Analysis of Microbial Communities and Pathogen Detection in Domestic Sewage Using Metagenomic Sequencing


Abstract
:
1. Introduction
2. Materials and Methods
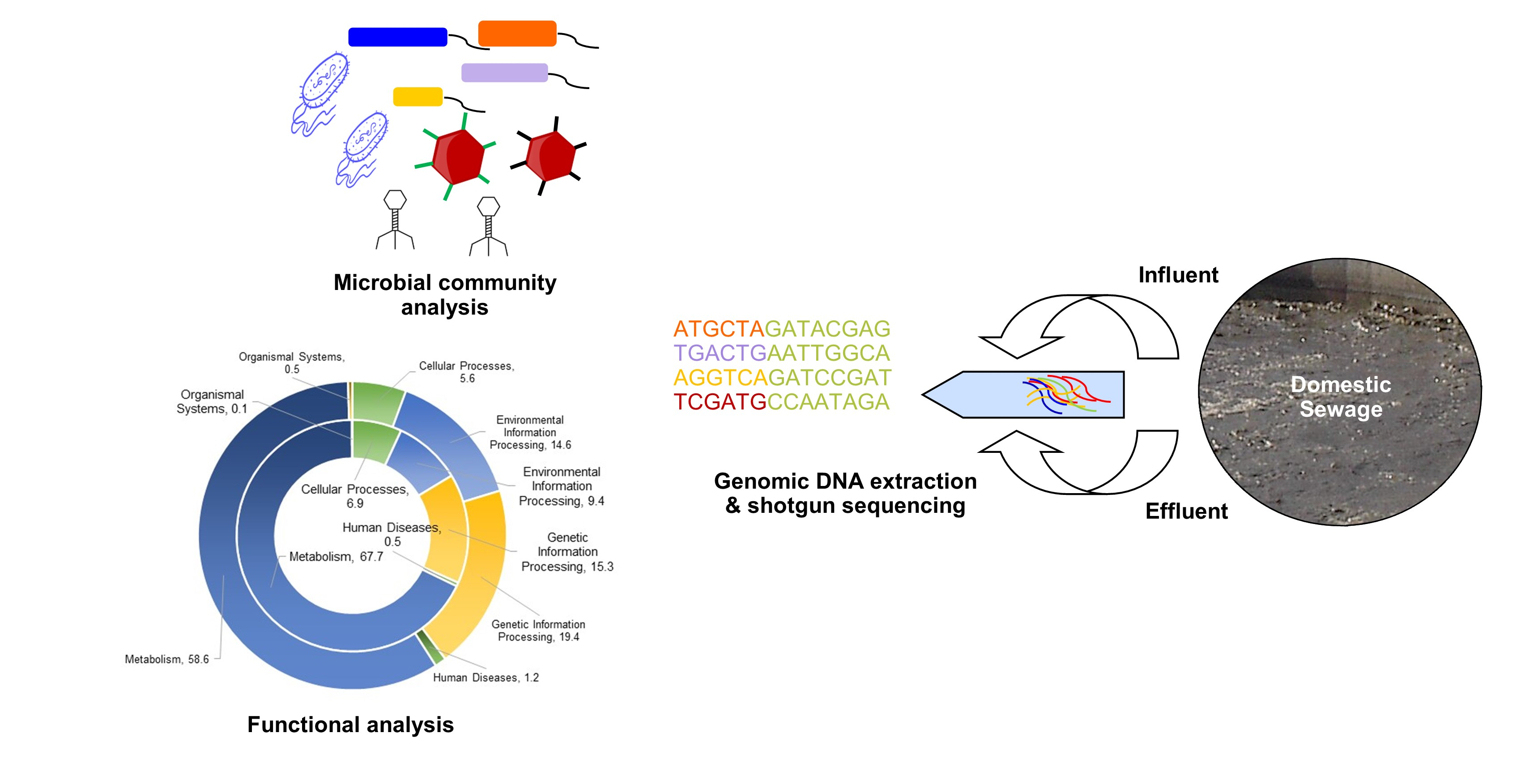
2.1. Sampling and Genomic DNA Extraction
2.2. Shotgun Sequencing and Data Analysis
3. Results and Discussion
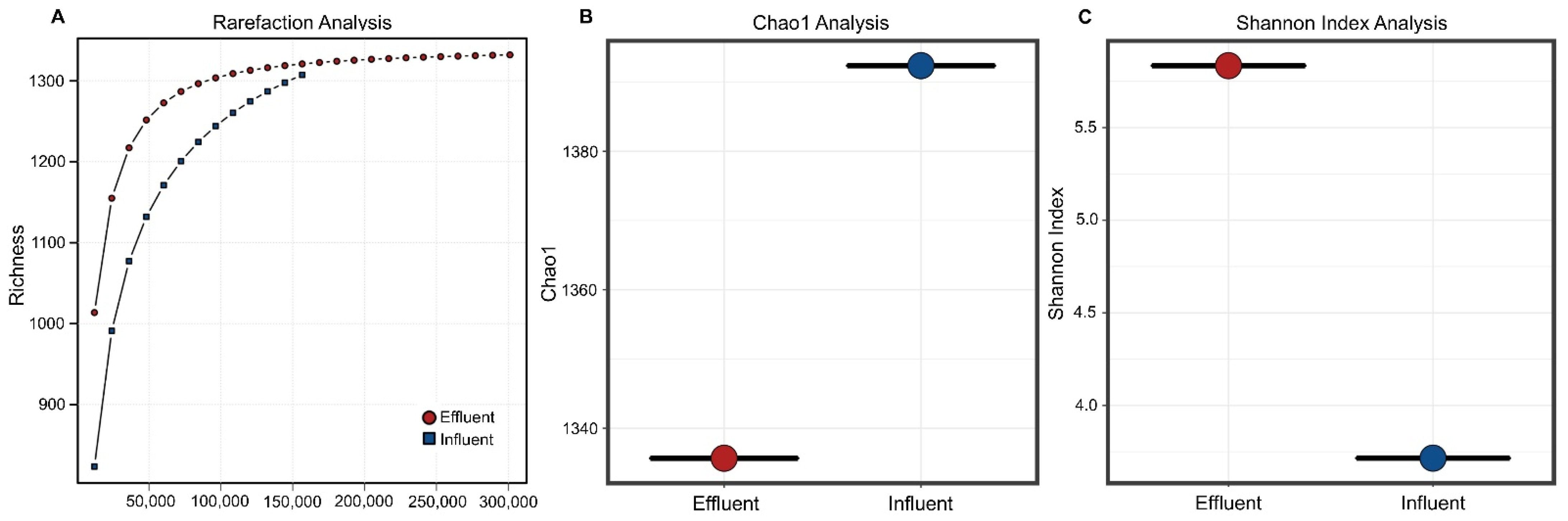
3.1. Microbial Diversity
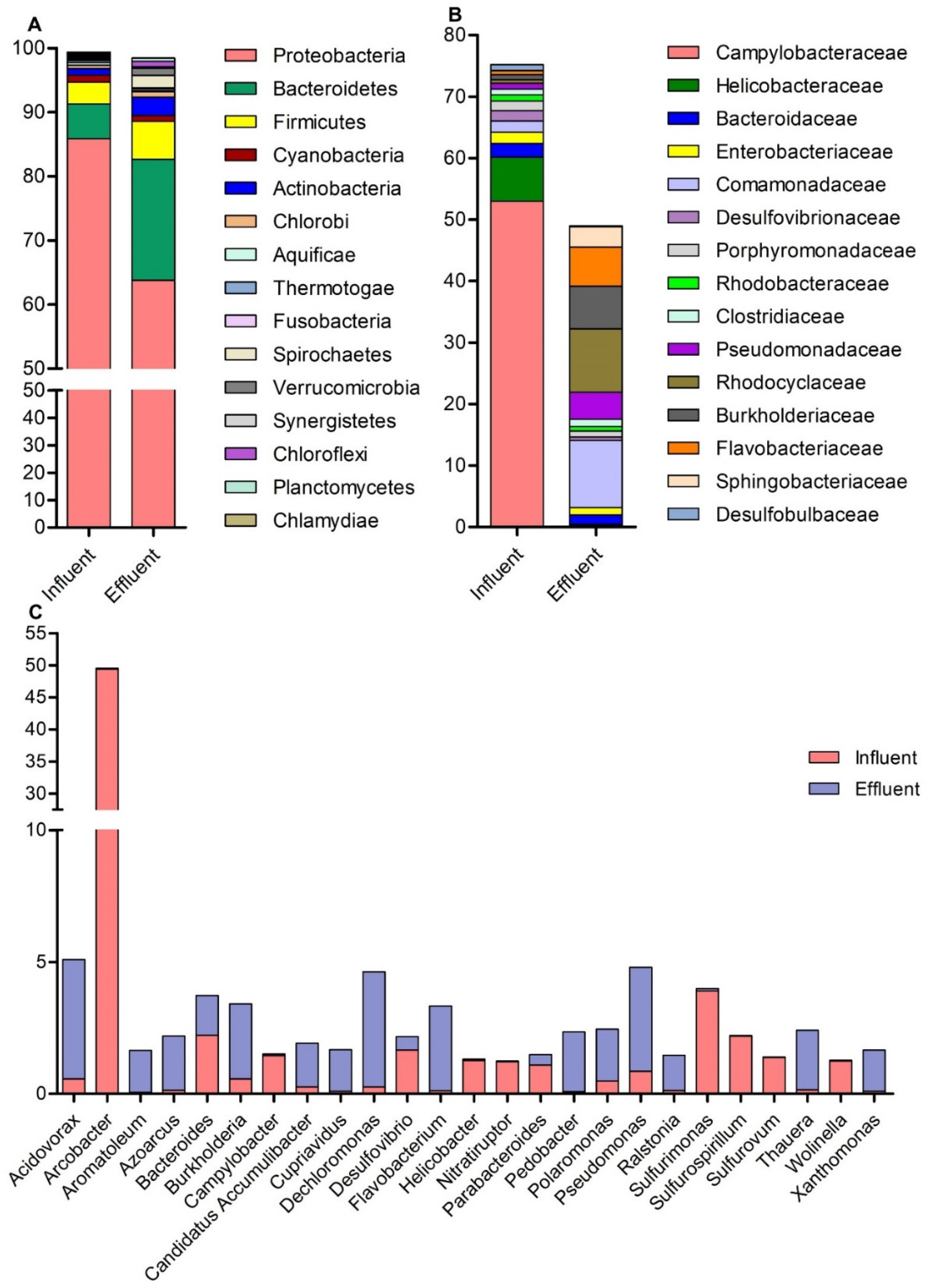
3.2. Bacteria
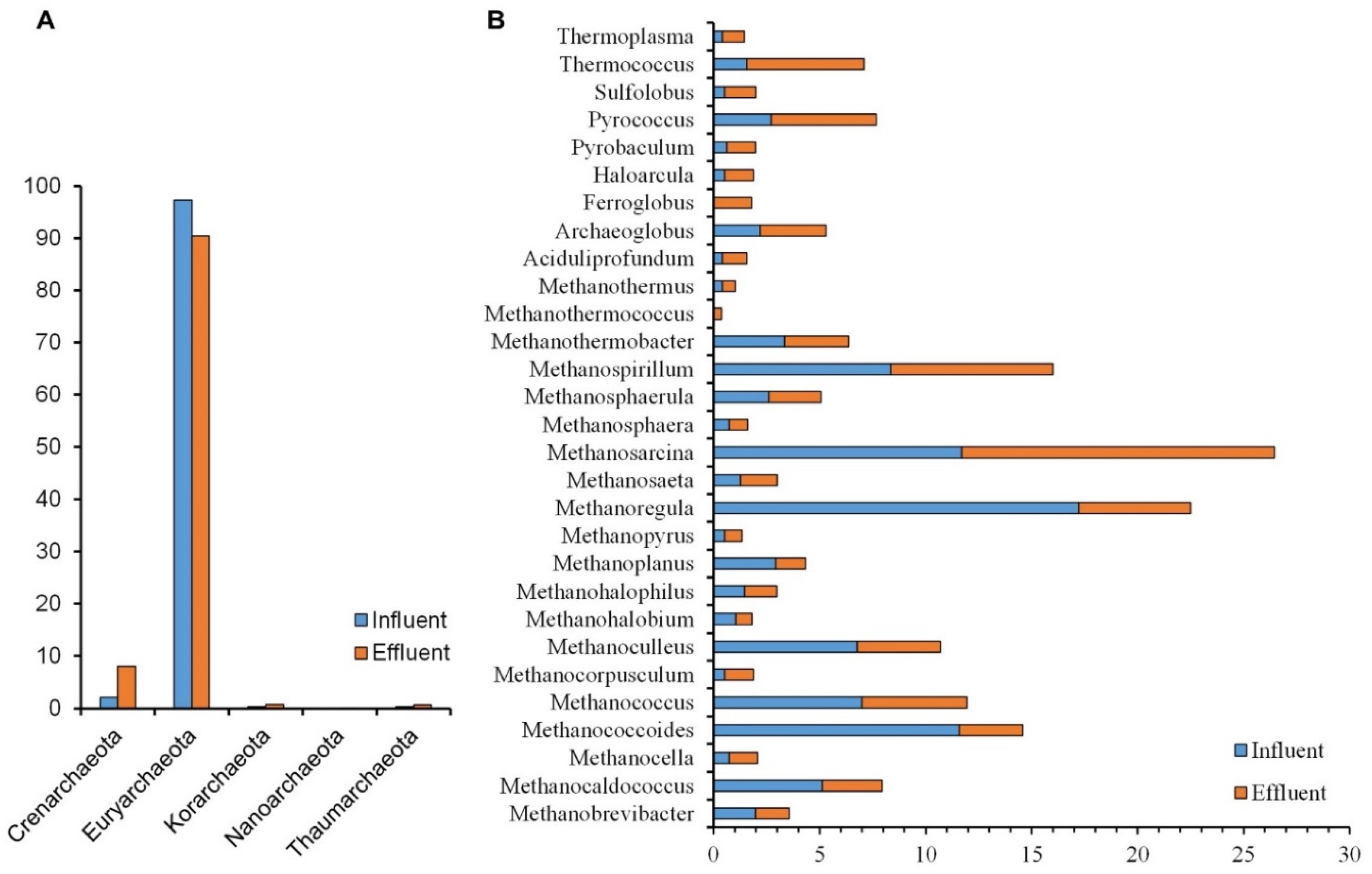
3.3. Archaea
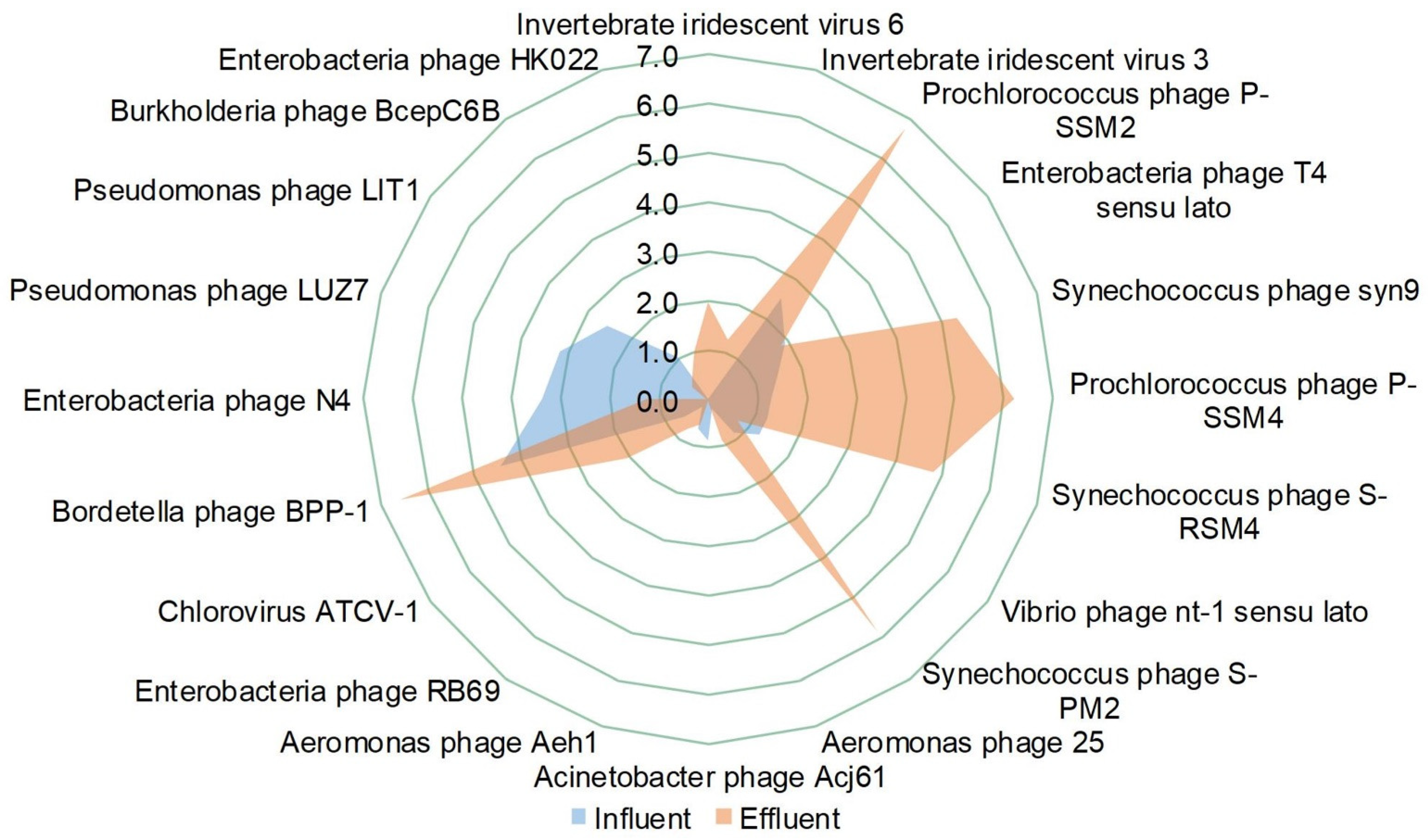
3.4. Virus
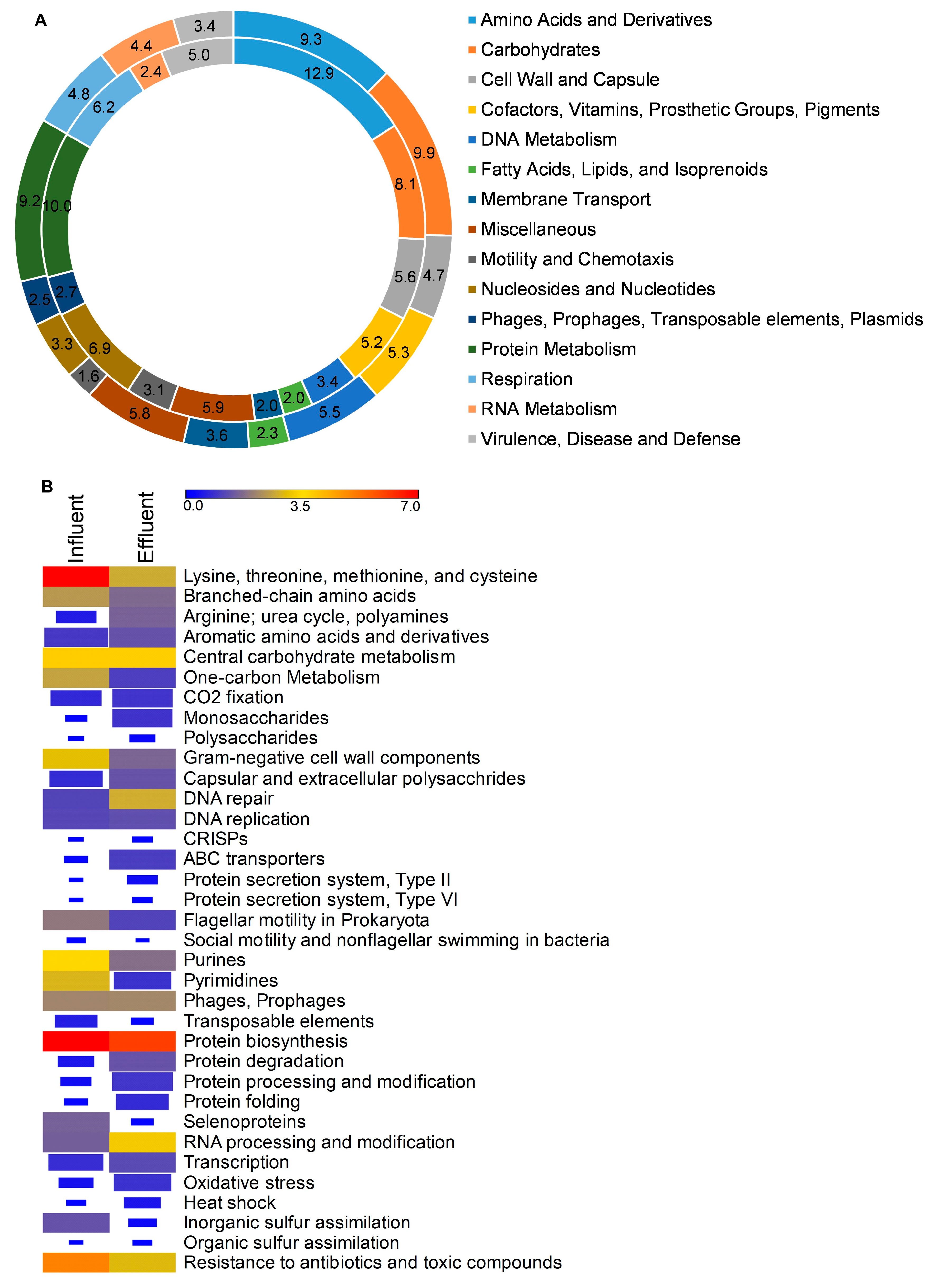
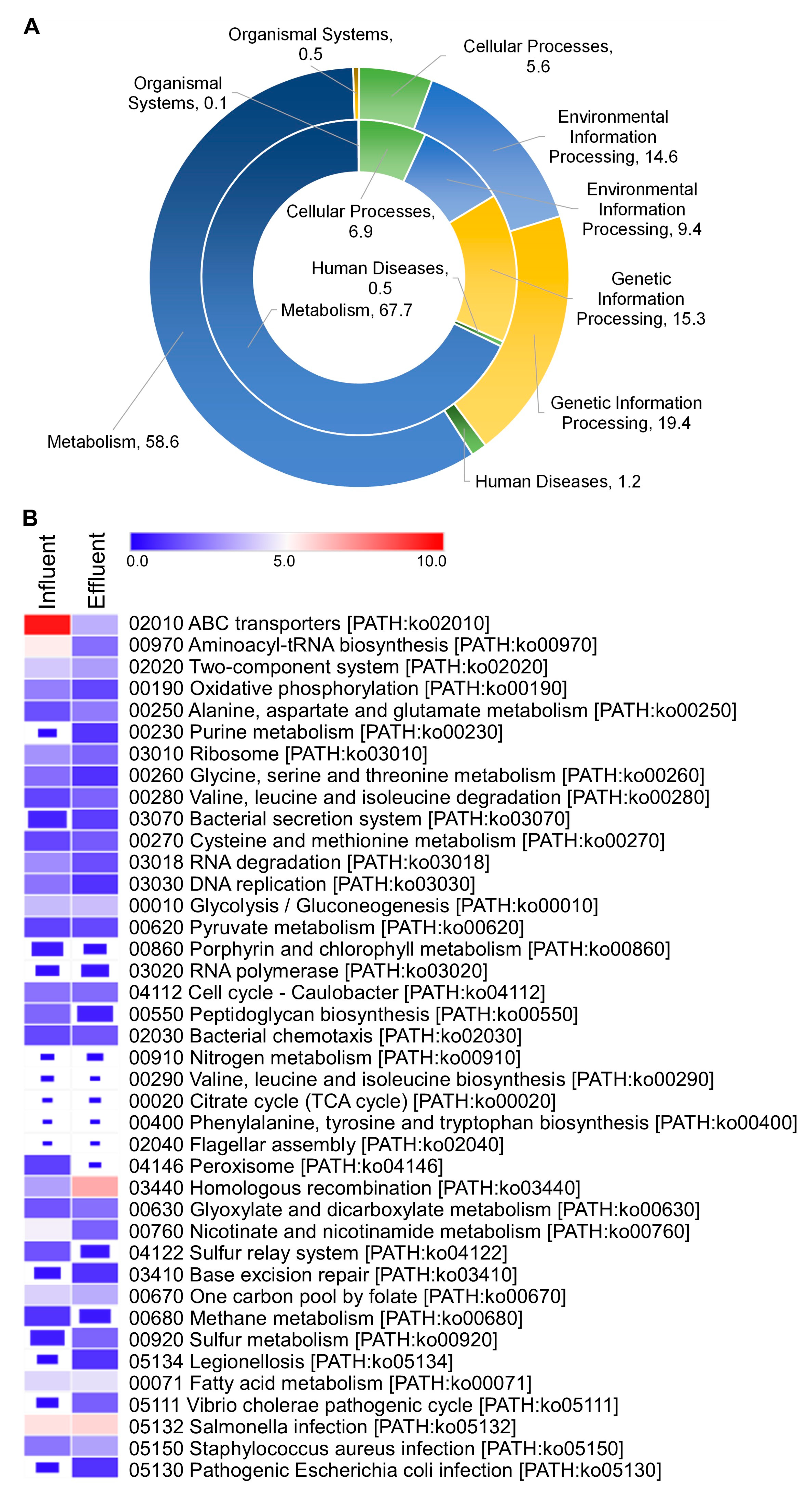
3.5. Metagenomes and Function Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mateo-Sagasta, J.; Raschid-Sally, L.; Thebo, A. Globel wastewater and sludge production, treatment and use. In Wastewater; Springer: Dordrecht, The Netherlands, 2015; pp. 15–38. [Google Scholar]
- Nascimento, A.L.; Souza, A.J.; Andrade, P.A.M.; Andreote, F.D.; Coscione, A.R.; Oliveira, F.C.; Regitano, J.B. Sewage Sludge Microbial Structures and Relations to Their Sources, Treatments, and Chemical Attributes. Front. Microbiol. 2018, 9, 1462. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Ning, D.; Zhang, B.; Li, Y.; Zhang, P.; Shan, X.; Zhang, Q.; Brown, M.R.; Li, Z.; Van Nostrand, J.D.; et al. Global diversity and biogeography of bacterial communities in wastewater treatment plants. Nat. Microbiol. 2019, 4, 1183–1195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chowdhary, P.; Raj, A.; Bharagava, R.N. Environmental pollution and health hazards from distillery wastewater and treatment approaches to combat the environmental threats: A review. Chemosphere 2018, 194, 229–246. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; Chandra, R. Pollutants released from the pulp paper industry: Aquatic toxicity and their health hazards. Aquat. Toxicol. 2019, 211, 202–216. [Google Scholar] [CrossRef]
- Al-Gheethi, A.A.; Efaq, A.N.; Bala, J.D.; Norli, I.; Abdel-Monem, M.O.; Kadir, M.O.A. Removal of pathogenic bacteria from sewage-treated effluent and biosolids for agricultural purposes. Appl. Water Sci. 2018, 8, 74. [Google Scholar] [CrossRef] [Green Version]
- Delforno, T.P.; Lacerda, G.V., Jr.; Sierra-Garcia, I.N.; Okada, D.Y.; Macedo, T.Z.; Varesche, M.B.; Oliveira, V.M. Metagenomic analysis of the microbiome in three different bioreactor configurations applied to commercial laundry wastewater treatment. Sci. Total Environ. 2017, 587–588, 389–398. [Google Scholar] [CrossRef]
- Ye, L.; Zhang, T. Pathogenic bacteria in sewage treatment plants as revealed by 454 pyrosequencing. Environ. Sci. Technol. 2011, 45, 7173–7179. [Google Scholar] [CrossRef]
- Kim, Y.K.; Yoo, K.; Kim, M.S.; Han, I.; Lee, M.; Kang, B.R.; Lee, T.K.; Park, J. The capacity of wastewater treatment plants drives bacterial community structure and its assembly. Sci. Rep. 2019, 9, 14809. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Shao, M.F.; Ye, L. 454 pyrosequencing reveals bacterial diversity of activated sludge from 14 sewage treatment plants. ISME J. 2012, 6, 1137–1147. [Google Scholar] [CrossRef]
- Al-Jassim, N.; Ansari, M.I.; Harb, M.; Hong, P.Y. Removal of bacterial contaminants and antibiotic resistance genes by conventional wastewater treatment processes in Saudi Arabia: Is the treated wastewater safe to reuse for agricultural irrigation? Water Res. 2015, 73, 277–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, C.; Tan, B.; Jiang, X.T.; Gu, X.; Chen, H.; Schmitz, B.W.; Haller, L.; Charles, F.R.; Zhang, T.; Gin, K. Metagenomic and Resistome Analysis of a Full-Scale Municipal Wastewater Treatment Plant in Singapore Containing Membrane Bioreactors. Front. Microbiol. 2019, 10, 172. [Google Scholar] [CrossRef] [PubMed]
- Tong, J.; Tang, A.; Wang, H.; Liu, X.; Huang, Z.; Wang, Z.; Zhang, J.; Wei, Y.; Su, Y.; Zhang, Y. Microbial community evolution and fate of antibiotic resistance genes along six different full-scale municipal wastewater treatment processes. Bioresour. Technol. 2019, 272, 489–500. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.A.; Patel, P.G.; Ganesh, A.G.; Rais, N.; Faheem, S.M.; Khan, S.T. Assessing Methanogenic Archaeal Community in Full Scale Anaerobic Sludge Digester Systems in Dubai, United Arab Emirates. Open Microbiol. J. 2018, 12, 123–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, X.; Zhang, X.X.; Wang, Z.; Huang, K.; Wang, Y.; Liang, W.; Tan, Y.; Liu, B.; Tang, J. Bacterial pathogens and community composition in advanced sewage treatment systems revealed by metagenomics analysis based on high-throughput sequencing. PLoS ONE 2015, 10, e0125549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandewalle, J.L.; Goetz, G.W.; Huse, S.M.; Morrison, H.G.; Sogin, M.L.; Hoffmann, R.G.; Yan, K.; McLellan, S.L. Acinetobacter, Aeromonas and Trichococcus populations dominate the microbial community within urban sewer infrastructure. Environ. Microbiol. 2012, 14, 2538–2552. [Google Scholar] [CrossRef]
- Shannon, K.E.; Lee, D.Y.; Trevors, J.T.; Beaudette, L.A. Application of real-time quantitative PCR for the detection of selected bacterial pathogens during municipal wastewater treatment. Sci. Total Environ. 2007, 382, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Elmund, G.K.; Allen, M.J.; Rice, E.W. Comparison of Escherichia coli, Total Coliform, and Fecal Coliform Populations as Indicators of Wastewater Treatment Efficiency. Water Environ. Res. 1999, 71, 332–339. [Google Scholar] [CrossRef] [Green Version]
- Ouda, O.K.M. Treated wastewater use in Saudi Arabia: Challenges and initiatives. Int. J. Water Res. Dev. 2016, 32, 799–809. [Google Scholar] [CrossRef]
- Pico, Y.; Alvarez-Ruiz, R.; Alfarhan, A.H.; El-Sheikh, M.A.; Alobaid, S.M.; Barcelo, D. Uptake and accumulation of emerging contaminants in soil and plant treated with wastewater under real-world environmental conditions in the Al Hayer area (Saudi Arabia). Sci. Total Environ. 2019, 652, 562–572. [Google Scholar] [CrossRef]
- Al-Saleh, I.; Elkhatib, R.; Al-Rajoudi, T.; Al-Qudaihi, G. Assessing the concentration of phthalate esters (PAEs) and bisphenol A (BPA) and the genotoxic potential of treated wastewater (final effluent) in Saudi Arabia. Sci. Total Environ. 2017, 578, 440–451. [Google Scholar] [CrossRef] [PubMed]
- Al Qarni, H.; Collier, P.; O’Keeffe, J.; Akunna, J. Investigating the removal of some pharmaceutical compounds in hospital wastewater treatment plants operating in Saudi Arabia. Environ. Sci. Pollut. Res. Int. 2016, 23, 13003–13014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasir, M.; Farman, M.; Shah, M.W.; Jiman-Fatani, A.A.; Othman, N.A.; Almasaudi, S.B.; Alawi, M.; Shakil, S.; Al-Abdullah, N.; Ismaeel, N.A.; et al. Genomic and antimicrobial resistance genes diversity in multidrug-resistant CTX-M-positive isolates of Escherichia coli at a health care facility in Jeddah. J. Infect. Public Health 2020, 13, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Meyer, F.; Paarmann, D.; D’Souza, M.; Olson, R.; Glass, E.M.; Kubal, M.; Paczian, T.; Rodriguez, A.; Stevens, R.; Wilke, A.; et al. The metagenomics RAST server—A public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinform. 2008, 9, 386. [Google Scholar] [CrossRef] [Green Version]
- Zakrzewski, M.; Proietti, C.; Ellis, J.J.; Hasan, S.; Brion, M.J.; Berger, B.; Krause, L. Calypso: A user-friendly web-server for mining and visualizing microbiome-environment interactions. Bioinformatics 2017, 33, 782–783. [Google Scholar] [CrossRef] [Green Version]
- Giwa, A.S.; Ali, N.; Athar, M.A.; Wang, K. Dissecting microbial community structure in sewage treatment plant for pathogens’ detection using metagenomic sequencing technology. Arch. Microbiol. 2020, 202, 825–833. [Google Scholar] [CrossRef]
- Shanks, O.C.; Newton, R.J.; Kelty, C.A.; Huse, S.M.; Sogin, M.L.; McLellan, S.L. Comparison of the microbial community structures of untreated wastewaters from different geographic locales. Appl. Environ. Microbiol. 2013, 79, 2906–2913. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Wang, L.; Xiang, F.; Zhao, L.; Qiao, Z. Activated Sludge Microbial Community and Treatment Performance of Wastewater Treatment Plants in Industrial and Municipal Zones. Int. J. Environ. Res. Public Health 2020, 17, 436. [Google Scholar] [CrossRef] [Green Version]
- Novo, A.; Andre, S.; Viana, P.; Nunes, O.C.; Manaia, C.M. Antibiotic resistance, antimicrobial residues and bacterial community composition in urban wastewater. Water Res. 2013, 47, 1875–1887. [Google Scholar] [CrossRef] [Green Version]
- Cydzik-Kwiatkowska, A.; Zielinska, M. Bacterial communities in full-scale wastewater treatment systems. World J. Microbiol. Biotechnol. 2016, 32, 66. [Google Scholar] [CrossRef] [Green Version]
- Yasir, M.; Angelakis, E.; Bibi, F.; Azhar, E.I.; Bachar, D.; Lagier, J.C.; Gaborit, B.; Hassan, A.M.; Jiman-Fatani, A.A.; Alshali, K.Z.; et al. Comparison of the gut microbiota of people in France and Saudi Arabia. Nutr. Diabetes 2015, 5, e153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angelakis, E.; Yasir, M.; Bachar, D.; Azhar, E.I.; Lagier, J.C.; Bibi, F.; Jiman-Fatani, A.A.; Alawi, M.; Bakarman, M.A.; Robert, C.; et al. Gut microbiome and dietary patterns in different Saudi populations and monkeys. Sci. Rep. 2016, 6, 32191. [Google Scholar] [CrossRef] [PubMed]
- Newton, R.J.; McLellan, S.L.; Dila, D.K.; Vineis, J.H.; Morrison, H.G.; Eren, A.M.; Sogin, M.L. Sewage reflects the microbiomes of human populations. mBio 2015, 6, e02574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shchegolkova, N.M.; Krasnov, G.S.; Belova, A.A.; Dmitriev, A.A.; Kharitonov, S.L.; Klimina, K.M.; Melnikova, N.V.; Kudryavtseva, A.V. Microbial Community Structure of Activated Sludge in Treatment Plants with Different Wastewater Compositions. Front. Microbiol. 2016, 7, 90. [Google Scholar] [CrossRef] [Green Version]
- Hu, M.; Wang, X.; Wen, X.; Xia, Y. Microbial community structures in different wastewater treatment plants as revealed by 454-pyrosequencing analysis. Bioresour. Technol. 2012, 117, 72–79. [Google Scholar] [CrossRef]
- Gonzalez-Martinez, A.; Rodriguez-Sanchez, A.; Lotti, T.; Garcia-Ruiz, M.J.; Osorio, F.; Gonzalez-Lopez, J.; van Loosdrecht, M.C. Comparison of bacterial communities of conventional and A-stage activated sludge systems. Sci. Rep. 2016, 6, 18786. [Google Scholar] [CrossRef]
- Xu, D.; Liu, S.; Chen, Q.; Ni, J. Microbial community compositions in different functional zones of Carrousel oxidation ditch system for domestic wastewater treatment. AMB Express 2017, 7, 40. [Google Scholar] [CrossRef] [Green Version]
- Webb, A.L.; Taboada, E.N.; Selinger, L.B.; Boras, V.F.; Inglis, G.D. Efficacy of wastewater treatment on Arcobacter butzleri density and strain diversity. Water Res. 2016, 105, 291–296. [Google Scholar] [CrossRef]
- Gao, P.; Xu, W.; Sontag, P.; Li, X.; Xue, G.; Liu, T.; Sun, W. Correlating microbial community compositions with environmental factors in activated sludge from four full-scale municipal wastewater treatment plants in Shanghai, China. Appl. Microbiol. Biotechnol. 2016, 100, 4663–4673. [Google Scholar] [CrossRef]
- Collado, L.; Figueras, M.J. Taxonomy, epidemiology, and clinical relevance of the genus Arcobacter. Clin. Microbiol. Rev. 2011, 24, 174–192. [Google Scholar] [CrossRef] [Green Version]
- Walker, A.W.; Duncan, S.H.; Louis, P.; Flint, H.J. Phylogeny, culturing, and metagenomics of the human gut microbiota. Trends Microbiol. 2014, 22, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Heylen, K.; Lebbe, L.; De Vos, P. Acidovorax caeni sp. nov., a denitrifying species with genetically diverse isolates from activated sludge. Int. J. Syst. Evol. Microbiol. 2008, 58, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Xie, B.; Liu, B.; Yi, Y.; Yang, L.; Liang, D.; Zhu, Y.; Liu, H. Microbiological mechanism of the improved nitrogen and phosphorus removal by embedding microbial fuel cell in Anaerobic-Anoxic-Oxic wastewater treatment process. Bioresour. Technol. 2016, 207, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Xu, X.; Zhu, L. Structure and function of the microbial consortia of activated sludge in typical municipal wastewater treatment plants in winter. Sci. Rep. 2017, 7, 17930. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Ju, F.; Cai, L.; Zhang, T. Profile and Fate of Bacterial Pathogens in Sewage Treatment Plants Revealed by High-Throughput Metagenomic Approach. Environ. Sci. Technol. 2015, 49, 10492–10502. [Google Scholar] [CrossRef]
- Qin, H.; Ji, B.; Zhang, S.; Kong, Z. Study on the bacterial and archaeal community structure and diversity of activated sludge from three wastewater treatment plants. Mar. Pollut. Bull. 2018, 135, 801–807. [Google Scholar] [CrossRef]
- Shapiro, O.H.; Kushmaro, A.; Brenner, A. Bacteriophage predation regulates microbial abundance and diversity in a full-scale bioreactor treating industrial wastewater. ISME J. 2010, 4, 327–336. [Google Scholar] [CrossRef] [Green Version]
- Delforno, T.P.; Lacerda Junior, G.V.; Noronha, M.F.; Sakamoto, I.K.; Varesche, M.B.A.; Oliveira, V.M. Microbial diversity of a full-scale UASB reactor applied to poultry slaughterhouse wastewater treatment: Integration of 16S rRNA gene amplicon and shotgun metagenomic sequencing. MicrobiologyOpen 2017, 6. [Google Scholar] [CrossRef]
- Gulino, K.; Rahman, J.; Badri, M.; Morton, J.; Bonneau, R.; Ghedin, E. Initial Mapping of the New York City Wastewater Virome. mSystems 2020, 5. [Google Scholar] [CrossRef]
- Sidhu, C.; Vikram, S.; Pinnaka, A.K. Unraveling the Microbial Interactions and Metabolic Potentials in Pre- and Post-treated Sludge from a Wastewater Treatment Plant Using Metagenomic Studies. Front. Microbiol. 2017, 8, 1382. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genus | Species | Influent | Effluent |
|---|---|---|---|
| Acinetobacter | Acinetobacter baumannii | 0.15 | 0.11 |
| Azotobacter | Azotobacter vinelandii | 0.02 | 0.37 |
| Bacteroides | Bacteroides fragilis | 0.40 | 0.24 |
| Bordetella | Bordetella bronchiseptica | 0.05 | 0.26 |
| Burkholderia | Burkholderia pseudomallei | 0.10 | 0.41 |
| Campylobacter | Campylobacter jejuni | 0.52 | 0.01 |
| Campylobacter | Campylobacter fetus | 0.38 | 0.01 |
| Clostridium | Clostridium botulinum | 0.05 | 0.12 |
| Enterococcus | Enterococcus faecium | 0.15 | 0.04 |
| Escherichia | Escherichia coli | 0.55 | 0.20 |
| Fusobacterium | Fusobacterium nucleatum | 0.15 | 0.04 |
| Helicobacter | Helicobacter pylori | 0.81 | 0.01 |
| Klebsiella | Klebsiella pneumoniae | 0.02 | 0.12 |
| Leptospira | Leptospira interrogans | 0.02 | 0.12 |
| Porphyromonas | Porphyromonas gingivalis | 0.18 | 0.19 |
| Pseudomonas | Pseudomonas aeruginosa | 0.30 | 1.19 |
| Stenotrophomonas | Stenotrophomonas maltophilia | 0.02 | 0.51 |
| Treponema | Treponema denticola | 0.01 | 0.40 |
| Vibrio | Vibrio parahaemolyticus | 0.33 | 0.03 |
| Vibrio | Vibrio cholerae | 0.11 | 0.08 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yasir, M. Analysis of Microbial Communities and Pathogen Detection in Domestic Sewage Using Metagenomic Sequencing. Diversity 2021, 13, 6. https://doi.org/10.3390/d13010006
Yasir M. Analysis of Microbial Communities and Pathogen Detection in Domestic Sewage Using Metagenomic Sequencing. Diversity. 2021; 13(1):6. https://doi.org/10.3390/d13010006
Chicago/Turabian StyleYasir, Muhammad. 2021. "Analysis of Microbial Communities and Pathogen Detection in Domestic Sewage Using Metagenomic Sequencing" Diversity 13, no. 1: 6. https://doi.org/10.3390/d13010006
APA StyleYasir, M. (2021). Analysis of Microbial Communities and Pathogen Detection in Domestic Sewage Using Metagenomic Sequencing. Diversity, 13(1), 6. https://doi.org/10.3390/d13010006