


New Insights into the Structural Requirements of Isatin-Derived Pro-Apoptotic Agents against Acute Myeloid Leukemia

 , ,
, ,  , and
, and 
Abstract
:1. Introduction
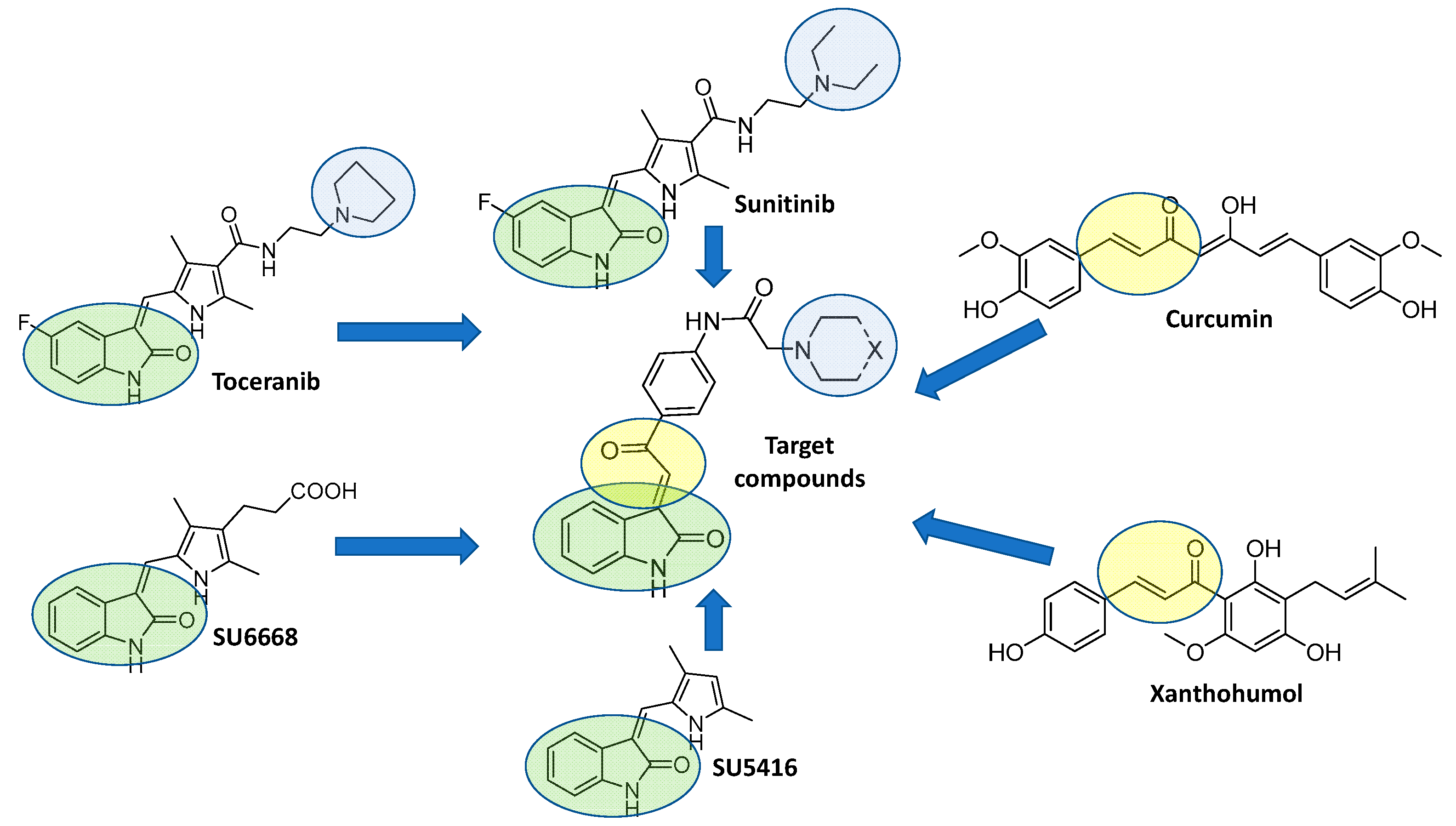
2. Rational Design
3. Results and Discussions
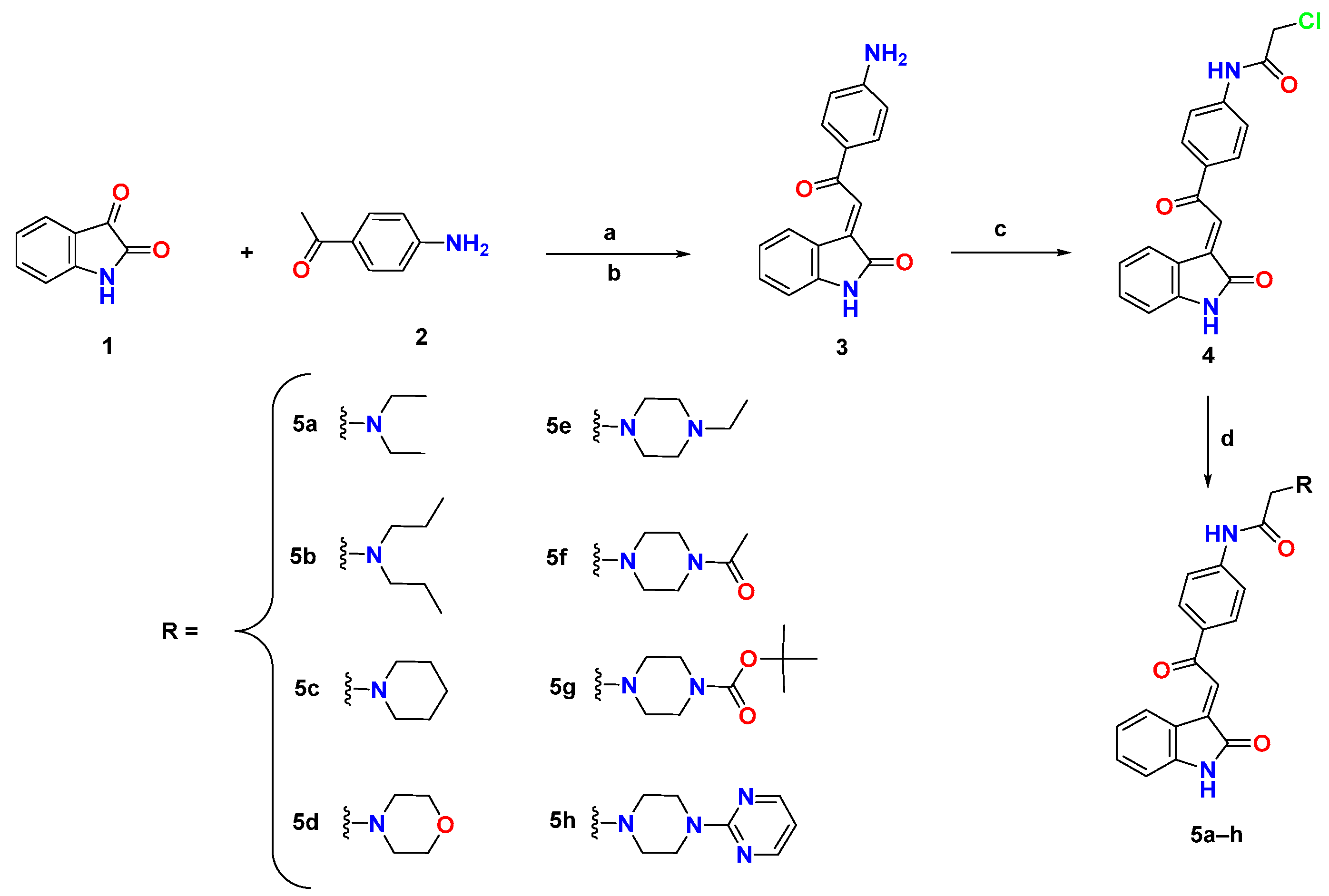
3.1. Chemistry
3.2. Biological Evaluation
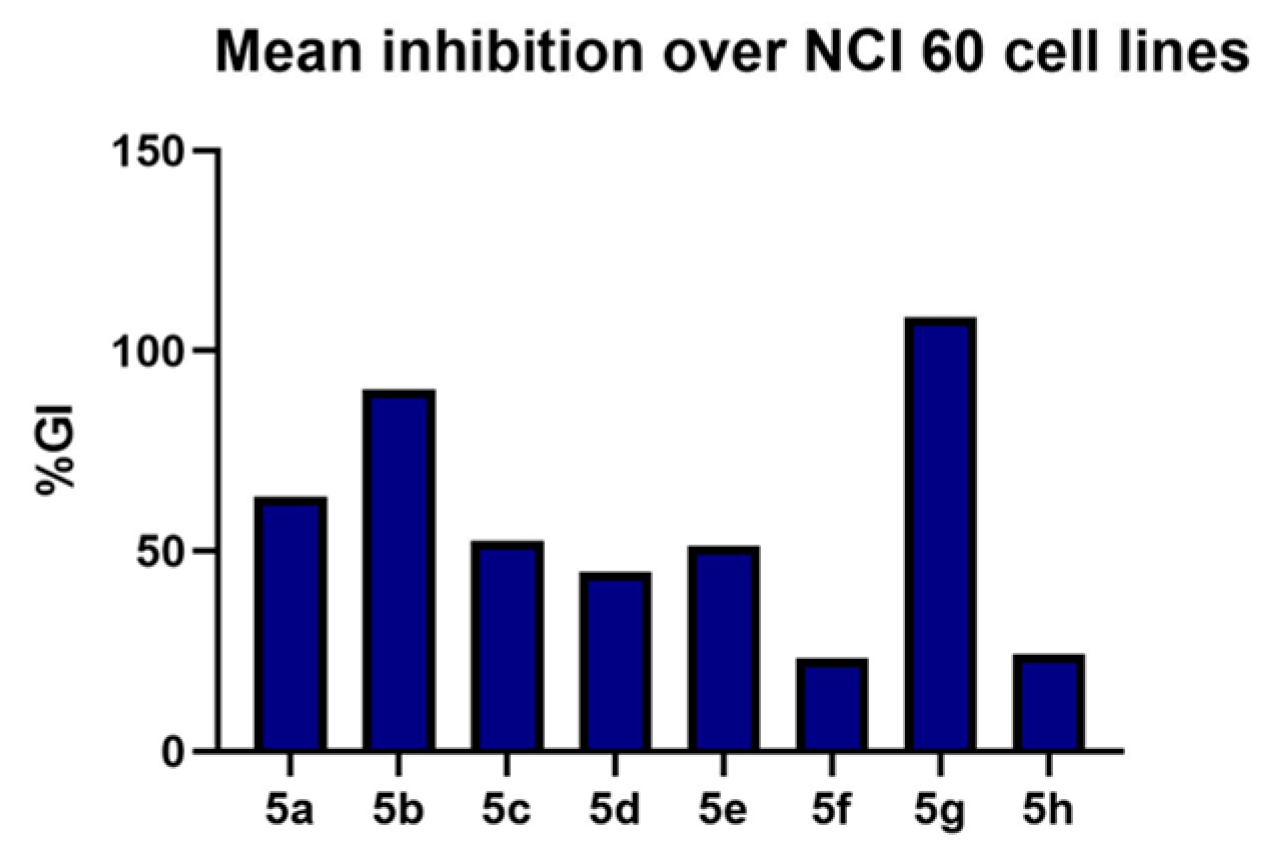
3.2.1. Evaluation of the In Vitro Cytotoxic Activity
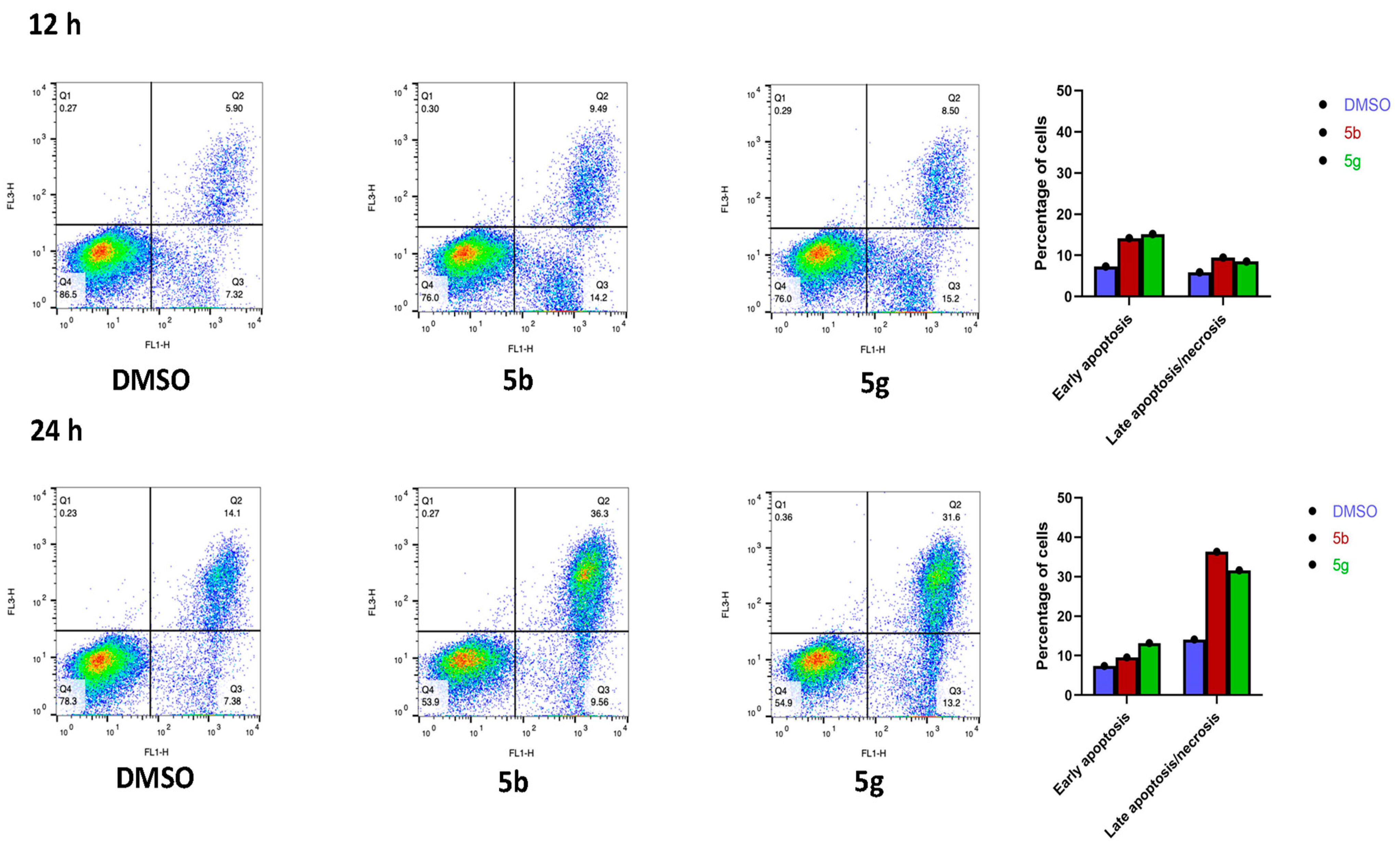
3.2.2. Analysis of Apoptosis/Necrosis Induction by 5b and 5g
3.2.3. Cell Cycle Analysis
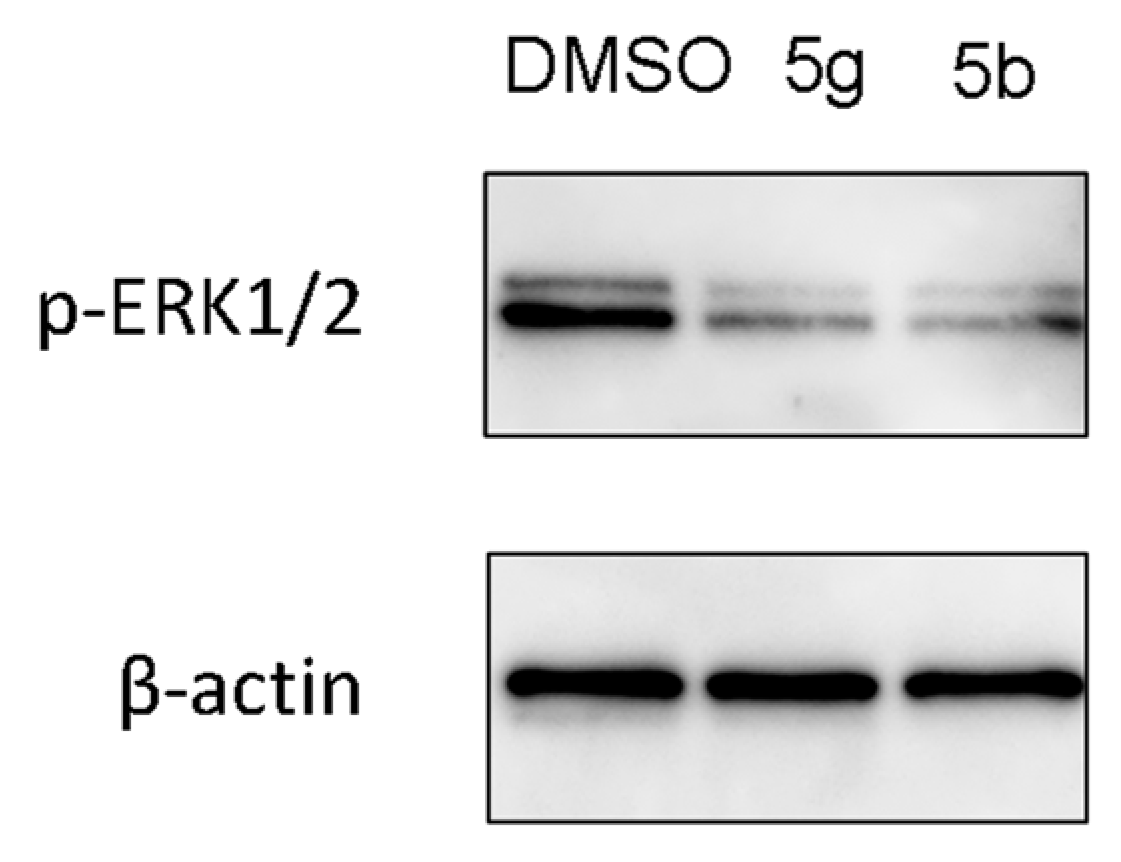
3.2.4. Western Blot Analysis
4. In Silico Screening Studies
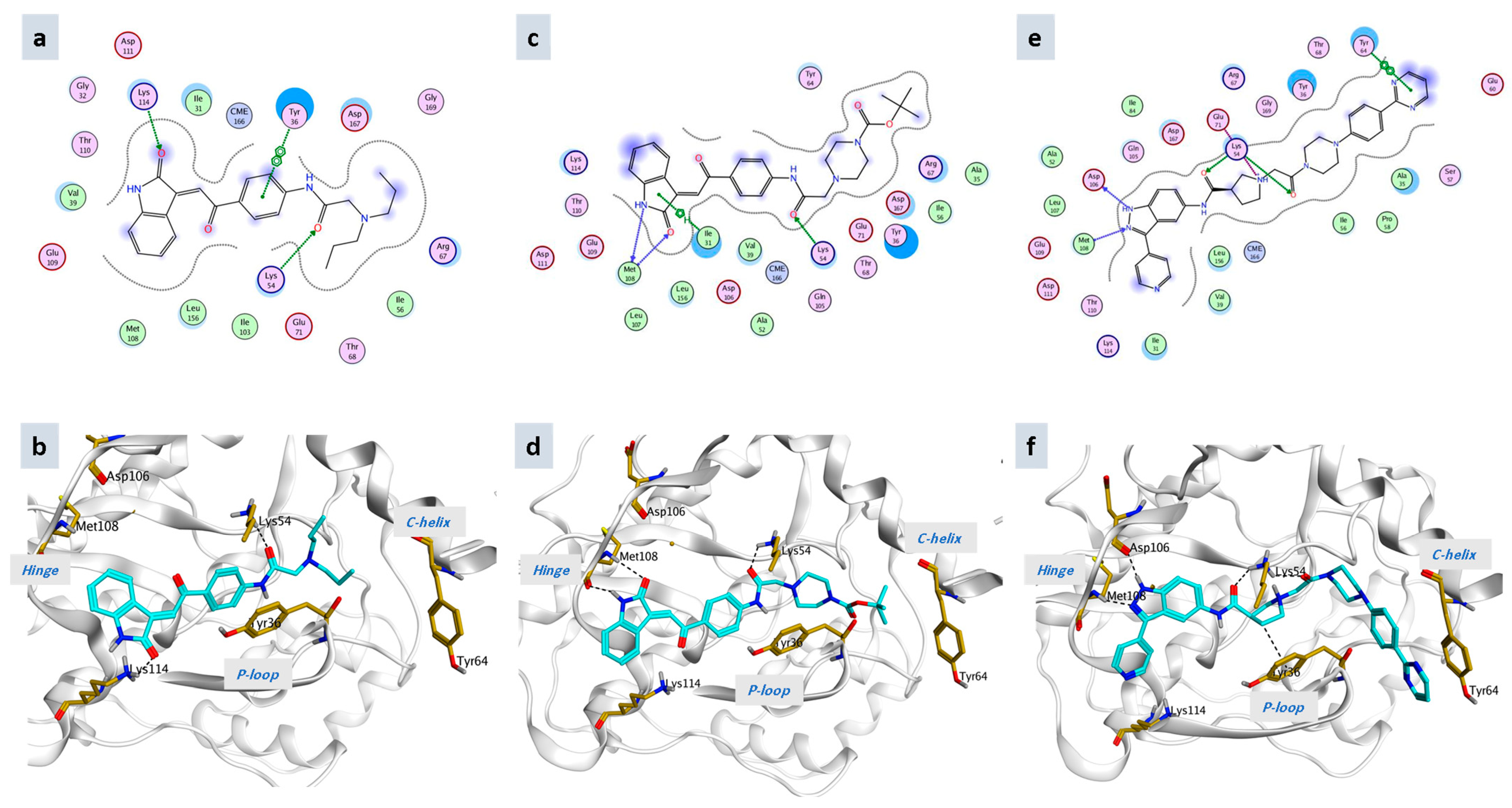
4.1. Molecular Docking
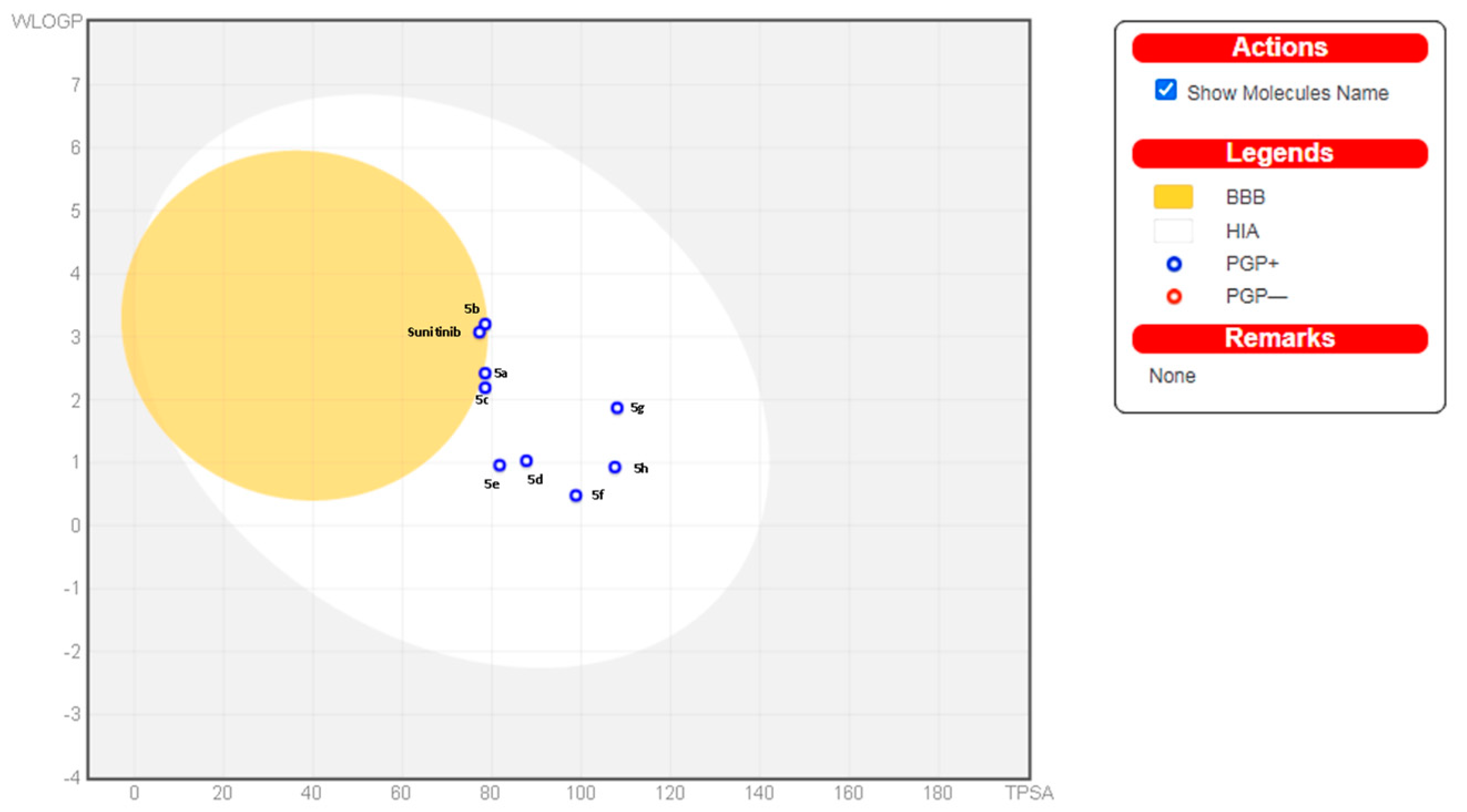
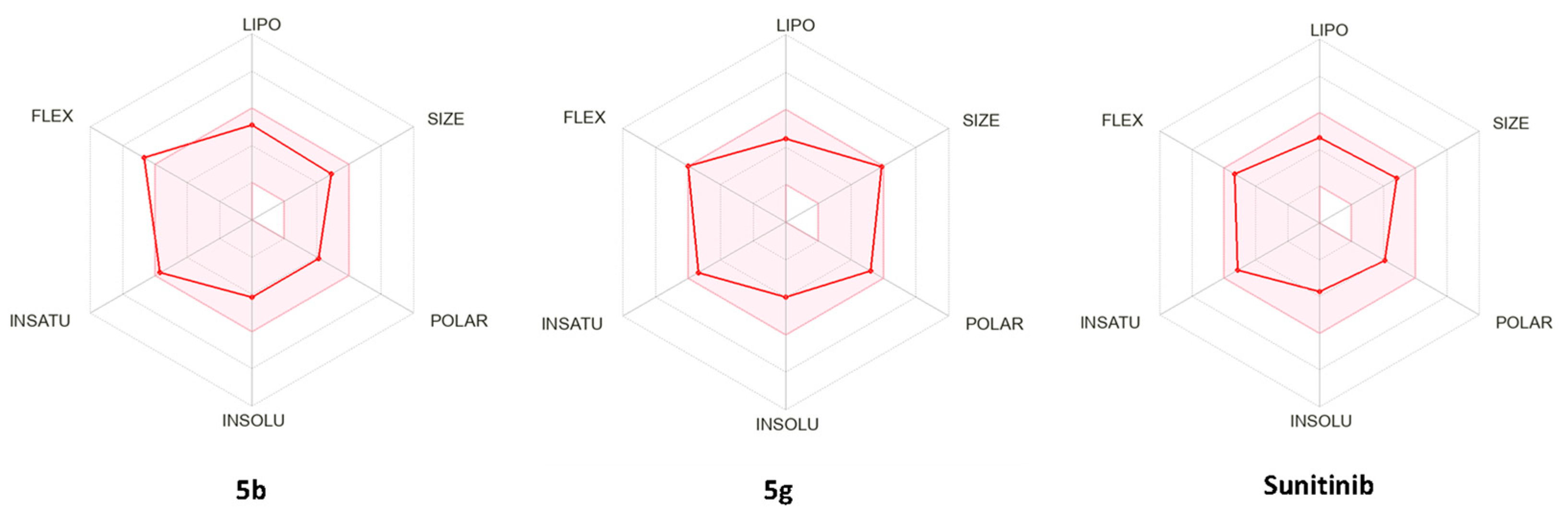
4.2. In Silico Prediction of Physicochemical and Pharmacokinetic Properties
5. Conclusions
6. Materials and Methods
6.1. Chemistry
6.1.1. Synthesis of (E)-3-(2-(4-Aminophenyl)-2-oxoethylidene)indolin-2-one (3)
6.1.2. Synthesis of (E)-2-Chloro-N-(4-(2-(2-oxoindolin-3-ylidene)acetyl)phenyl)acetamide (4)
6.1.3. General Procedure for Synthesis of 5a–h
6.1.4. (E)-2-(Diethylamino)-N-(4-(2-(2-oxoindolin-3-ylidene)acetyl)phenyl)acetamide (5a)
6.1.5. (E)-2-(Dipropylamino)-N-(4-(2-(2-oxoindolin-3-ylidene)acetyl)phenyl)acetamide (5b)
6.1.6. (E)-N-(4-(2-(2-Oxoindolin-3-ylidene)acetyl)phenyl)-2-(piperidin-1-yl)acetamide (5c)
6.1.7. (E)-2-Morpholino-N-(4-(2-(2-oxoindolin-3-ylidene)acetyl)phenyl)acetamide (5d)
6.1.8. (E)-2-(4-Ethylpiperazin-1-yl)-N-(4-(2-(2-oxoindolin-3-ylidene)acetyl)phenyl) acetamide (5e)
6.1.9. (E)-2-(4-Acetylpiperazin-1-yl)-N-(4-(2-(2-oxoindolin-3-ylidene)acetyl)phenyl)acetamide (5f)
6.1.10. Tert-butyl (E)-4-(2-oxo-2-((4-(2-(2-oxoindolin-3-ylidene)acetyl)phenyl)amino) ethyl) piperazine-1-carboxylate (5g)
6.1.11. (E)-N-(4-(2-(2-Oxoindolin-3-ylidene)acetyl)phenyl)-2-(4-(pyrimidin-2-yl)piperazin-1-yl)acetamide (5h)
6.2. Biology
6.2.1. Evaluation of In Vitro Antiproliferative Activity for Compounds by NCI
6.2.2. Evaluation of Antiproliferative Activity for Compounds against HL-60 and PBMC
6.2.3. Cell Cycle Analysis and Apoptosis Analysis
6.2.4. Western Blot
6.3. In Silico Screening
6.3.1. Molecular Docking
6.3.2. Prediction of Physicochemical Properties and Pharmacokinetics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yun, D.; Yao, J.; Fu, W.; Huang, F.; Chen, L.; Wei, T.; Yu, C.; Xu, H.; Zhou, X.; et al. Design, synthesis and QSAR study of novel isatin analogues inspired Michael acceptor as potential anticancer compounds. Eur. J. Med. Chem. 2018, 144, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Key Statistics for Acute Myeloid Leukemia (AML), (n.d.). Available online: https://www.cancer.org/cancer/acute-myeloid-leukemia/about/key-statistics.html (accessed on 15 November 2022).
- Jastaniah, W.; Al Ghemlas, I.; Al Daama, S.; Ballourah, W.; Bayoumy, M.; Al-Anzi, F.; al Shareef, O.; Alsultan, A.; Abrar, M.B.; Al Sudairy, R. Clinical characteristics and outcome of childhood de novo acute myeloid leukemia in Saudi Arabia: A multicenter SAPHOS leukemia group study. Leuk. Res. C 2016, 49, 66–72. [Google Scholar] [CrossRef] [PubMed]
- What Is Acute Myeloid Leukemia (AML)?|What Is AML? (n.d.). Available online: https://www.cancer.org/cancer/acute-myeloid-leukemia/about/what-is-aml.html (accessed on 15 November 2022).
- Ismail, M.; Abdullatif, N.; Nagdy, N.; Al Shaikh, A. Double Hematological and Solid Malignancy Diagnosed from Bone Marrow Studies: Case Report, Laboratory View. J. Umm Al-Qura Univ. Med. Sci. 2020, 6, 4–7. [Google Scholar] [CrossRef]
- Amin, A.H.; Al Sharifi, L.M.; Kakhharov, A.J.; Opulencia, M.J.C.; Alsaikhan, F.; Bokov, D.O.; Majdi, H.S.; Jawad, M.A.; Hammid, A.T.; Shalaby, M.N.; et al. Role of Acute Myeloid Leukemia (AML)-Derived exosomes in tumor progression and survival. Biomed. Pharmacother. 2022, 150, 113009. [Google Scholar] [CrossRef]
- Im, D.; Jun, J.; Baek, J.; Kim, H.; Kang, D.; Bae, H.; Cho, H.; Hah, J.M. Rational design and synthesis of 2-(1H-indazol-6-yl)-1H-benzo[d]imidazole derivatives as inhibitors targeting FMS-like tyrosine kinase 3 (FLT3) and its mutants. J. Enzym. Inhib. Med. Chem. 2022, 37, 472–486. [Google Scholar] [CrossRef]
- Varun; Sonam; Kakkar, R. Isatin and its derivatives: A survey of recent syntheses, reactions, and applications. Medchemcomm 2019, 10, 351–368. [Google Scholar] [CrossRef]
- Al-Warhi, T.; Elimam, D.M.; Elsayed, Z.M.; Abdel-Aziz, M.M.; Maklad, R.M.; Al-Karmalawy, A.A.; Afarinkia, K.; Abourehab, M.A.S.; Abdel-Aziz, H.A.; Eldehna, W.M. Development of novel isatin thiazolyl-pyrazoline hybrids as promising antimicrobials in MDR pathogens. RSC Adv. 2022, 12, 31466–31477. [Google Scholar] [CrossRef]
- Abdalla, A.N.; Di Stefano, M.; Poli, G.; Tuccinardi, T.; Bader, A.; Vassallo, A.; Abdallah, M.E.; El-Readi, M.Z.; Refaat, B.; Algarni, A.S.; et al. Co-Inhibition of P-gp and Hsp90 by an Isatin-Derived Compound Contributes to the Increase of the Chemosensitivity of MCF7/ADR-Resistant Cells to Doxorubicin. Molecules 2021, 27, 90. [Google Scholar] [CrossRef]
- Morabito, A.; De Maio, E.; Di Maio, M.; Normanno, N.; Perrone, F. Tyrosine Kinase Inhibitors of Vascular Endothelial Growth Factor Receptors in Clinical Trials: Current Status and Future Directions. Oncologist 2006, 11, 753–764. [Google Scholar] [CrossRef]
- Chuma, M.; Terashita, K.; Sakamoto, N. New molecularly targeted therapies against advanced hepatocellular carcinoma: From molecular pathogenesis to clinical trials and future directions. Hepatol. Res. 2015, 45, E1–E11. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, F.; Al-Oteibi, M.; Layati, S.; Kadi, F.; Chaudhary, A.; Gari, M.; Al-Qahtani, M. Sunitinib effectively reduces clonogenic acute myeloid leukemia cells in vitro. BMC Genom. 2014, 15, P67. [Google Scholar] [CrossRef] [Green Version]
- Fiedler, W.; Kayser, S.; Kebenko, M.; Janning, M.; Krauter, J.; Schittenhelm, M.; Götze, K.; Weber, D.; Göhring, G.; Teleanu, V.; et al. A phase I/II study of sunitinib and intensive chemotherapy in patients over 60 years of age with acute myeloid leukaemia and activating FLT3 mutations. Br. J. Haematol. 2015, 169, 694–700. [Google Scholar] [CrossRef] [PubMed]
- Gowda, K.; Annageldiyev, C.; Desai, D.; Amin, S.; Claxton, D.; Sharma, A. Abstract 795: Isatin analog for the treatment of acute myeloid leukemia. Cancer Res. 2018, 78, 795. [Google Scholar] [CrossRef]
- Ding, Z.; Zhou, M.; Zeng, C. Recent advances in isatin hybrids as potential anticancer agents. Arch. Pharm. 2020, 353, 1900367. [Google Scholar] [CrossRef]
- Medvedev, A.; Buneeva, O.; Gnedenko, O.; Ershov, P.; Ivanov, A. Isatin, an endogenous nonpeptide biofactor: A review of its molecular targets, mechanisms of actions, and their biomedical implications. Biofactors 2018, 44, 95–108. [Google Scholar] [CrossRef]
- Singh, S.N.; Regati, S.; Paul, A.K.; Layek, M.; Jayaprakash, S.; Reddy, K.V.; Deora, G.S.; Mukherjee, S.; Pal, M. Cu-mediated 1,3-dipolar cycloaddition of azomethine ylides with dipolarophiles: A faster access to spirooxindoles of potential pharmacological interest. Tetrahedron Lett. 2013, 54, 5448–5452. [Google Scholar] [CrossRef]
- Fenton, M.S.; Marion, K.M.; Salem, A.K.; Hogen, R.; Naeim, F.; Hershman, J.M. Sunitinib Inhibits MEK/ERK and SAPK/JNK Pathways and Increases Sodium/Iodide Symporter Expression in Papillary Thyroid Cancer. Thyroid 2010, 20, 965–974. [Google Scholar] [CrossRef]
- Jackson, P.A.; Widen, J.C.; Harki, D.A.; Brummond, K.M. Covalent Modifiers: A Chemical Perspective on the Reactivity of α,β-Unsaturated Carbonyls with Thiols via Hetero-Michael Addition Reactions. J. Med. Chem. 2017, 60, 839. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.; Heiss, E.H.; Schachner, D.; Jiang, B.; Liu, W.; Breuss, J.M.; Dirsch, V.M.; Atanasov, A.G. Xanthohumol Blocks Proliferation and Migration of Vascular Smooth Muscle Cells in Vitro and Reduces Neointima Formation in Vivo. J. Nat. Prod. 2017, 80, 2146–2150. [Google Scholar] [CrossRef]
- Esatbeyoglu, T.; Huebbe, P.; Ernst, I.M.A.; Chin, D.; Wagner, A.E.; Rimbach, G. Curcumin—From Molecule to Biological Function. Angew. Chem. Int. Ed. 2012, 51, 5308–5332. [Google Scholar] [CrossRef] [PubMed]
- Sahu, N.K.; Balbhadra, S.S.; Choudhary, J.; Kohli, D.V. Exploring pharmacological significance of chalcone scaffold: A review. Curr. Med. Chem. 2012, 19, 209–225. [Google Scholar] [CrossRef] [PubMed]
- Farombi, E.O.; Shrotriya, S.; Na, H.K.; Kim, S.H.; Surh, Y.J. Curcumin attenuates dimethylnitrosamine-induced liver injury in rats through Nrf2-mediated induction of heme oxygenase-1. Food Chem. Toxicol. 2008, 46, 1279–1287. [Google Scholar] [CrossRef] [PubMed]
- Shankaraiah, N.; Nekkanti, S.; Brahma, U.R.; Kumar, N.P.; Deshpande, N.; Prasanna, D.; Senwar, K.R.; Lakshmi, U.J. Synthesis of different heterocycles-linked chalcone conjugates as cytotoxic agents and tubulin polymerization inhibitors. Bioorg. Med. Chem. 2017, 25, 4805–4816. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, L.P.; Dai, F.; Yan, W.J.; Wang, H.B.; Tu, Z.S.; Zhou, B. Hexamethoxylated Monocarbonyl Analogues of Curcumin Cause G2/M Cell Cycle Arrest in NCI-H460 Cells via Michael Acceptor-Dependent Redox Intervention. J. Agric. Food Chem. 2015, 63, 7731–7742. [Google Scholar] [CrossRef] [PubMed]
- Gersch, M.; Kreuzer, J.; Sieber, S.A. Electrophilic natural products and their biological targets. Nat. Prod. Rep. 2012, 29, 659–682. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Wu, L.; Dai, H.; Gao, M.; Rashid, H.U.; Wang, H.; Xie, P.; Liu, X.; Jiang, J.; Wang, L. Novel α, β-Unsaturated Sophoridinic Derivatives: Design, Synthesis, Molecular Docking and Anti-Cancer Activities. Molecules 2017, 22, 1967. [Google Scholar] [CrossRef] [Green Version]
- Shaveta; Mishra, S.; Singh, P. Hybrid molecules: The privileged scaffolds for various pharmaceuticals. Eur. J. Med. Chem. 2016, 124, 500–536. [Google Scholar] [CrossRef]
- Kerru, N.; Singh, P.; Koorbanally, N.; Raj, R.; Kumar, V. Recent advances (2015–2016) in anticancer hybrids. Eur. J. Med. Chem. 2017, 142, 179–212. [Google Scholar] [CrossRef]
- Radwan, M.O.; Ciftci, H.I.; Ali, T.F.S.; Koga, R.; Tateishi, H.; Nakata, A.; Ito, A.; Yoshida, M.; Fujita, M.; Otsuka, M. Structure activity study of S-trityl-cysteamine dimethylaminopyridine derivatives as SIRT2 inhibitors: Improvement of SIRT2 binding and inhibition. Bioorg. Med. Chem. Lett. 2020, 30, 127458. [Google Scholar] [CrossRef]
- Radwan, M.O.; Koga, R.; Hida, T.; Ejima, T.; Kanemaru, Y.; Tateishi, H.; Okamoto, Y.; Inoue, J.; Fujita, M.; Otsuka, M. Minimum structural requirements for inhibitors of the zinc finger protein TRAF6. Bioorg. Med. Chem. Lett. 2019, 29, 2162–2167. [Google Scholar] [CrossRef]
- Nakagawa, R.; Tateishi, H.; Radwan, M.O.; Chinen, T.; Ciftci, H.; Iwamaru, K.; Baba, N.; Tominaga, Y.; Koga, R.; Toma, T.; et al. A New 1,2-Naphthoquinone Derivative with Anti-lung Cancer Activity. Chem. Pharm. Bull. 2022, 70, 477–482. [Google Scholar] [CrossRef] [PubMed]
- Goler, A.M.Y.; Jannuzzi, A.T.; Bayrak, N.; Ylldlz, M.; Ylldlrlm, H.; Otsuka, M.; Fujita, M.; Radwan, M.O.; Tuyun, A.F. In Vitro and in Silico Study to Assess Toxic Mechanisms of Hybrid Molecules of Quinone-Benzocaine as Plastoquinone Analogues in Breast Cancer Cells. ACS Omega 2022, 7, 30250–30264. [Google Scholar] [CrossRef] [PubMed]
- Ciftci, H.I.; Radwan, M.O.; Sever, B.; Hamdy, A.K.; Emirdağ, S.; Ulusoy, N.G.; Sozer, E.; Can, M.; Yayli, N.; Araki, N.; et al. EGFR-Targeted Pentacyclic Triterpene Analogues for Glioma Therapy. Int. J. Mol. Sci. 2021, 22, 10945. [Google Scholar] [CrossRef] [PubMed]
- Alkahtani, H.M.; Alanazi, M.M.; Aleanizy, F.S.; Alqahtani, F.Y.; Alhoshani, A.; Alanazi, F.E.; Almehizia, A.A.; Abdalla, A.N.; Alanazi, M.G.; El-Azab, A.S.; et al. Synthesis, anticancer, apoptosis-inducing activities and EGFR and VEGFR2 assay mechanistic studies of 5,5-diphenylimidazolidine-2,4-dione derivatives: Molecular docking studies. Saudi Pharm. J. 2019, 27, 682–693. [Google Scholar] [CrossRef]
- Motzer, R.J.; Escudier, B.; Gannon, A.; Figlin, R.A. Sunitinib: Ten Years of Successful Clinical Use and Study in Advanced Renal Cell Carcinoma. Oncologist 2017, 22, 41–52. [Google Scholar] [CrossRef] [Green Version]
- Mctigue, M.; Murray, B.W.; Chen, J.H.; Deng, Y.-L.; Solowiej, J.; Kania, R.S. Molecular conformations, interactions, and properties associated with drug efficiency and clinical performance among VEGFR TK inhibitors. Proc. Natl. Acad. Sci. USA 2012, 109, 18281–18289. [Google Scholar] [CrossRef] [Green Version]
- NCI-60 Screening Methodology|NCI-60 Human Tumor Cell Lines Screen|Discovery & Development Services|Developmental Therapeutics Program (DTP), (n.d.). Available online: https://dtp.cancer.gov/discovery_development/nci-60/methodology.htm (accessed on 4 November 2022).
- Nishimura, N.; Radwan, M.O.; Amano, M.; Endo, S.; Fujii, E.; Hayashi, H.; Ueno, S.; Ueno, N.; Tatetsu, H.; Hata, H.; et al. Novel p97/VCP inhibitor induces endoplasmic reticulum stress and apoptosis in both bortezomib-sensitive and -resistant multiple myeloma cells. Cancer Sci. 2019, 110, 3275–3287. [Google Scholar] [CrossRef] [Green Version]
- Daina, A.; Michielin, O.; Zoete, V. SwissTargetPrediction: Updated data and new features for efficient prediction of protein targets of small molecules. Nucleic Acids Res. 2019, 47, W357–W3664. [Google Scholar] [CrossRef] [Green Version]
- Kidger, A.M.; Sipthorp, J.; Cook, S.J. ERK1/2 inhibitors: New weapons to inhibit the RAS-regulated RAF-MEK1/2-ERK1/2 pathway. Pharmacol Ther. 2018, 187, 45–60. [Google Scholar] [CrossRef]
- Lu, Y.; Liu, B.; Liu, Y.; Yu, X.; Cheng, G. Dual effects of active ERK in cancer: A potential target for enhancing radiosensitivity. Oncol. Lett. 2020, 20, 993. [Google Scholar] [CrossRef] [PubMed]
- Sammons, R.M.; Ghose, R.; Tsai, K.Y.; Dalby, K.N. Targeting ERK beyond the boundaries of the kinase active site in melanoma. Mol. Carcinog. 2019, 58, 1551–1570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Madhoun, N.Y.; Gadhoum, S.Z.; Merzaban, J.S. ERK1/2 Pathway Is Required for Differentiation of AML Triggered by Anti-CD44 Monoclonal Antibodies. Blood 2012, 120, 4334. [Google Scholar] [CrossRef]
- Heightman, T.D.; Berdini, V.; Braithwaite, H.; Buck, I.M.; Cassidy, M.; Castro, J.; Courtin, A.; Day, J.E.H.; East, C.; Fazal, L.; et al. Fragment-Based Discovery of a Potent, Orally Bioavailable Inhibitor That Modulates the Phosphorylation and Catalytic Activity of ERK1/2. J. Med. Chem. 2018, 61, 4978–4992. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Pei, J.; Wang, A.; Shuai, W.; Feng, L.; Bu, F.; Zhu, Y.; Zhang, L.; Wang, G.; Ouyang, L. Development of small molecule extracellular signal-regulated kinases (ERKs) inhibitors for cancer therapy. Acta Pharm. Sin. B 2022, 12, 2171–2192. [Google Scholar] [CrossRef]
- RCSB PDB—6GDM: Fragment-Based Discovery of a Highly Potent, Orally Bioavailable Inhibitor which Modulates the Phosphorylation and Catalytic Activity of ERK1/2, (n.d.). Available online: https://www.rcsb.org/structure/6GDM (accessed on 16 November 2022).
- Daina, A.; Zoete, V. A BOILED-Egg to Predict Gastrointestinal Absorption and Brain Penetration of Small Molecules. ChemMedChem 2016, 11, 1117–1121. [Google Scholar] [CrossRef] [Green Version]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Ciftci, H.I.; Ozturk, S.E.; Ali, T.F.S.; Radwan, M.O.; Tateishi, H.; Koga, R.; Ocak, Z.; Can, M.; Otsuka, M.; Fujita, M. The First Pentacyclic Triterpenoid Gypsogenin Derivative Exhibiting Anti-ABL1 Kinase and Anti-chronic Myelogenous Leukemia Activities. Biol. Pharm. Bull. 2018, 41, 570–574. [Google Scholar] [CrossRef] [Green Version]
- Toma, T.; Tateishi, H.; Kawakami, K.; Ali, T.F.S.; Kamo, M.; Monde, K.; Nakashima, Y.; Fujita, M.; Otsuka, M. Novel Inhibitor for Downstream Targeting of Transforming Growth Factor-β Signaling to Suppress Epithelial to Mesenchymal Transition and Cell Migration. Int. J. Mol. Sci. 2022, 23, 5047. [Google Scholar] [CrossRef]
- Yıldırım, H.; Yıldız, M.; Bayrak, N.; Mataracı-Kara, E.; Özbek-Çelik, B.; Otsuka, M.; Fujita, M.; Radwan, M.O.; Tuyun, A.F. Natural-product-inspired design and synthesis of thiolated coenzyme Q analogs as promising agents against Gram-positive bacterial strains: Insights into structure–activity relationship, activity profile, mode of action, and molecular docking. RSC Adv. 2022, 12, 20507–20518. [Google Scholar] [CrossRef]
- Yıldırım, H.; Yıldız, M.; Bayrak, N.; Mataracı-Kara, E.; Radwan, M.O.; Jannuzzi, A.T.; Otsuka, M.; Fujita, M.; Tuyun, A.F. Promising Antibacterial and Antifungal Agents Based on Thiolated Vitamin K3 Analogs: Synthesis, Bioevaluation, Molecular Docking. Pharmaceuticals 2022, 15, 586. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. ILOGP: A simple, robust, and efficient description of n-octanol/water partition coefficient for drug design using the GB/SA approach. J. Chem. Inf. Model. 2014, 54, 3284–3301. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Panel | Cell Line | Growth Inhibition (GI) Percent at 10 µM | |||||||
|---|---|---|---|---|---|---|---|---|---|
| 5a | 5b | 5c | 5d | 5e | 5f | 5g | 5h | ||
| Leukemia | CCRF-CEM | −0.58 | −2.47 | −25.57 | −11.86 | 95.63 | −4.06 | −22.12 | −22.54 |
| HL-60 (TB) | −45.21 | −43.91 | −46.85 | −32.82 | −42.41 | 92.45 | −46.44 | 87.43 | |
| K-562 | −25.82 | −35.03 | 99.15 | 97.72 | −14.55 | 75.31 | −22.75 | 73.96 | |
| MOLT-4 | −22.48 | −31.26 | −20.73 | −16.45 | 97.87 | 96.34 | −44.85 | 96.53 | |
| RPMI-8226 | −13.65 | −26.17 | 99.56 | 87.8 | 89.17 | 51.88 | −28.80 | 57.83 | |
| Non-Small Cell Lung Cancer | A549/ATCC | 7.65 | 15.65 | 5.75 | 8.73 | 5.40 | 3.91 | 25.07 | - |
| EKVX | 64.47 | 97.33 | 58.57 | 52.43 | 55.57 | 34.27 | −20.50 | 29.54 | |
| HOP-62 | 2.47 | 5.92 | 10.48 | 4.68 | 1.90 | 1.32 | 13.62 | 2.03 | |
| HOP-92 | 72.22 | −30.46 | 51.94 | 34.82 | 61.49 | 16.95 | −40.81 | 6.74 | |
| NCI-H226 | 90.40 | −32.95 | 68.41 | 62.8 | 89.46 | 20.85 | −75.77 | 30.22 | |
| NCI-H23 | 31.40 | 49.02 | 35.91 | 25.51 | 28.69 | 14.27 | 87.95 | 13.07 | |
| NCI-H322M | 7.28 | 2.41 | 9.22 | 8.40 | 1.08 | 2.44 | 13.76 | - | |
| NCI-H460 | 22.85 | 32.52 | 27.57 | 13.56 | 17.94 | - | 31.57 | 5.68 | |
| NCI-H522 | −51.49 | −75.35 | 65.98 | 72.97 | 89.06 | 64.81 | −81.87 | 50.89 | |
| Colon Cancer | COLO 205 | −44.48 | −68.73 | 76.69 | 72.46 | 96.60 | 25.69 | −94.90 | 25.62 |
| HCC-2998 | - | - | - | - | - | - | 14.35 | - | |
| HCT-116 | −88.52 | −88.15 | −75.60 | −23.15 | −66.61 | 55.51 | −96.98 | 58.67 | |
| HCT-15 | −10.90 | −51.56 | −17.35 | 91.63 | 90.36 | 31.60 | −61.56 | 28.88 | |
| HT29 | −32.52 | −47.12 | 79.2 | 51.52 | 80.76 | 19.20 | −50.13 | 13.02 | |
| KM12 | 4.34 | 17.72 | 5.98 | 4.3 | 7.44 | - | 26.35 | - | |
| SW-620 | −62.38 | −76.08 | −11.10 | 97.61 | −17.31 | 52.79 | −88.72 | 58.93 | |
| CNS Cancer | SF-268 | 27.17 | 25.01 | 6.29 | 21.48 | 24.54 | - | 34.98 | 7.61 |
| SF-295 | - | 3.44 | 4.19 | 1.03 | - | - | 13.25 | - | |
| SF-539 | 58.59 | −35.29 | 51.98 | 33.45 | 47.06 | 19.08 | −72.33 | 18.57 | |
| SNB-19 | 18.59 | 29.18 | 16.02 | 13.30 | 14.21 | 5.03 | 29.92 | 1.38 | |
| SNB-75 | 24.89 | 32.00 | 8.81 | 23.84 | 24.28 | - | - | 38.98 | |
| U251 | 20.79 | 36.19 | 16.26 | 13.18 | 13.21 | 1.68 | 32.48 | 4.09 | |
| Melanoma | LOX IMVI | −61.45 | −98.96 | −59.39 | −32.88 | −33.15 | 55.70 | −100 | 59.67 |
| MALME-3M | −51.93 | −89.79 | −11.41 | −30.57 | −30.35 | 98.15 | −81.40 | 80.97 | |
| M14 | 55.53 | −57.59 | 43.48 | 36.56 | 41.68 | 21.33 | −48.57 | 23.73 | |
| MDA-MB-435 | 18.23 | 32.15 | 16.21 | 14.82 | 16.54 | 0.65 | 28.10 | 5.04 | |
| SK-MEL-2 | 1.35 | 26.27 | 11.26 | 3.60 | - | 3.62 | 29.01 | 5.84 | |
| SK-MEL-28 | 26.14 | 76.26 | 23.69 | 10.76 | 9.36 | 4.05 | −7.77 | 5.89 | |
| SK-MEL-5 | 22.84 | 29.75 | 16.39 | 16.76 | 12.88 | 0.23 | 92.07 | 2.24 | |
| UACC-257 | 35.99 | 40.23 | 26.66 | 18.85 | 23.92 | 13.54 | 74.24 | 14.06 | |
| UACC-62 | 44.45 | 64.56 | 40.53 | 40.72 | 28.94 | 27.21 | 78.70 | 22.70 | |
| Ovarian cancer | IGROV1 | 20.65 | 40.83 | 16.87 | 14.78 | 11.68 | - | 74.06 | - |
| OVCAR-3 | −45.16 | −72.98 | −32.20 | −35.23 | −31.09 | 56.64 | −83.94 | 51.82 | |
| OVCAR-4 | 45.75 | 48.46 | 30.51 | 25.41 | 37.02 | 10.47 | −7.98 | 13.22 | |
| OVCAR-5 | 30.72 | 38.39 | 28.57 | 23.16 | 27.63 | 6.67 | −33.88 | - | |
| OVCAR-8 | 42.34 | −48.41 | 41.34 | 26.49 | 27.92 | 1.19 | −39.58 | 9.37 | |
| NCI/ADR-RES | 13.50 | 74.59 | 40.33 | 10.56 | 11.44 | 0.23 | 92.02 | 2.54 | |
| SK-OV-3 | 1.01 | 6.23 | 11.06 | 8.00 | 1.87 | - | 6.75 | 4.52 | |
| Renal Cancer | 786–0 | 97.75 | −87.01 | 42.78 | 30.65 | 61.43 | 17.68 | −90.34 | 17.63 |
| A498 | - | - | - | - | - | - | - | - | |
| ACHN | −27.51 | −97.53 | 98.59 | 97.21 | −23.79 | 24.89 | −97.14 | 21.01 | |
| CAKI-1 | 66.71 | 87.63 | 39.91 | 42.13 | 59.4 | 28.32 | −20.53 | 21.56 | |
| RXF 393 | −50.71 | −93.84 | 99.46 | 81.83 | −10.63 | 45.27 | −96.23 | 42.73 | |
| SN12C | 47.76 | 71.39 | 42.9 | 30.75 | 51.07 | 22.72 | −37.69 | 11.87 | |
| TK-10 | 41.12 | −27.68 | 5.04 | 5.76 | 29.77 | - | −25.96 | - | |
| Prostate Cancer | PC-3 | 20.15 | 68.93 | 48.74 | 9.24 | 16.48 | 4.42 | 74.73 | 14.82 |
| DU-145 | 68.03 | 93.75 | 32.78 | 48.07 | 43.57 | - | 92.09 | 3.35 | |
| Breast Cancer | MCF7 | 97.26 | −26.39 | 88.56 | 77.09 | 86.96 | 74.61 | −86.32 | 66.02 |
| HS 578T | 28.86 | 39.6 | 30.69 | 22.04 | 24.84 | 12.17 | 61.59 | 14.34 | |
| BT-549 | 17.90 | 40.49 | 22.76 | - | 10.65 | - | −21.27 | 4.81 | |
| T-47D | 59.34 | −12.28 | 53.94 | 46.40 | 60.89 | 43.43 | −66.39 | 47.61 | |
| MDA-MB-468 | 68.69 | 76.47 | 57.59 | 51.50 | 53.78 | 49.41 | −67.01 | 50.04 | |
| Mean | 63.66 | 90.35 | 52.52 | 44.76 | 51.47 | 23.22 | −8.44 | 24.36 | |
| Cells | 5a | 5b | 5c | 5d | 5e | 5f | 5g | 5h | Staurosporine |
|---|---|---|---|---|---|---|---|---|---|
| HL60 | 0.72 ± 0.09 | 0.38 ± 0.08 | 1.06 ± 0.10 | 1.43 ± 0.16 | 1.07 ± 0.13 | 2.57 ± 0.29 | 0.57 ± 0.05 | 0.79 ± 0.08 | 0.013 ± 0.002 |
| PBMC | - | 14.17 ± 1.13 | - | - | - | - | - | - | - |
| SI | - | 37.2 | - | - | - | - | - | - | - |
| Sample | Cell Cycle Distribution (%) | ||
|---|---|---|---|
| %G0-G1 | %S | %G2/M | |
| DMSO/HL-60 | 61 | 16.7 | 21.5 |
| 5b/HL-60 | 68.8 | 10.3 | 20.1 |
| 5g/HL-60 | 67.5 | 15.6 | 16.7 |
| 5a | 5b | 5c | 5d | 5e | 5f | 5g | 5h | Sunitinib | |
|---|---|---|---|---|---|---|---|---|---|
| Mwt | 377.44 | 405.49 | 389.45 | 391.42 | 418.49 | 432.47 | 490.55 | 468.51 | 398.47 |
| Fraction Csp3 | 0.23 | 0.29 | 0.26 | 0.23 | 0.29 | 0.25 | 0.33 | 0.19 | 0.36 |
| Num. rotatable bonds | 8 | 10 | 6 | 6 | 7 | 7 | 9 | 7 | 8 |
| Num. H-bond acceptors | 4 | 4 | 4 | 5 | 5 | 5 | 6 | 6 | 4 |
| Num. H-bond donors | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 3 |
| Molar Refractivity | 113.01 | 122.63 | 119.62 | 115.90 | 131.24 | 131.44 | 147.37 | 142.26 | 116.31 |
| TPSA | 78.51 | 78.51 | 78.51 | 87.74 | 81.75 | 98.82 | 108.05 | 107.53 | 77.23 |
| Log P | 2.25 | 2.61 | 3.05 | 2.75 | 3.24 | 2.82 | 3.57 | 2.83 | 3.55 |
| GI absorption | High | High | High | High | High | High | High | High | High |
| BBB penetration | No | Yes | No | No | No | No | No | No | Yes |
| Lipinski violation | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Bioavailability score | 0.55 | 0.55 | 0.55 | 0.55 | 0.55 | 0.55 | 0.55 | 0.55 | 0.55 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hamdy, A.K.; Sakamoto, T.; Toma, T.; Sakamoto, M.; Abourehab, M.A.S.; Otsuka, M.; Fujita, M.; Tateishi, H.; Radwan, M.O. New Insights into the Structural Requirements of Isatin-Derived Pro-Apoptotic Agents against Acute Myeloid Leukemia. Pharmaceuticals 2022, 15, 1579. https://doi.org/10.3390/ph15121579
Hamdy AK, Sakamoto T, Toma T, Sakamoto M, Abourehab MAS, Otsuka M, Fujita M, Tateishi H, Radwan MO. New Insights into the Structural Requirements of Isatin-Derived Pro-Apoptotic Agents against Acute Myeloid Leukemia. Pharmaceuticals. 2022; 15(12):1579. https://doi.org/10.3390/ph15121579
Chicago/Turabian StyleHamdy, Ahmed K., Takashi Sakamoto, Tsugumasa Toma, Masaharu Sakamoto, Mohammed A. S. Abourehab, Masami Otsuka, Mikako Fujita, Hiroshi Tateishi, and Mohamed O. Radwan. 2022. "New Insights into the Structural Requirements of Isatin-Derived Pro-Apoptotic Agents against Acute Myeloid Leukemia" Pharmaceuticals 15, no. 12: 1579. https://doi.org/10.3390/ph15121579
APA StyleHamdy, A. K., Sakamoto, T., Toma, T., Sakamoto, M., Abourehab, M. A. S., Otsuka, M., Fujita, M., Tateishi, H., & Radwan, M. O. (2022). New Insights into the Structural Requirements of Isatin-Derived Pro-Apoptotic Agents against Acute Myeloid Leukemia. Pharmaceuticals, 15(12), 1579. https://doi.org/10.3390/ph15121579