, L.D.; Van den Broeck, L.; Lin, Z.; Ramakrishna, P.; van de Cotte, B.; Gaudinier, A.; Goh, T.; Slane, D.; Beeckman, T.; et al. RALFL34 regulates formative cell divisions in Arabidopsis pericycle during lateral root initiation. J. Exp. Bot. 2016, 67, 4863–4875. [Google Scholar] [CrossRef] [PubMed]
- Abarca, A.; Franck, C.M.; Zipfel, C. Family-wide evaluation of RAPID ALKALINIZATION FACTOR peptides. Plant Physiol. 2021, 187, 996–1010. [Google Scholar] [CrossRef] [PubMed]
- Combier, J.-P.; Küster, H.; Journet, E.-P.; Hohnjec, N.; Gamas, P.; Niebel, A. Evidence for the involvement in nodulation of the two small putative regulatory peptide-encoding genes MtRALFL1 and MtDVL1. Mol. Plant-Microbe Interact. 2008, 21, 1118–1127. [Google Scholar] [CrossRef]
- Ge, Z.; Bergonci, T.; Zhao, Y.; Zou, Y.; Du, S.; Liu, M.-C.; Luo, X.; Ruan, H.; García-Valencia, L.E.; Zhong, S.; et al. Arabidopsis pollen tube integrity and sperm release are regulated by RALF-mediated signaling. Science 2017, 358, 1596–1600. [Google Scholar] [CrossRef]
- Zhong, S.; Li, L.; Wang, Z.; Ge, Z.; Li, Q.; Bleckmann, A.; Wang, J.; Song, Z.; Shi, Y.; Liu, T.; et al. RALF peptide signaling controls the polytubey block in Arabidopsis. Science 2022, 375, 290–296. [Google Scholar] [CrossRef]
- Germain, H.; Chevalier, É.; Caron, S.; Matton, D.P. Characterization of five RALF-like genes from Solanum chacoense provides support for a developmental role in plants. Planta 2005, 220, 447–454. [Google Scholar] [CrossRef]
- Negrini, F.; O’Grady, K.; Hyvӧnen, M.; Folta, K.; Baraldi, E. Genomic structure and transcript analysis of the Rapid Alkalinization Factor (RALF) gene family during host-pathogen crosstalk in Fragaria vesca and Fragaria x ananassa strawberry. PLoS ONE 2020, 15, e0226448. [Google Scholar] [CrossRef]
- Zhang, X.; Peng, H.; Zhu, S.; Xing, J.; Li, X.; Zhu, Z.; Zheng, J.; Wang, L.; Wang, B.; Chen, J.; et al. Nematode-encoded RALF peptide mimics facilitate parasitism of plants through the FERONIA receptor kinase. Mol. Plant 2020, 13, 1434–1454. [Google Scholar] [CrossRef] [PubMed]
- De Rybel, B.; Vassileva, V.; Parizot, B.; Demeulenaere, M.; Grunewald, W.; Audenaert, D.; Van Campenhout, J.; Overvoorde, P.; Jansen, L.; Vanneste, S.; et al. A novel Aux/IAA28 signaling cascade activates GATA23-dependent specification of lateral root founder cell identity. Curr. Biol. 2010, 20, 1697–1706. [Google Scholar] [CrossRef] [PubMed]
- Hirakawa, Y. Non-cell-autonomous control of vascular stem cell fate by a CLE peptide/receptor system. Proc. Natl. Acad. Sci. USA 2008, 105, 15208–15213. [Google Scholar] [CrossRef] [PubMed]
- Chevalier, E.; Loubert-Hudon, A.; Matton, D.P. ScRALF3, a secreted RALF-like peptide involved in cell-cell communication between the sporophyte and the female gametophyte in a solanaceous species. Plant J. 2013, 73, 1019–1033. [Google Scholar] [CrossRef] [PubMed]
- Kiryushkin, A.S.; Ilina, E.L.; Guseva, E.D.; Pawlowski, K.; Demchenko, K.N. Lateral root initiation in cucumber (Cucumis sativus): What does the expression pattern of Rapid Alkalinization Factor 34 (RALF34) tell us? Int. J. Mol. Sci. 2023, 23. submitted for publication. [Google Scholar]
- Ge, Z.; Dresselhaus, T.; Qu, L.-J. How CrRLK1L receptor complexes perceive RALF signals. Trends Plant Sci. 2019, 24, 978–981. [Google Scholar] [CrossRef] [PubMed]
- Solis-Miranda, J.; Quinto, C. The CrRLK1L subfamily: One of the keys to versatility in plants. Plant Physiol. Biochem. 2021, 166, 88–102. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Fu, Q.; Xu, F.; Zheng, H.; Yu, F. New paradigms in cell adaptation: Decades of discoveries on the CrRLK1L receptor kinase signalling network. New Phytol. 2021, 232, 1168–1183. [Google Scholar] [CrossRef]
- Xie, Y.; Sun, P.; Li, Z.; Zhang, F.; You, C.; Zhang, Z. FERONIA receptor kinase integrates with hormone signaling to regulate plant growth, development, and responses to environmental stimuli. Int. J. Mol. Sci. 2022, 23, 3730. [Google Scholar] [CrossRef]
- Baez, L.A.; Tichá, T.; Hamann, T. Cell wall integrity regulation across plant species. Plant Mol. Biol. 2022, 109, 483–504. [Google Scholar] [CrossRef]
- Abd Algfoor, Z.; Shahrizal Sunar, M.; Abdullah, A.; Kolivand, H. Identification of metabolic pathways using pathfinding approaches: A systematic review. Brief. Funct. Genom. 2016, 16, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Gargallo-Garriga, A.; Sardans, J.; Pérez-Trujillo, M.; Rivas-Ubach, A.; Oravec, M.; Vecerova, K.; Urban, O.; Jentsch, A.; Kreyling, J.; Beierkuhnlein, C.; et al. Opposite metabolic responses of shoots and roots to drought. Sci. Rep. 2014, 4, 6829. [Google Scholar] [CrossRef] [PubMed]
- Novák, J.; Černý, M.; Pavlů, J.; Zemánková, J.; Skalák, J.; Plačková, L.; Brzobohatý, B. Roles of proteome dynamics and cytokinin signaling in root to hypocotyl ratio changes induced by shading roots of Arabidopsis seedlings. Plant Cell Physiol. 2015, 56, 1006–1018. [Google Scholar] [CrossRef] [PubMed]
- Hildreth, S.B.; Foley, E.E.; Muday, G.K.; Helm, R.F.; Winkel, B.S.J. The dynamic response of the Arabidopsis root metabolome to auxin and ethylene is not predicted by changes in the transcriptome. Sci. Rep. 2020, 10, 679. [Google Scholar] [CrossRef]
- Gogolev, Y.V.; Ahmar, S.; Akpinar, B.A.; Budak, H.; Kiryushkin, A.S.; Gorshkov, V.Y.; Hensel, G.; Demchenko, K.N.; Kovalchuk, I.; Mora-Poblete, F.; et al. OMICs, epigenetics, and genome editing techniques for food and nutritional security. Plants 2021, 10, 1423. [Google Scholar] [CrossRef]
- Berka, M.; Luklová, M.; Dufková, H.; Berková, V.; Novák, J.; Saiz-Fernández, I.; Rashotte, A.M.; Brzobohatý, B.; Černý, M. Barley root proteome and metabolome in response to cytokinin and abiotic stimuli. Front. Plant Sci. 2020, 11, 590337. [Google Scholar] [CrossRef]
- Ludovic, C.G.; Alexander, L.; Ruedi, A. Mass spectrometry applied to bottom-up proteomics: Entering the high-throughput era for hypothesis testing. Annu. Rev. Anal. Chem. 2016, 9, 449–472. [Google Scholar] [CrossRef]
- Danko, K.; Lukasheva, E.; Zhukov, V.A.; Zgoda, V.; Frolov, A. Detergent-assisted protein digestion—On the way to avoid the key bottleneck of shotgun bottom-up proteomics. Int. J. Mol. Sci. 2022, 23, 13903. [Google Scholar] [CrossRef]
- Mergner, J.; Frejno, M.; List, M.; Papacek, M.; Chen, X.; Chaudhary, A.; Samaras, P.; Richter, S.; Shikata, H.; Messerer, M.; et al. Mass-spectrometry-based draft of the Arabidopsis proteome. Nature 2020, 579, 409–414. [Google Scholar] [CrossRef]
- Frolov, A.; Mamontova, T.; Ihling, C.; Lukasheva, E.; Bankin, M.; Chantseva, V.; Vikhnina, M.; Soboleva, A.; Shumilina, J.; Mavropolo-Stolyarenko, G.; et al. Mining seed proteome: From protein dynamics to modification profiles. Biol. Commun. 2018, 63, 43–58. [Google Scholar] [CrossRef]
- Wang, L.; Yang, T.; Wang, B.; Lin, Q.; Zhu, S.; Li, C.; Ma, Y.; Tang, J.; Xing, J.; Li, X.; et al. RALF1-FERONIA complex affects splicing dynamics to modulate stress responses and growth in plants. Sci. Adv. 2020, 6, eaaz1622. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Zhang, D.; Zhuang, J.; Huang, Y.; Mu, Y.; Lv, S. Study on the correlation between gene expression and enzyme activity of seven key enzymes and ginsenoside content in ginseng in over time in Ji’an, China. Int. J. Mol. Sci. 2017, 18, 2682. [Google Scholar] [CrossRef]
- Cantarello, C.; Volpe, V.; Azzolin, C.; Bertea, C. Modulation of enzyme activities and expression of genes related to primary and secondary metabolism in response to UV-B stress in cucumber (Cucumis sativus L.). J. Plant Interact. 2005, 1, 151–161. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, J.; Yao, L.; Li, B.; Ma, X.; Si, E.; Yang, K.; Li, C.; Shang, X.; Meng, Y.; et al. Combined proteomic and metabolomic analysis of the molecular mechanism underlying the response to salt stress during seed germination in barley. Int. J. Mol. Sci. 2022, 23, 10515. [Google Scholar] [CrossRef]
- Paudel, G.; Bilova, T.; Schmidt, R.; Greifenhagen, U.; Berger, R.; Tarakhovskaya, E.; Stöckhardt, S.; Balcke, G.U.; Humbeck, K.; Brandt, W.; et al. Osmotic stress is accompanied by protein glycation in Arab. Thaliana J. Exp. Bot. 2016, 67, 6283–6295. [Google Scholar] [CrossRef]
- Kiryushkin, A.S.; Ilina, E.L.; Puchkova, V.A.; Guseva, E.D.; Pawlowski, K.; Demchenko, K.N. Lateral root initiation in the parental root meristem of cucurbits: Old players in a new position. Front. Plant Sci. 2019, 10, 365. [Google Scholar] [CrossRef]
- Frolov, A.; Bluher, M.; Hoffmann, R. Glycation sites of human plasma proteins are affected to different extents by hyperglycemic conditions in type 2 diabetes mellitus. Anal. Bioanal. Chem. 2014, 406, 5755–5763. [Google Scholar] [CrossRef] [PubMed]
- Shimotohno, A.; Aki, S.S.; Takahashi, N.; Umeda, M. Regulation of the plant cell cycle in response to hormones and the environment. Annu. Rev. Plant Biol. 2021, 72, 273–296. [Google Scholar] [CrossRef]
- Dekomah, S.D.; Bi, Z.; Dormatey, R.; Wang, Y.; Haider, F.U.; Sun, C.; Yao, P.; Bai, J. The role of CDPKs in plant development, nutrient and stress signaling. Front. Genet. 2022, 13, 996203. [Google Scholar] [CrossRef]
- Asano, T.; Hayashi, N.; Kobayashi, M.; Aoki, N.; Miyao, A.; Mitsuhara, I.; Ichikawa, H.; Komatsu, S.; Hirochika, H.; Kikuchi, S.; et al. A rice calcium-dependent protein kinase OsCPK12 oppositely modulates salt-stress tolerance and blast disease resistance. Plant J. 2012, 69, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Nikonorova, N.; Murphy, E.; De Lima, C.F.F.; Zhu, S.; Van de Cotte, B.; Vu, L.D.; Balcerowicz, D.; Li, L.; Kong, X.; De Rop, G.; et al. The Arabidopsis root tip (phospho)proteomes at growth-promoting versus growth-repressing conditions reveal novel root growth regulators. Cells 2021, 10, 1665. [Google Scholar] [CrossRef] [PubMed]
- De Veylder, L.; Beeckman, T.; Beemster, G.T.S.; de Almeida Engler, J.; Ormenese, S.; Maes, S.; Naudts, M.; Van Der Schueren, E.; Jacqmard, A.; Engler, G.; et al. Control of proliferation, endoreduplication and differentiation by the Arabidopsis E2Fa–DPa transcription factor. EMBO J. 2002, 21, 1360–1368. [Google Scholar] [CrossRef] [PubMed]
- Berckmans, B.; Vassileva, V.; Schmid, S.P.C.; Maes, S.; Parizot, B.; Naramoto, S.; Magyar, Z.; Kamei, C.L.A.; Koncz, C.; Bogre, L.; et al. Auxin-dependent cell cycle reactivation through transcriptional regulation of Arabidopsis E2Fa by lateral organ boundary proteins. Plant Cell 2011, 23, 3671–3683. [Google Scholar] [CrossRef] [PubMed]
- Shumilina, J.; Gorbach, D.; Popova, V.; Tsarev, A.; Kuznetsova, A.; Grashina, M.; Dorn, M.; Lukasheva, E.; Osmolovskaya, N.; Romanovskaya, E.; et al. Protein glycation and drought response of pea (Pisum sativum L.) root nodule proteome: A proteomics approach. Biol. Commun. 2021, 66, 210–224. [Google Scholar] [CrossRef]
- Mamontova, T.; Lukasheva, E.; Mavropolo-Stolyarenko, G.; Proksch, C.; Bilova, T.; Kim, A.; Babakov, V.; Grishina, T.; Hoehenwarter, W.; Medvedev, S.; et al. Proteome map of pea (Pisum sativum L.) embryos containing different amounts of residual chlorophylls. Int. J. Mol. Sci. 2018, 19, 4066. [Google Scholar] [CrossRef] [PubMed]
- Frolov, A.; Bilova, T.; Paudel, G.; Berger, R.; Balcke, G.U.; Birkemeyer, C.; Wessjohann, L.A. Early responses of mature Arabidopsis thaliana plants to reduced water potential in the agar-based polyethylene glycol infusion drought model. J. Plant Physiol. 2017, 208, 70–83. [Google Scholar] [CrossRef]
- Haruta, M.; Constabel, C.P. Rapid alkalinization factors in poplar cell cultures. Peptide isolation, cDNA cloning, and differential expression in leaves and methyl jasmonate-treated cells. Plant Physiol. 2003, 131, 814–823. [Google Scholar] [CrossRef]
- Strzalka, W.; Ziemienowicz, A. Proliferating cell nuclear antigen (PCNA): A key factor in DNA replication and cell cycle regulation. Ann. Bot. 2011, 107, 1127–1140. [Google Scholar] [CrossRef]
- Moldovan, G.-L.; Pfander, B.; Jentsch, S. PCNA, the maestro of the replication fork. Cell 2007, 129, 665–679. [Google Scholar] [CrossRef]
- Stegmann, M.; Monaghan, J.; Smakowska-Luzan, E.; Rovenich, H.; Lehner, A.; Holton, N.; Belkhadir, Y.; Zipfel, C. The receptor kinase FER is a RALF-regulated scaffold controlling plant immune signaling. Science 2017, 355, 287–289. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Osmolovskaya, N.; Shumilina, J.; Kim, A.; Didio, A.; Grishina, T.; Bilova, T.; Keltsieva, O.A.; Zhukov, V.; Tikhonovich, I.; Tarakhovskaya, E.; et al. Methodology of drought stress research: Experimental setup and physiological characterization. Int. J. Mol. Sci. 2018, 19, 4089. [Google Scholar] [CrossRef] [PubMed]
- Muthukumar, K.; Rajakumar, S.; Sarkar, M.N.; Nachiappan, V. Glutathione peroxidase3 of Saccharomyces cerevisiae protects phospholipids during cadmium-induced oxidative stress. Antonie Van Leeuwenhoek 2011, 99, 761–771. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.; Lim, C.W.; Lee, S.C. Core components of abscisic acid signaling and their post-translational modification. Front. Plant Sci. 2022, 13, 895698. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Wilson, A.J.; Zhang, X.C.; Thoms, D.; Sohrabi, R.; Song, S.; Geissmann, Q.; Liu, Y.; Walgren, L.; He, S.Y.; et al. FERONIA restricts Pseudomonas in the rhizosphere microbiome via regulation of reactive oxygen species. Nat. Plants 2021, 7, 644–654. [Google Scholar] [CrossRef]
- Shi, S.; Li, S.; Asim, M.; Mao, J.; Xu, D.; Ullah, Z.; Liu, G.; Wang, Q.; Liu, H. The Arabidopsis calcium-dependent protein kinases (CDPKs) and their roles in plant growth regulation and abiotic stress responses. Int. J. Mol. Sci. 2018, 19, 1900. [Google Scholar] [CrossRef]
- Haruta, M.; Sabat, G.; Stecker, K.; Minkoff, B.B.; Sussman, M.R. A peptide hormone and its receptor protein kinase regulate plant cell expansion. Science 2014, 343, 408–411. [Google Scholar] [CrossRef]
- Chen, J.; Yu, F.; Liu, Y.; Du, C.; Li, X.; Zhu, S.; Wang, X.; Lan, W.; Rodriguez, P.L.; Liu, X.; et al. FERONIA interacts with ABI2-type phosphatases to facilitate signaling cross-talk between abscisic acid and RALF peptide in Arabidopsis. Proc. Natl. Acad. Sci. USA 2016, 113, E5519–E5527. [Google Scholar] [CrossRef]
- Pirrung, M.C. Ethylene biosynthesis from 1-aminocyclopropanecarboxylic acid. Acc. Chem. Res. 1999, 32, 711–718. [Google Scholar] [CrossRef]
- Mao, D.; Yu, F.; Li, J.; Van de poel, B.; Tan, D.; Li, J.; Liu, Y.; Li, X.; Dong, M.; Chen, L.; et al. FERONIA receptor kinase interacts with S-adenosylmethionine synthetase and suppresses S-adenosylmethionine production and ethylene biosynthesis in Arabidopsis. Plant Cell Environ. 2015, 38, 2566–2574. [Google Scholar] [CrossRef]
- Deslauriers, S.D.; Larsen, P.B. FERONIA is a key modulator of brassinosteroid and ethylene responsiveness in Arabidopsis hypocotyls. Mol. Plant 2010, 3, 626–640. [Google Scholar] [CrossRef] [PubMed]
- Mariconti, L.; Pellegrini, B.; Cantoni, R.; Stevens, R.; Bergounioux, C.; Cella, R.; Albani, D. The E2F family of transcription factors from Arabidopsis thaliana: Novel and conserved components of the RETINOBLASTOMA/E2F pathway in plants. J. Biol. Chem. 2002, 277, 9911–9919. [Google Scholar] [CrossRef] [PubMed]
- Magyar, Z.; Atanassova, A.; De Veylder, L.; Rombauts, S.; Inzé, D. Characterization of two distinct DP-related genes from Arabidopsis thaliana. FEBS Lett. 2000, 486, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Berardini, T.Z.; Reiser, L.; Li, D.; Mezheritsky, Y.; Muller, R.; Strait, E.; Huala, E. The arabidopsis information resource: Making and mining the “gold standard” annotated reference plant genome. Genesis 2015, 53, 474–485. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, Z.; Yan, P.; Huang, S.; Fei, Z.; Lin, K. RNA-Seq improves annotation of protein-coding genes in the cucumber genome. BMC Genom. 2011, 12, 540. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Wu, S.; Bai, Y.; Sun, H.; Jiao, C.; Guo, S.; Zhao, K.; Blanca, J.; Zhang, Z.; Huang, S.; et al. Cucurbit Genomics Database (CuGenDB): A central portal for comparative and functional genomics of cucurbit crops. Nucleic Acids Res. 2019, 47, D1128–D1136. [Google Scholar] [CrossRef]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Yang, Z. Maximum likelihood phylogenetic estimation from DNA sequences with variable rates over sites: Approximate methods. J. Mol. Evol. 1994, 39, 306–314. [Google Scholar] [CrossRef]
- Jones, D.T.; Taylor, W.R.; Thornton, J.M. The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci. 1992, 8, 275–282. [Google Scholar] [CrossRef]
- Wan, H.; Zhao, Z.; Qian, C.; Sui, Y.; Malik, A.A.; Chen, J. Selection of appropriate reference genes for gene expression studies by quantitative real-time polymerase chain reaction in cucumber. Anal. Biochem. 2010, 399, 257–261. [Google Scholar] [CrossRef] [PubMed]
- Dubrovsky, J.G.; Soukup, A.; Napsucialy-Mendivil, S.; Jeknic, Z.; Ivanchenko, M.G. The lateral root initiation index: An integrative measure of primordium formation. Ann. Bot. 2009, 103, 807–817. [Google Scholar] [CrossRef] [PubMed]
- Ilina, E.L.; Kiryushkin, A.S.; Semenova, V.A.; Demchenko, N.P.; Pawlowski, K.; Demchenko, K.N. Lateral root initiation and formation within the parental root meristem of Cucurbita pepo: Is auxin a key player? Ann. Bot. 2018, 122, 873–888. [Google Scholar] [CrossRef] [PubMed]
- Demchenko, K.N.; Demchenko, N.P. Changes of root structure in connection with the development of lateral root primordia in wheat and pumpkins. In Recent Advances of Plant Root Structure and Function; Developments in Plant and Soil Sciences; Gašparíková, O., Čiamporová, M., Mistrík, I., Baluška, F., Eds.; Springer: Dordrecht, The Netherlands, 2001; Volume 90, pp. 39–47. [Google Scholar] [CrossRef]
- Hoagland, D.R.; Arnon, D.T. The water-culture method for growing plants without soil. In California Agriculture Experiment Station. Circular 347; University of California: Berkeley, CA, USA, 1938; pp. 1–39. [Google Scholar]
- Gancheva, M.S.; Dodueva, I.E.; Lebedeva, M.A.; Tvorogova, V.E.; Tkachenko, A.A.; Lutova, L.A. Identification, expression, and functional analysis of CLE genes in radish (Raphanus sativus L.) storage root. BMC Plant Biol. 2016, 16, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Morato do Canto, A.; Ceciliato, P.H.O.; Ribeiro, B.; Ortiz Morea, F.A.; Franco Garcia, A.A.; Silva-Filho, M.C.; Moura, D.S. Biological activity of nine recombinant AtRALF peptides: Implications for their perception and function in Arabidopsis. Plant Physiol. Biochem. 2014, 75, 45–54. [Google Scholar] [CrossRef]
- Limpens, E.; Ramos, J.; Franken, C.; Raz, V.; Compaan, B.; Franssen, H.; Bisseling, T.; Geurts, R. RNA interference in Agrobacterium rhizogenes-transformed roots of Arabidopsis and Medicago truncatula. J. Exp. Bot. 2004, 55, 983–992. [Google Scholar] [CrossRef]
- Hornung, E.; Krueger, C.; Pernstich, C.; Gipmans, M.; Porzel, A.; Feussner, I. Production of (10E,12Z)-conjugated linoleic acid in yeast and tobacco seeds. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2005, 1738, 105–114. [Google Scholar] [CrossRef]
- Ilina, E.L.; Logachov, A.A.; Laplaze, L.; Demchenko, N.P.; Pawlowski, K.; Demchenko, K.N. Composite Cucurbita pepo plants with transgenic roots as a tool to study root development. Ann. Bot. 2012, 110, 479–489. [Google Scholar] [CrossRef]
- Chantseva, V.; Bilova, T.; Smolikova, G.; Frolov, A.; Medvedev, S. 3D-clinorotation induces specific alterations in metabolite profiles of germinating Brassica napus L. seeds. Biol. Commun. 2019, 64, 55–74. [Google Scholar] [CrossRef]
- Soboleva, A.; Frolova, N.; Bureiko, K.; Shumilina, J.; Balcke, G.U.; Zhukov, V.A.; Tikhonovich, I.A.; Frolov, A. Dynamics of reactive carbonyl species in pea root nodules in response to polyethylene glycol (PEG)-induced osmotic stress. Int. J. Mol. Sci. 2022, 23, 2726. [Google Scholar] [CrossRef]
- Leonova, T.; Popova, V.; Tsarev, A.; Henning, C.; Antonova, K.; Rogovskaya, N.; Vikhnina, M.; Baldensperger, T.; Soboleva, A.; Dinastia, E.; et al. Does protein glycation impact on the drought-related changes in metabolism and nutritional properties of mature pea (Pisum sativum L.) seeds? Int. J. Mol. Sci. 2020, 21, 567. [Google Scholar] [CrossRef] [PubMed]
- Pang, Z.; Chong, J.; Zhou, G.; de Lima Morais, D.A.; Chang, L.; Barrette, M.; Gauthier, C.; Jacques, P.-É.; Li, S.; Xia, J. MetaboAnalyst 5.0: Narrowing the gap between raw spectra and functional insights. Nucleic Acids Res. 2021, 49, W388–W396. [Google Scholar] [CrossRef] [PubMed]
- Frolov, A.; Didio, A.; Ihling, C.; Chantzeva, V.; Grishina, T.; Hoehenwarter, W.; Sinz, A.; Smolikova, G.; Bilova, T.; Medvedev, S. The effect of simulated microgravity on the Brassica napus seedling proteome. Funct. Plant Biol. 2018, 45, 440–452. [Google Scholar] [CrossRef] [PubMed]
- Greifenhagen, U.; Frolov, A.; Bluher, M.; Hoffmann, R. Plasma proteins modified by advanced glycation end products (AGEs) reveal site-specific susceptibilities to glycemic control in patients with type 2 diabetes. J. Biol. Chem. 2016, 291, 9610–9616. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Spiller, S.; Frolov, A.; Hoffmann, R. Quantification of specific glycation sites in human serum albumin as prospective type 2 diabetes mellitus biomarkers. Protein Peptide Lett. 2017, 24, 887–896. [Google Scholar] [CrossRef]
- Eng, J.K.; McCormack, A.L.; Yates, J.R. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 1994, 5, 976–989. [Google Scholar] [CrossRef]
- Eng, J.K.; Fischer, B.; Grossmann, J.; MacCoss, M.J. A fast SEQUEST cross correlation algorithm. J. Proteome Res. 2008, 7, 4598–4602. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing. Available online: https://www.R-project.org (accessed on 28 March 2023).
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Schwacke, R.; Ponce-Soto, G.Y.; Krause, K.; Bolger, A.M.; Arsova, B.; Hallab, A.; Gruden, K.; Stitt, M.; Bolger, M.E.; Usadel, B. MapMan4: A refined protein classification and annotation framework applicable to multi-omics data analysis. Mol. Plant 2019, 12, 879–892. [Google Scholar] [CrossRef]
- Lohse, M.; Nagel, A.; Herter, T.; MAY, P.; Schroda, M.; Zrenner, R.; Tohge, T.; Fernie, A.R.; Stitt, M.; Usadel, B. Mercator: A fast and simple web server for genome scale functional annotation of plant sequence data. Plant Cell Environ. 2014, 37, 1250–1258. [Google Scholar] [CrossRef] [PubMed]
- Horton, P.; Park, K.-J.; Obayashi, T.; Fujita, N.; Harada, H.; Adams-Collier, C.J.; Nakai, K. WoLF PSORT: Protein localization predictor. Nucleic Acids Res. 2007, 35, W585–W587. [Google Scholar] [CrossRef] [PubMed]
- Bu, D.; Luo, H.; Huo, P.; Wang, Z.; Zhang, S.; He, Z.; Wu, Y.; Zhao, L.; Liu, J.; Guo, J.; et al. KOBAS-i: Intelligent prioritization and exploratory visualization of biological functions for gene enrichment analysis. Nucleic Acids Res. 2021, 49, W317–W325. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Regulated Metabolites | FC * | log2(FC) | padjusted | −lg(p) | |
|---|---|---|---|---|---|
| DOWNREGULATED | D-mannose | 0.343 | −1.542 | 0.029 | 1.538 |
| cGMP | 0.391 | −1.356 | 0.006 | 2.242 | |
| gluconic/galacturonic acid | 0.648 | −0.625 | 0.047 | 1.324 | |
| UPREGULATED | nicotinic acid | 1.665 | 0.735 | 0.006 | 2.242 |
| adenosine | 1.745 | 0.803 | 0.009 | 2.053 | |
| glucose-1-phosphate | 1.813 | 0.858 | 0.049 | 1.310 | |
| arginine | 1.825 | 0.868 | 0.020 | 1.703 | |
| orotidine 5′-monophosphate | 1.916 | 0.938 | 0.049 | 1.310 | |
| ornithine | 2.151 | 1.105 | 0.011 | 1.965 | |
| 6-phosphogluconic acid | 2.346 | 1.230 | 0.029 | 1.538 | |
| Protein Name | Accession a | Direction of Alterations b | PC1 | FC c | padjustedd | Functional Annotation | Prediction of Localization |
|---|---|---|---|---|---|---|---|
| Thioredoxin | Csa2g362500.1 | up | 0.084 | 17.8 | 5.3 × 10–35 | redox homeostasis | cytosol |
| Glutathione peroxidase | Csa5g154200.1 | up | 0.081 | 17.2 | 4.4 × 10–35 | redox homeostasis | cytosol, mitochondrion |
| Ribosomal protein L19 | Csa4g001980.1 | up | 0.081 | 17.0 | 5.6 × 10–35 | protein biosynthesis | cytosol |
| CHP-rich zinc finger protein-like | Csa3g850660.1 | up | 0.081 | 17.2 | 6.8 × 10–35 | not assigned | cytosol |
| 40S ribosomal protein S24 | Csa2g012120.1 | up | 0.080 | 16.7 | 3.2 × 10–34 | protein biosynthesis | cytosol |
| Carnitine operon protein CaiE | Csa1g001460.1 | up | 0.079 | 16.6 | 5.6 × 10–35 | cellular respiration | mitochondrion |
| Wound/stress protein | Csa6g500520.1 | up | 0.078 | 16.5 | 4.4 × 10–35 | not assigned | endoplasmic reticulum |
| Porin/voltage-dependent anion-selective channel protein | Csa6g404150.1 | up | 0.078 | 16.5 | 5.6 × 10–35 | solute transport | mitochondrion |
| 60S ribosomal protein L18a | Csa4g608120.1 | up | 0.077 | 16.4 | 6.8 × 10–35 | protein biosynthesis | cytosol |
| Phosphofructokinase | Csa1g575110.1 | up | 0.077 | 16.0 | 8.7 × 10–35 | cellular respiration | cytosol |
| Eukaryotic translation initiation factor 3 subunit C | Csa3g223320.1 | up | 0.077 | 16.3 | 1.4 × 10–32 | protein biosynthesis | nucleus |
| Pleckstrin homology domain-containing family A member | Csa3g134900.1 | up | 0.077 | 16.3 | 5.0 × 10–16 | lipid metabolism | plasma membrane |
| 3-oxoacyl-[acyl-carrier-protein] synthase 3 | Csa5g160250.1 | up | 0.076 | 16.3 | 9.0 × 10–35 | lipid metabolism | plastid |
| RNA-binding protein 8A | Csa1g616290.1 | up | 0.076 | 16.1 | 8.3 × 10–35 | RNA processing | nucleus |
| Transmembrane 9 superfamily protein member | Csa1g256720.1 | up | 0.075 | 15.3 | 9.5 × 10–35 | not assigned | plasma membrane |
| 70 kDa heat shock protein | Csa2g070310.1 | down | −0.100 | 21.1 | 1.7 × 10–35 | protein homeostasis | cytosol |
| Ubiquitin-like domain-containing CTD phosphatase | Csa5g587150.1 | down | −0.089 | 18.7 | 4.5 × 10–35 | protein modification | nucleus |
| Phloem lectin | Csa4g500830.1 | down | −0.086 | 18.2 | 4.8 × 10–35 | not assigned | plastid |
| AP-4 complex accessory subunit tepsin | Csa3g462690.1 | down | −0.083 | 17.6 | 2.4 × 10–34 | vesicle trafficking | Golgi apparatus |
| 10 kDa chaperonin | Csa5g570400.1 | down | −0.083 | 17.6 | 4.8 × 10–35 | protein homeostasis | plastid |
| Pentatricopeptide repeat-containing protein | Csa3g840460.1 | down | −0.082 | 17.5 | 1.7 × 10–35 | not assigned | plastid |
| 60S ribosomal protein L32 | Csa1g462020.2 | down | −0.082 | 17.3 | 2.0 × 10–30 | protein biosynthesis | cytosol |
| Subtilisin-like serine protease | Csa5g187860.1 | down | −0.081 | 17.4 | 8.3 × 10–35 | protein homeostasis | vacuole |
| Protein MEMO1 | Csa2g382410.1 | down | −0.081 | 17.2 | 6.8 × 10–35 | not assigned | mitochondrion |
| NADH dehydrogenase 1 alpha subcomplex subunit 13 | Csa2g169710.1 | down | −0.079 | 16.9 | 5.6 × 10–35 | cellular respiration | mitochondrion |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shumilina, J.; Kiryushkin, A.S.; Frolova, N.; Mashkina, V.; Ilina, E.L.; Puchkova, V.A.; Danko, K.; Silinskaya, S.; Serebryakov, E.B.; Soboleva, A.; et al. Integrative Proteomics and Metabolomics Analysis Reveals the Role of Small Signaling Peptide Rapid Alkalinization Factor 34 (RALF34) in Cucumber Roots. Int. J. Mol. Sci. 2023, 24, 7654. https://doi.org/10.3390/ijms24087654
Shumilina J, Kiryushkin AS, Frolova N, Mashkina V, Ilina EL, Puchkova VA, Danko K, Silinskaya S, Serebryakov EB, Soboleva A, et al. Integrative Proteomics and Metabolomics Analysis Reveals the Role of Small Signaling Peptide Rapid Alkalinization Factor 34 (RALF34) in Cucumber Roots. International Journal of Molecular Sciences. 2023; 24(8):7654. https://doi.org/10.3390/ijms24087654
Chicago/Turabian StyleShumilina, Julia, Alexey S. Kiryushkin, Nadezhda Frolova, Valeria Mashkina, Elena L. Ilina, Vera A. Puchkova, Katerina Danko, Svetlana Silinskaya, Evgeny B. Serebryakov, Alena Soboleva, and et al. 2023. "Integrative Proteomics and Metabolomics Analysis Reveals the Role of Small Signaling Peptide Rapid Alkalinization Factor 34 (RALF34) in Cucumber Roots" International Journal of Molecular Sciences 24, no. 8: 7654. https://doi.org/10.3390/ijms24087654
APA StyleShumilina, J., Kiryushkin, A. S., Frolova, N., Mashkina, V., Ilina, E. L., Puchkova, V. A., Danko, K., Silinskaya, S., Serebryakov, E. B., Soboleva, A., Bilova, T., Orlova, A., Guseva, E. D., Repkin, E., Pawlowski, K., Frolov, A., & Demchenko, K. N. (2023). Integrative Proteomics and Metabolomics Analysis Reveals the Role of Small Signaling Peptide Rapid Alkalinization Factor 34 (RALF34) in Cucumber Roots. International Journal of Molecular Sciences, 24(8), 7654. https://doi.org/10.3390/ijms24087654