
Phylogeography and Genetic Structure of Sand Dune Specialist Stilpnolepis centiflora (Asteraceae) in Northwest China Revealed by Molecular Data




Abstract
:1. Introduction
2. Materials and Methods
2.1. Sampling Methods
2.2. Laboratory Procedures
2.3. CpDNA and ITS Sequence Analysis
3. Results
3.1. Genetic Variation Based on Plastid Sequence
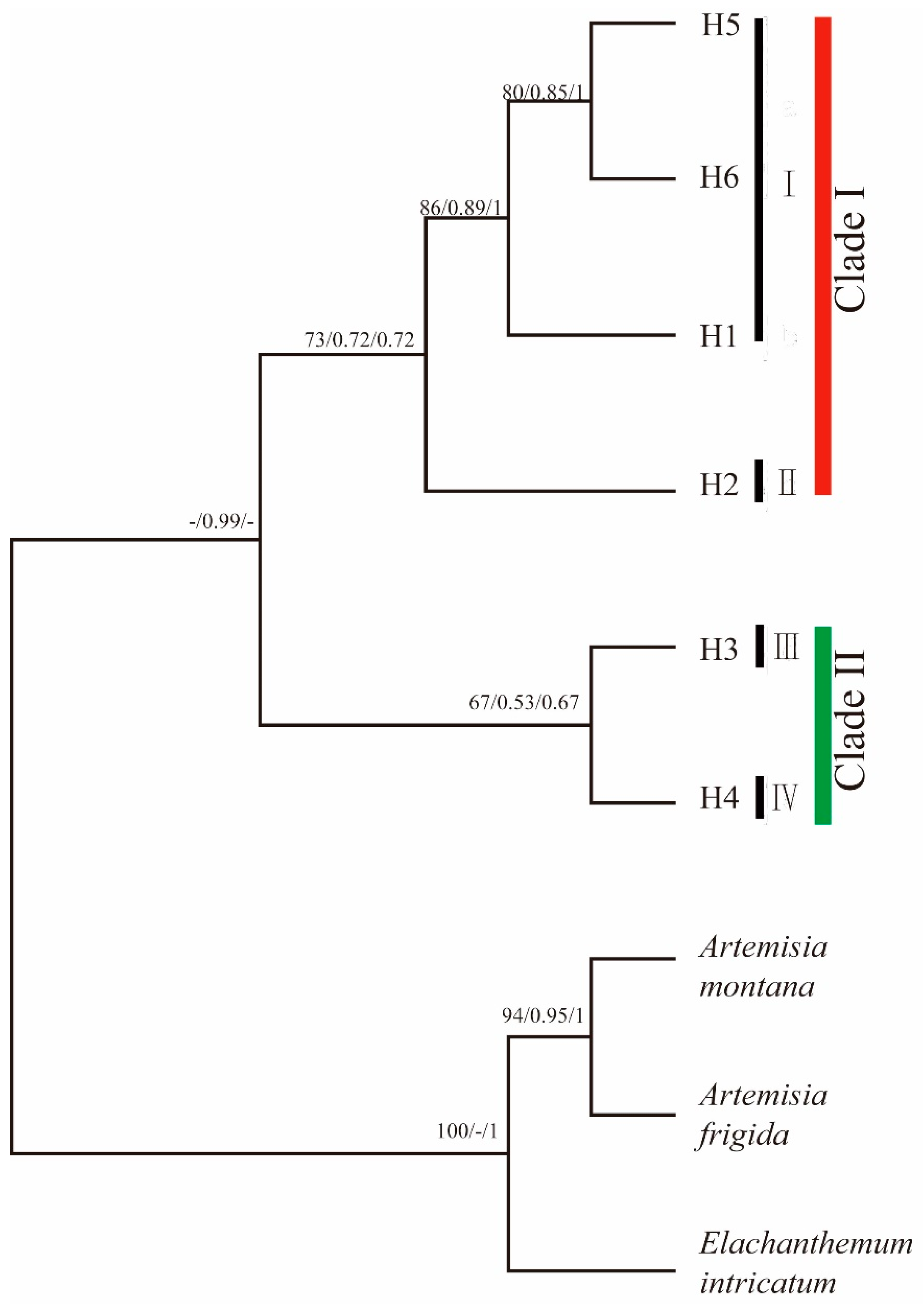
3.2. Genetic Variation, Ribotype Distribution, Population Structure, and Phylogenetic Relationships between Ribotypes Based on ITS Sequence
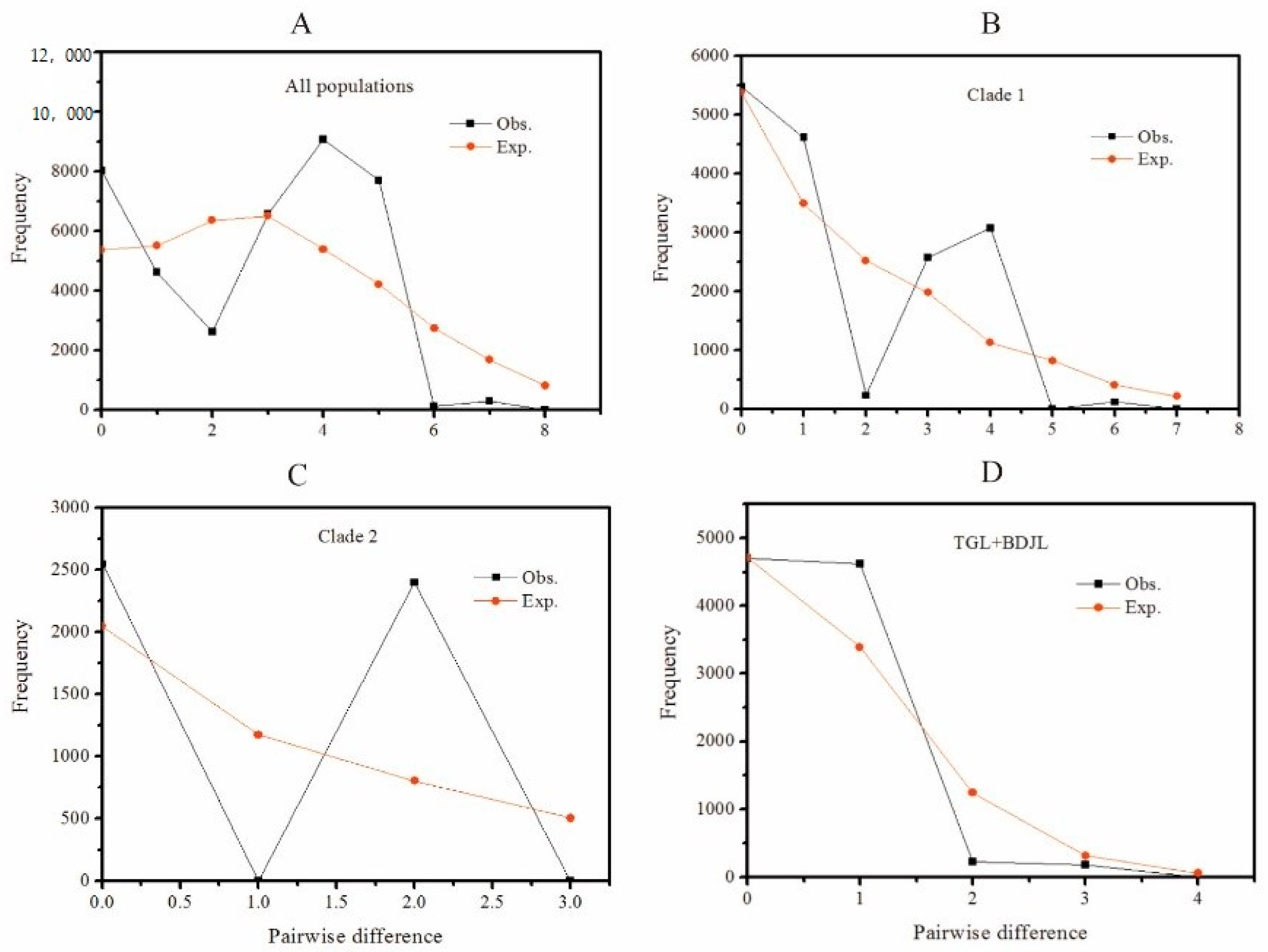
3.3. Demographic History
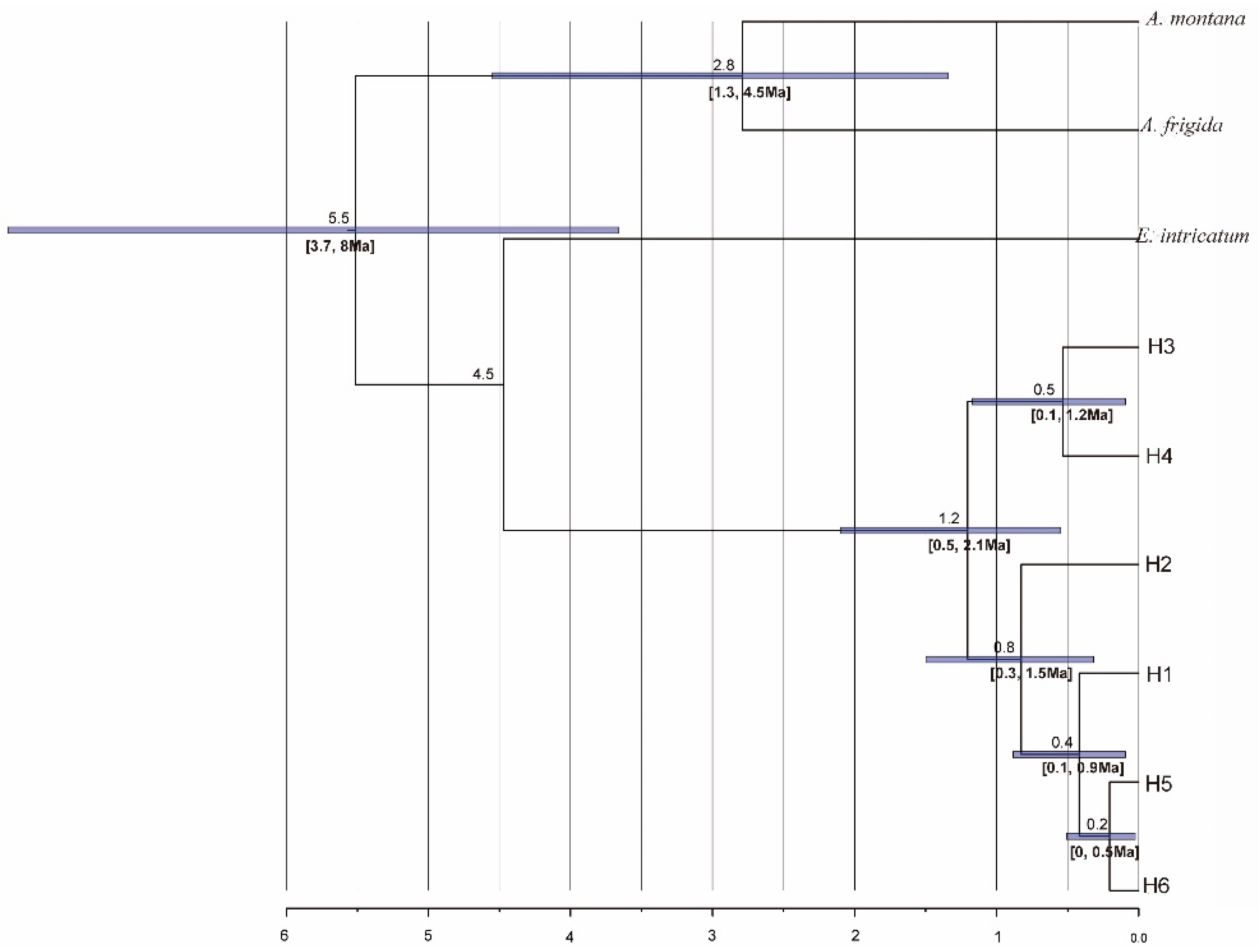
3.4. Phylogeny-Based Estimations of Divergence Times
4. Discussion
4.1. Genetic Diversity and Genetic Differentiation
4.2. Identifying a Contact Zone between Deserts
4.3. nrDNA and cpDNA Variation
4.4. Relationship between Allopatric Divergence of S. centiflora and the Evolution of Deserts
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Manel, S.; Holderegger, R. Ten years of landscape genetics. Trends Ecol. Evol. 2013, 28, 614–621. [Google Scholar] [CrossRef] [PubMed]
- Avise, J. Phylogeography: The History and Formation of Species; Harvard University Press: Cambridge, MA, USA, 2000. [Google Scholar]
- Hewitt, G.M. The genetic legacy of the Quaternary ice ages. Nature 2000, 405, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, G.M. Genetic consequences of climatic oscillations in the Quaternary. Philos. Trans. R. Soc. B Biol. Sci. 2004, 359, 183–195. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.M.; Zhang, X.; Li, Y.; Zhang, L.; Xiong, Y.C.; Wang, G. Spatial distribution patterns of the soil seed bank of Stipagrostis pennata (Trin.) de Winter in the Gurbantonggut Desert of north-west China. J. Arid. Environ. 2005, 63, 203–222. [Google Scholar] [CrossRef]
- Zheng, B.; Xu, Q.; Shen, Y. The relationship between climate change and Quaternary glacial cycles on the Qinghai–Tibetan Plateau: Review and speculation. Quat. Int. 2002, 97–98, 93–101. [Google Scholar] [CrossRef]
- Li, Z.-H.; Chen, J.; Zhao, G.-F.; Guo, Y.-P.; Kou, Y.-X.; Ma, Y.-Z.; Wang, G.; Ma, X.-F. Response of a desert shrub to past geological and climatic change: A phylogeographic study of Reaumuria soongarica (Tamaricaceae) in western China. J. Syst. Evol. 2012, 50, 351–361. [Google Scholar] [CrossRef]
- Su, Z.; Zhang, M. Evolutionary response to Quaternary climate aridification and oscillations in north-western China revealed by chloroplast phylogeography of the desert shrub Nitraria sphaerocarpa (Nitrariaceae). Biol. J. Linn. Soc. 2013, 109, 757–770. [Google Scholar] [CrossRef] [Green Version]
- Riddle, B.R.; Honeycutt, R.L. Historical biogeography in North-American arid regions—An approach using mitochondrial-dna phylogeny in grasshopper mice (genus Onychomys). Evolution 1990, 44, 1–15. [Google Scholar]
- Riddle, B.R.; Hafner, D.J.; Alexander, L.F. Comparative Phylogeography of Baileys’ Pocket Mouse (Chaetodipus baileyi) and the Peromyscus eremicus Species Group: Historical Vicariance of the Baja California Peninsular Desert. Mol. Phylogenetics Evol. 2000, 17, 161–172. [Google Scholar] [CrossRef] [Green Version]
- Riddle, B.R.; Hafner, D.J.; Alexander, L.F. Phylogeography and Systematics of the Peromyscus eremicus Species Group and the Historical Biogeography of North American Warm Regional Deserts. Mol. Phylogenetics Evol. 2000, 17, 145–160. [Google Scholar] [CrossRef] [Green Version]
- Leache, A.D.; Mulcahy, D.G. Phylogeny, divergence times and species limits of spiny lizards (Sceloporus magister species group) in western North American deserts and Baja California. Mol. Ecol. 2007, 16, 5216–5233. [Google Scholar] [CrossRef]
- Jaeger, J.R.; Riddle, B.R.; Bradford, D.F. Cryptic Neogene vicariance and Quaternary dispersal of the red-spotted toad (Bufo punctatus): Insights on the evolution of North American warm desert biotas. Mol. Ecol. 2005, 14, 3033–3048. [Google Scholar] [CrossRef]
- Guo, Y.-P.; Zhang, R.; Chen, C.-Y.; Zhou, D.-W.; Liu, J.-Q. Allopatric divergence and regional range expansion of Juniperus sabina in China. J. Syst. Evol. 2010, 48, 153–160. [Google Scholar] [CrossRef]
- Dong, G.R.; Li, B.S.; Gao, S.Y.; Wu, Z.; Shao, Y.J. Discovery of Quaternary ancient eolian sands and its significance in the Ordos Plateau. Chin. Sci. Bull. 1983, 4, 998–1001. [Google Scholar]
- Dong, G.R.; Chen, H.Z.; Jin, J.; Wang, G.Y. Resent advancing on Quaternary research in desert area, North China. Prog. Geo. 1991, 6, 29–32. [Google Scholar]
- Dong, G.; Li, S.; Li, B.S.; Wang, Y.; Yan, M.C. A preliminary study on the formation and evolution of deserts in China. J. Desert Res. 1991, 11, 23–32. [Google Scholar]
- Wang, Y.; Li, S.; Wang, J.H.; Yan, M.C. The uplift of the Qinghai-Xizang (Tibetan) Plateau and its effect on the formation and evolution of Chinese deserts. Arid Zone Res. 1996, 13, 20–24. [Google Scholar]
- Zheng, H.B.; Powell, C.M.; Butcher, K.; Cao, J.J. Late Neogene loess deposition in southern Tarim Basin: Tectonic and palaeo-environmental implications. Tectonophysics 2003, 375, 49–59. [Google Scholar] [CrossRef]
- Sun, J.; Zhang, L.; Deng, C.; Zhu, R. Evidence for enhanced aridity in the Tarim Basin of China since 5.3Ma. Quat. Sci. Rev. 2008, 27, 1012–1023. [Google Scholar] [CrossRef]
- Fang, X.; Lü, L.; Yang, S.; Li, J.; An, Z.; Jiang, P.; Chen, X. Loess in Kunlun Mountains and its implications on desert development and Tibetan Plateau uplift in west China. Sci. China Ser. D Earth Sci. 2002, 45, 289–299. [Google Scholar] [CrossRef]
- Sun, J. Source Regions and Formation of the Loess Sediments on the High Mountain Regions of Northwestern China. Quat. Res. 2002, 58, 341–351. [Google Scholar] [CrossRef]
- Yang, X.; Rost, K.T.; Lehmkuhl, F.; Zhenda, Z.; Dodson, J. The evolution of dry lands in northern China and in the Republic of Mongolia since the Last Glacial Maximum. Quat. Int. 2004, 118–119, 69–85. [Google Scholar] [CrossRef]
- Hoskin, C.J.; Higgie, M.; McDonald, K.R.; Moritz, C. Reinforcement drives rapid allopatric speciation. Nature 2005, 437, 1353–1356. [Google Scholar] [CrossRef]
- Ma, S.; Zhang, M. Phylogeography and conservation genetics of the relic Gymnocarpos przewalskii (Caryophyllaceae) restricted to northwestern China. Conserv. Genet. 2012, 13, 1531–1541. [Google Scholar] [CrossRef]
- Shi, Z.; Humphries, C.J.; Gilbert, M.G. Asteraceae (Compositae). In Flora of China; Wu, Z.Y., Raven, P.H., Eds.; Science Press: Beijing, China; Missouri Botanical Garden Press: St. Louis, MO, USA, 2011; Volume 20–21, pp. 758–759. [Google Scholar]
- Zhao, Y.Z. The area and floristic grographic element of Stilpnolepis centiflora. J. Inn. Mong. Univ. 1996, 27, 662–663. [Google Scholar]
- Pellicer, J.; Hidalgo, O.; Garcia, S.; Garnatje, T.; Korobkov, A.A.; Valles, J.; Martin, J. Palynological study of Ajania and related genera (Asteraceae, Anthemideae). Bot. J. Linn. Soc. 2009, 161, 171–189. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, S.-X.; Ji, P.-Z.; Gao, L.-Z. Phylogeography of Camellia taliensis (Theaceae) inferred from chloroplast and nuclear DNA: Insights into evolutionary history and conservation. BMC Evol. Biol. 2012, 12, 92. [Google Scholar] [CrossRef] [Green Version]
- Turchetto-Zolet, A.C.; Cruz, F.; Vendramin, G.G.; Simon, M.F.; Salgueiro, F.; Margis-Pinheiro, M.; Margis, R. Large-scale phylogeography of the disjunct Neotropical tree species Schizolobium parahyba (Fabaceae-Caesalpinioideae). Mol. Phylogenet Evol. 2012, 65, 174–182. [Google Scholar] [CrossRef]
- Cullings, K. Design and testing of a plant-specific PCR primer for ecological and evolutionary studies. Mol. Ecol. 1992, 1, 233–240. [Google Scholar] [CrossRef]
- Shaw, J.; Lickey, E.B.; Beck, J.T.; Farmer, S.B.; Liu, W.; Miller, J.; Siripun, K.C.; Winder, C.T.; Schilling, E.E.; Small, R.L. The tortoise and the hare II: Relative utility of 21 noncoding chloroplast DNA sequences for phylogenetic analysis. Am. J. Bot. 2005, 92, 142–166. [Google Scholar] [CrossRef] [Green Version]
- Shaw, J.; Lickey, E.B.; Schilling, E.E.; Small, R.L. Comparison of whole chloroplast genome sequences to choose noncoding regions for phylogenetic studies in angiosperms: The tortoise and the hare III. Am. J. Bot. 2007, 94, 275–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, T.J.; Burns, T.; Lee, S.; Taylor, J. Amplification and Direct Sequencing of Fungal Ribosomal RNA Genes for Phylogenetics; Academic Press: New York, NY, USA, 1990. [Google Scholar]
- Thompson, J.D.; Gibson, T.J.; Plewniak, F.; Jeanmougin, F.; Higgins, D.G. The CLUSTAL_X windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997, 25, 4876–4882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garrick, R.C.; Sunnucks, P.; Dyer, R.J. Nuclear gene phylogeography using PHASE: Dealing with unresolved genotypes, lost alleles, and systematic bias in parameter estimation. BMC Evol. Biol. 2010, 10, 118. [Google Scholar] [CrossRef] [Green Version]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef]
- Bandelt, H.J.; Forster, P.; Rohl, A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 1999, 16, 37–48. [Google Scholar] [CrossRef]
- Cassens, I.; Mardulyn, P.; Milinkovitch, M.C. Evaluating intraspecific “Network” construction methods using simulated se-quence data: Do existing algorithms outperform the global maximum parsimony approach? Syst. Biol. 2005, 54, 363–372. [Google Scholar] [CrossRef] [Green Version]
- Excoffier, L.; Lischer, H.E.L. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef]
- Miller, M.P. Alleles In Space (AIS): Computer software for the joint analysis of interindividual spatial and genetic information. J. Hered. 2005, 96, 722–724. [Google Scholar] [CrossRef]
- Pons, O.; Petit, R.J. Measuring and Testing Genetic Differentiation with Ordered versus Unordered Alleles. Genetics 1996, 144, 1237–1245. [Google Scholar] [CrossRef]
- Dupanloup, I.; Schneider, S.; Excoffier, L. A simulated annealing approach to define the genetic structure of populations. Mol. Ecol. 2002, 11, 2571–2581. [Google Scholar] [CrossRef]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [Green Version]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef]
- Earl, D.A.; Vonholdt, B.M. Structure Harvester: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Excoffier, L.; Smouse, P.E.; Quattro, J.M. Analysis of Molecular Variance Inferred from Metric Distances among DNA Haplo-types—Application to Human Mitochondrial-DNA Restriction Data. Genetics 1992, 131, 479–491. [Google Scholar] [CrossRef]
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007, 7, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.-F.; Gong, X.; Chiang, Y.-C.; Kuroda, C. Phylogenetic patterns and disjunct distribution in Ligularia hodgsonii Hook. (Asteraceae). J. Biogeogr. 2013, 40, 1741–1754. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A.J. Tracer v. 1.5. 2009. Available online: http://tree.bio.ed.ac.uk/software/tracer/ (accessed on 25 March 2014).
- Rambaut, A. FigTree v. 1.3.1. 2009. Available online: http://tree.bio.ed.ac.uk/software/FigTree/ (accessed on 5 May 2012).
- Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1989, 123, 585–595. [Google Scholar] [CrossRef]
- Fu, Y.-X. Statistical Tests of Neutrality of Mutations Against Population Growth, Hitchhiking and Background Selection. Genetics 1997, 147, 915–925. [Google Scholar] [CrossRef]
- Slatkin, M.; Hudson, R.R. Pairwise comparisons of mitochondrial DNA sequences in stable and exponentially growing populations. Genetics 1991, 129, 555–562. [Google Scholar] [CrossRef]
- Schneider, S.; Excoffier, L. Estimation of past demographic parameters from the distribution of pairwise differences when the mutation rates very among sites: Application to human mitochondrial DNA. Genetics 1999, 152, 1079–1089. [Google Scholar] [CrossRef]
- Wolfe, K.H.; Li, W.-H.; Sharp, P.M. Rates of nucleotide substitution vary greatly among plant mitochondrial, chloroplast, and nuclear DNAs. Proc. Natl. Acad. Sci. USA 1987, 84, 9054–9058. [Google Scholar] [CrossRef] [Green Version]
- Petit, R.J.; Duminil, J.; Fineschi, S.; Hampe, A.; Salvini, D.; Vendramin, G.G. Comparative organization of chloroplast, mitochondrial and nuclear diversity in plant populations. Mol. Ecol. 2005, 14, 689–701. [Google Scholar] [CrossRef] [Green Version]
- Birky, C.W., Jr.; Fuerst, P.; Maruyama, T. Organelle gene diversity under migration, mutation, and drift: Equilibrium expectations, approach to equilibrium, effects of heteroplasmic cells, and comparison to nuclear genes. Genetics 1989, 121, 613–627. [Google Scholar] [CrossRef]
- Petit, R.J.; Aguinagalde, I.; de Beaulieu, J.-L.; Bittkau, C.; Brewer, S.; Cheddadi, R.; Ennos, R.; Fineschi, S.; Grivet, D.; Lascoux, M.; et al. Glacial Refugia: Hotspots but Not Melting Pots of Genetic Diversity. Science 2003, 300, 1563–1565. [Google Scholar] [CrossRef] [Green Version]
- Qiu, Y.-X.; Fu, C.-X.; Comes, H.P. Plant molecular phylogeography in China and adjacent regions: Tracing the genetic imprints of Quaternary climate and environmental change in the world’s most diverse temperate flora. Mol. Phylogenetics Evol. 2011, 59, 225–244. [Google Scholar] [CrossRef]
- Liu, J.Q.; Sun, Y.S.; Ge, X.J.; Gao, L.M.; Qiu, Y.X. Phylogeographic studies of plants in China: Advances in the past and directions in the future. J. Syst. Evol. 2012, 50, 267–275. [Google Scholar] [CrossRef]
- Alvarez, I.; Wendel, J.F. Ribosomal ITS sequences and plant phylogenetic inference. Mol. Phylogenet Evol. 2003, 29, 417–434. [Google Scholar] [CrossRef] [Green Version]
- Lorenz-Lemke, A.P.; Muschner, V.C.; Bonatto, S.L.; Cervi, A.C.; Salzano, F.M.; Freitas, L.B. Phylogeographic inferences concerning evolution of Brazilian Passiflora actinia and P. elegans (Passifloraceae) based on ITS (nrDNA) variation. Ann. Bot. 2005, 95, 799–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaal, B.A.; Hayworth, D.A.; Olsen, K.M.; Rauscher, J.T.; Smith, W.A. Phylogeographic studies in plants: Problems and pro-spects. Mol. Ecol. 1998, 7, 465–474. [Google Scholar] [CrossRef]
- Meng, H.-H.; Zhang, M.-L. Diversification of plant species in arid Northwest China: Species-level phylogeographical history of Lagochilus Bunge ex Bentham (Lamiaceae). Mol. Phylogenetics Evol. 2013, 68, 398–409. [Google Scholar] [CrossRef] [PubMed]
- Li, J.J.; Shi, Y.F.; Li, B.Y. Uplift of the Qinghai-Xizang (Tibet) Plateau and Global Change Lanzhou; Lanzhou University Press: Lanzhou, China, 1995. [Google Scholar]
- Shi, Y.F.; Zheng, B.X.; Li, S.J. Last glaciation and maximum glaciation in Qinghai-Xizang (Tibet) plateau. J. Glaciol. Geocryol. 1990, 12, 1–15. [Google Scholar]
- Zhou, S.; Li, J. The sequence of Quaternary glaciation in the Bayan Har Mountains. Quat. Int. 1998, 45–46, 135–142. [Google Scholar] [CrossRef]
- Yang, S.-J.; Yin, Z.-H.; Ma, X.-M.; Lei, F.-M. Phylogeography of ground tit (Pseudopodoces humilis) based on mtDNA: Evidence of past fragmentation on the Tibetan Plateau. Mol. Phylogenetics Evol. 2006, 41, 257–265. [Google Scholar] [CrossRef]
- Qi, D.; Guo, S.; Zhao, X.; Yang, J.; Tang, W. Genetic diversity and historical population structure of Schizopygopsis pylzovi (Teleostei: Cyprinidae) in the Qinghai?Tibetan Plateau. Freshw. Biol. 2007, 52, 1090–1104. [Google Scholar] [CrossRef]
- Jin, Y.T.; Brown, R.P.; Liu, N.F. Cladogenesis and phylogeography of the lizard Phrynocephalus vlangalii (Agamidae) on the Tibetan plateau (vol 17, pg 1971, 2008). Mol. Ecol. 2008, 17, 3033–3034. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Code/Location | Latitude/Longitude (N/E) | cpDNA | ITS | ||||||
|---|---|---|---|---|---|---|---|---|---|
| N | Haplotype | h | π | N | Ribotypes | h | π | ||
| Total | 280 | 0.7944 | 0.0020 | 250 | 0.8140 | 0.0030 | |||
| Kubuqi Desert | |||||||||
| 1 SHK | 39.64°/106.60° | 10 | H2 | 0 | 0 | 22 | R1, R2, R3, R4 | 0.6623 | 0.0028 |
| 2 DGN | 40.71°/108.51° | 10 | H2 | 0 | 0 | 20 | R1, R2, R5, R6, R7 | 0.6632 | 0.0034 |
| 3 DGTL | 40.49°/108.67° | 10 | H2 | 0 | 0 | 18 | R1, R2, R5, R6, R8 | 0.7451 | 0.0026 |
| 4 DLT | 40.28°/109.93° | 10 | H2 | 0 | 0 | 18 | R1, R2, R8 | 0.5817 | 0.0026 |
| Mu Us Desert | |||||||||
| 5 AZQ | 40.33°/109.39° | 10 | H3 | 0 | 0 | 16 | R2, R8, R9, R10, R13 | 0.8250 | 0.0020 |
| 6 BJT | 38.05°/107.68° | 10 | H3 | 0 | 0 | 12 | R6, R14, | 0.3030 | 0.0025 |
| 7 YC | 37.93°/106.41° | 10 | H3 | 0 | 0 | 14 | R1, R8, R10 | 0.4835 | 0.0021 |
| 8 JL | 37.46°/105.01° | 10 | H3 | 0 | 0 | 20 | R1, R8, R9, R10, R11, R12, R13 | 0.6895 | 0.0025 |
| Badain Jaran Desert | |||||||||
| 9 YBA | 39.35°/102.34° | 10 | H5 H6 | 0.3556 | 0.0005 | 12 | R5, R6, R7, R17 | 0.7143 | 0.0012 |
| 10 YBB | 39.55°/102.53° | 10 | H5 H6 | 0.2000 | 0.0003 | 14 | R6, R7, R17 | 0.4394 | 0.0007 |
| 11 YQA | 39.56°/102.60° | 10 | H5 | 0 | 0 | 24 | R5, R6, R7, R17 | 0.4312 | 0.0007 |
| 12 YQB | 39.64°/102.58° | 10 | H5 | 0 | 0 | 24 | R5, R6, R7, R17, R31 | 0.7391 | 0.0014 |
| Ulan Buh Desert | |||||||||
| 13 WHA | 39.78°/106.86° | 10 | H1 | 0 | 0 | 14 | R5, R6, R15, R17 | 0.6923 | 0.0012 |
| 14 WHB | 39.78°/106.85° | 10 | H1 | 0 | 0 | 20 | R5, R6, R15, R17 | 0.6105 | 0.0010 |
| 15 WHC | 39.64°/106.63° | 10 | H5 | 0 | 0 | 20 | R5, R6, R7, R15, R24 | 0.6263 | 0.0013 |
| 16 WHD | 39.64°/106.6° | 10 | H4 | 0 | 0 | 20 | R5, R6, R7, R17, R24 | 0.4421 | 0.0008 |
| 17 WHE | 39.91°/106.66° | 10 | H4 | 0 | 0 | 16 | R6, R7, R24 | 0.6583 | 0.0016 |
| 18 WHF | 39.82°/106.69° | 10 | H4 | 0 | 0 | 16 | R6, R17 | 0.5000 | 0.0007 |
| 19 WHG | 40.03°/106.63° | 10 | H4 | 0 | 0 | 16 | R6, R17 | 0.1250 | 0.0002 |
| 20 WST | 38.16°/107.51° | 10 | H4 | 0 | 0 | 12 | R6, R15, R16, R17 | 0.5606 | 0.0009 |
| Tengger Desert | |||||||||
| 21 ZQA | 38.71°/105.33° | 10 | H1 | 0 | 0 | 22 | R5, R6, R7, R17, R29, R30 | 0.7619 | 0.0015 |
| 22 ZQB | 38.69°/105.40° | 10 | H5 | 0 | 0 | 20 | R5, R6, R7, R17, R23, R27, R28 | 0.6895 | 0.0017 |
| 23 ZWA | 37.57°/105.10° | 10 | H5 | 0 | 0 | 16 | R6, R7, R17, R23, R24 | 0.7667 | 0.0017 |
| 24 ZWB | 37.59°/104.6° | 10 | H5 | 0 | 0 | 20 | R6, R7, R17, R25 | 0.7105 | 0.00140 |
| 25 ZQC | 38.55°/105.35° | 10 | H4 | 0 | 0 | 10 | R6, R7, R17, R26 | 0.7333 | 0.0013 |
| 26 MJW | 37.89°/107.58° | 10 | H1 | 0 | 0 | 12 | R7, R17 | 0.5455 | 0.0015 |
| 27 SPT | 37.45°/104.93° | 10 | H1 | 0 | 0 | 28 | R6, R7, R17, R18, R19, R20, R21 | 0.8042 | 0.0018 |
| 28 ZQDS | 38.30°/103.72° | 10 | H1 | 0 | 0 | 22 | R5, R6, R7, R17, R22 | 0.7792 | 0.0015 |
| Date | HS | HT | GST | NST |
|---|---|---|---|---|
| cpDNA | 0.020 (0.014) | 0.820 (0.018) | 0.976 (0.018) | 0.986 (0.010) |
| ITS | 0.617 (0.031) | 0.817 (0.037) | 0.244 (0.042) | 0.485 (0.053) |
| Source of Variation | cpDNA | ITS | ||||
|---|---|---|---|---|---|---|
| d.f. | PV (%) | Fixation Index | d.f. | PV (%) | Fixation Index | |
| Among populations | 27 | 98.6 | FST = 0.986 | 27 | 46.68 | FST = 0.467 |
| Within populations | 252 | 1.4 | 470 | 53.32 | ||
| Total | 279 | 497 | ||||
| Five deserts | ||||||
| Among deserts | 4 | 92.9 | FSC = 0.841 ** | 4 | 35.17 | FSC = 0.240 ** |
| Among populations within deserts | 23 | 5.97 | FST = 0.989 ** | 23 | 15.58 | FST = 0.508 ** |
| Within populations | 252 | 1.13 | FCT = 0.929 ** | 470 | 49.25 | FCT = 0.352 ** |
| Total | 279 | 497 | ||||
| Four lineages | ||||||
| Among lineages | 3 | 91.73 | FSC = 0.879 ** | |||
| Among populations within lineages | 24 | 7.27 | FST = 0.990 ** | |||
| Within populations | 252 | 1 | FCT = 0.917 ** | |||
| Total | 279 | |||||
| Populations | τ | SSD (p Value) | HRag (p Value) | Tajima’s D (p Value) | Fu’s Fs (p Value) |
|---|---|---|---|---|---|
| Overall | 4.379 | 0.034 (0.110) | 0.064 (0.220) | 1.956 (0.970) | 5.793 (0.952) |
| Clade 1 | 4.326 | 0.043 (0.280) | 0.136 (0.320) | 1.119 (0.893) | 4.028 (0.935) |
| Clade 2 | 2.742 | 0.183 (0.120) | 0.736 (0.040) | 2.322 (0.993) | 4.467 (0.965) |
| Lineage I | 0.703 | 0.026 (0.020) | 0.204 (0.010) | 0.105 (0.616) | 1.296 (0.740) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, X.; Jiang, X.; Zhang, H.; Qiu, J. Phylogeography and Genetic Structure of Sand Dune Specialist Stilpnolepis centiflora (Asteraceae) in Northwest China Revealed by Molecular Data. Diversity 2022, 14, 104. https://doi.org/10.3390/d14020104
Shi X, Jiang X, Zhang H, Qiu J. Phylogeography and Genetic Structure of Sand Dune Specialist Stilpnolepis centiflora (Asteraceae) in Northwest China Revealed by Molecular Data. Diversity. 2022; 14(2):104. https://doi.org/10.3390/d14020104
Chicago/Turabian StyleShi, Xiaojun, Xiaolong Jiang, Hongxiang Zhang, and Juan Qiu. 2022. "Phylogeography and Genetic Structure of Sand Dune Specialist Stilpnolepis centiflora (Asteraceae) in Northwest China Revealed by Molecular Data" Diversity 14, no. 2: 104. https://doi.org/10.3390/d14020104
APA StyleShi, X., Jiang, X., Zhang, H., & Qiu, J. (2022). Phylogeography and Genetic Structure of Sand Dune Specialist Stilpnolepis centiflora (Asteraceae) in Northwest China Revealed by Molecular Data. Diversity, 14(2), 104. https://doi.org/10.3390/d14020104