3. Discussion
NSC-34 is a cell line widely used as a motor neuron model. It is suitable for the testing of different disease-like conditions, like neurotoxicity [
18] as well as neurodegeneration [
19]. In this work, using the scratch-injury technique [
20,
21], we want to mimic the effect of an injury on differentiated NSC-34 cells to answer if the treatment of CBG influences the outcome of traumatic damage and neuronal regeneration. We exposed the cells at CBG at pre-treatment and at post-treatment, after the cells already suffered traumatic injury. The following transcriptomic analysis was performed to elucidate the effects of CBG treatments on the differential expression of motor neuron genes after scratch injury.
As previously reported, oxidative stress and calcium waves are responsible for creating an unfavorable environment for the neurons, which were spared from physical injury, leading them to death. For these reasons, an evaluation on the stress status of the cells and on the main hallmarks of apoptosis was done.
The process of apoptosis is attenuated, but not yet abolished in the pre-treatment group.
Bax gene expression does not seem to be different compared to scratched cells, where it was upregulated, but after the pre-treatment the pro-apoptotic gene
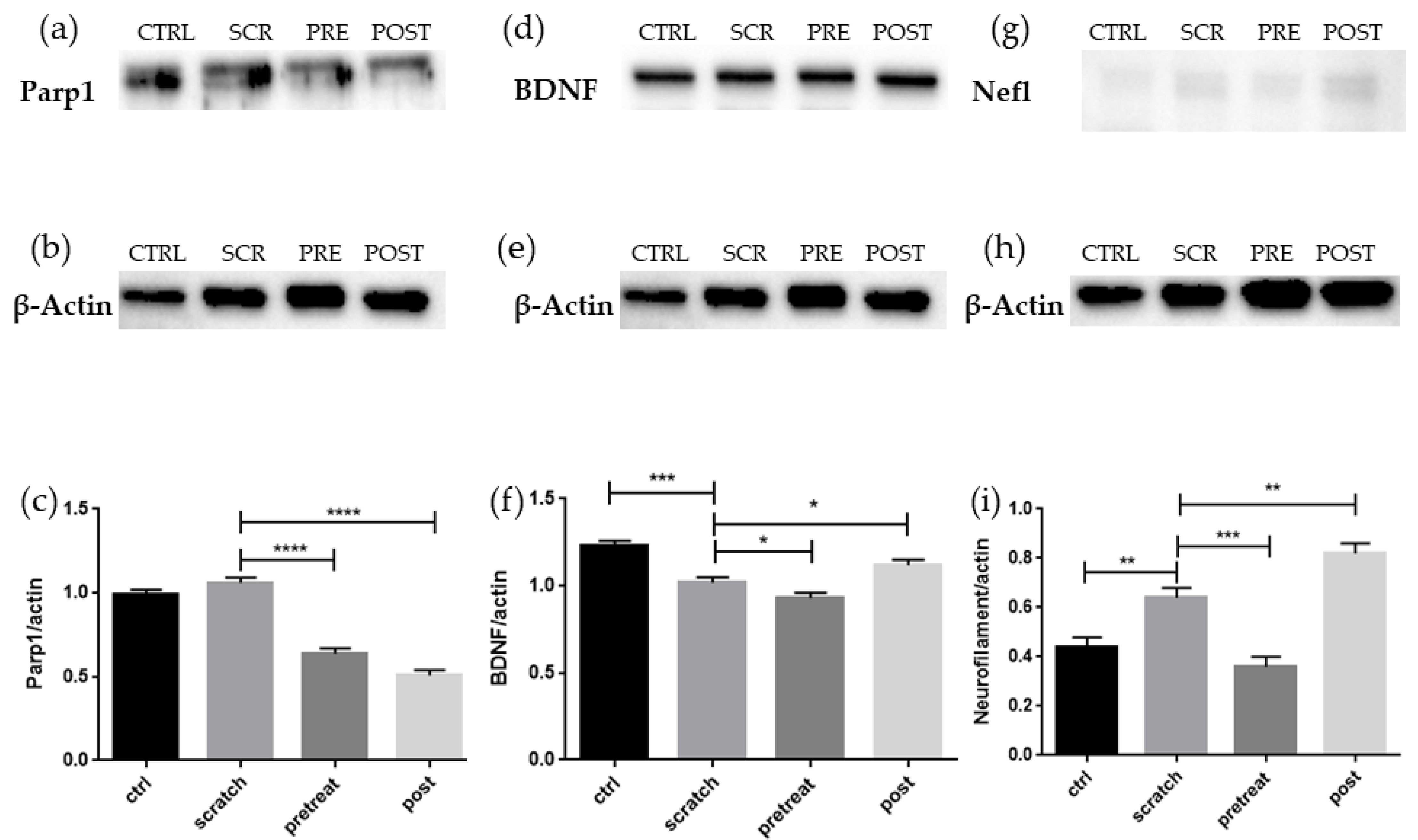
Parp1 was downregulated [
22]. Parp1 protein level does not appear to be significantly different between the control and scratched cells, probably because cell harvest occurred 48 h after the injury was made. However, there is an increasing trend between control and scratched cells. As in the transcriptomic data, Parp1 protein is significantly reduced in pre-treated cells, but it is even more decreased in post-treated cells. Previous studies suggest that inhibition of Parp1 promotes axonal regrowth, but the translation of this model from invertebrate to vertebrates fails to demonstrate the connection between
Parp1 and axonal regrowth, indicating that more in-depth analyses are required [
23].
Pidd1, encoding for p53-induced death domain protein 1 [
24], is downregulated as well and, along with the upregulation of pro-survival genes, like
Birc2 [
25], indicates a possible role of CBG pre-treatment in limit of apoptosis signaling. No upregulation of Caspases genes has taken place. Also, comparing the scratched cells with the ones which received CBG as post-treatment, the
Bax gene does not vary its expression, but
Pidd1 expression is strongly downregulated, indicating that the increase of
Trp53, encoding for the protein p53, may not be direct to apoptosis. The increased expression of the anti-apoptotic gene
Birc2 level seems to support this hypothesis, along with no increased expression of apoptotic Caspases. The level of
Ddit3 gene, encoding for CHOP, is increased in the post-treatment cells: CHOP increase is usually correlated with ER stress and apoptosis, along with an increased level of
Jun that, in this comparison, is downregulated [
26]; an experiment on primary neurons connects an increase of BDNF level with the subsequent increase of CHOP, indicating an expanded selection of functions for CHOP over apoptosis. The strong increased level of
Xbp1, indicated as the target of BDNF action via IRE-1α for neurite outgrowth, supports the role of CHOP in BDNF-mediated regeneration, while the downregulation of
Atf4, part of the apoptotic signal cascade mediated by CHOP, point toward a block of this pathway [
27].
There is a link between mitochondrial dysfunction and the onset of ALS, where motor neurons undergo degeneration; since it seems that mitochondrial calcium overload and oxidative stress, among other conditions, can favor the permeability of the mitochondrial membrane, thus leading to cell death [
28]. Genes implicated in the calcium flux and oxidative stress were investigated. The expression of
Cycs does not differ between scratched cells, where it was found upregulated, and pre-treated ones.
Cycs is a key regulator of mitochondria activity and is suggested to represent an early apoptotic marker [
29]. There seemed to be a tentativeness of the cells to balance the expression of the different mitochondria complexes; Cx II is downregulated while Cx IV is upregulated, showing an opposite trend than the one after scratch injury. The expression of the voltage channel coded from the gene
Vdac3 remains upregulated.
Vdac3 seems to have poor control over the redox state of the cells, in contrast its functions seem to be controlled by the redox state [
30].
Mcu expression is reduced, so that the flow of calcium entering mitochondria could be decreased compared to the scratched cells, where
Mcu was upregulated.
Gpx4 is downregulated after the scratch injury but, even if
Gpx4 is an antioxidant enzyme and its downregulation might mean a significant increase in oxidative stress [
31], the expression of
Nos1 is reduced.
Nos1 encodes for neuronal nitric oxide synthase, so decreasing its level of expression can indicate a cell response by attenuating continuous oxidative stress [
32]. Observing the results of post-treated cells’ transcriptome analyses,
Cycs expression is downregulated, so it might indicate that the level of cytochrome C in the mitochondria is on the way to return to the level before the scratch. To support this evidence, also the expression of almost all mitochondrial complex Cxs is downregulated, suggesting a slight return to normality after the upregulation following scratch injury. Neither
Mcu nor
Vdac3 expression levels are different from scratched injured cells, and so it is for oxidative stress genes
Gpx4 and
Nos1.A pathway involved in SCI is the FoxO one. In the pre-treated cells there is a slight increase of
Foxo3 and
Prkab1, a subunit of AMPK, which plays its direct role in the FoxO signaling pathway. For this reason, the FoxO pathway involving AMPK action was inspected. Normally AMPK pathway will lead to cell death, caused by the activation of the pro-apoptotic gene
Bcl2l11, encoding for Bim [
33]. However, in our experimental set, Bim is not overexpressed and AP-1, encoded by
Jun and necessary for apoptosis, is downregulated; therefore, other pathways might be followed [
34]. It has been shown that an axis connecting AMPK activation with FoxO3 and their managing of intracellular oxidative stress by enhancing Thioredoxin, a potent antioxidant of the cell [
35]; indeed the expression of
Txn1, encoding for the cytoplasmic Thioredoxin, is strongly upregulated in our experimental set. This is also supported by the evidence that the signal AMPK-FoxO3 is neuroprotective [
36]. It was also suggested that FoxO3 can play a role in the management of oxidative stress by inducing autophagy as a mechanism of cell survival [
37], so the genes involved in this pathway were also inspected.
Mapk8 is upregulated and seems to be the responsible for the initiation of the FoxO3 signaling towards autophagy;
Bnip3 was shown to protect neuronal cells during oxidative stress by inducing the digestion of unwanted material, which accumulate in the cells [
38], and it was found upregulated. Other players in the autophagy process were found upregulated, such as
Atg3,
Atg4,
Atg7 and
Atg12, [
39] as well as
Becn1. The autophagy properties of
Becn1, encoding for the Beclin-1, can be inhibited by
Bcl2, downregulated in this comparison, while
Uvrag is upregulated and its known to interact with Beclin-1 to form the autophagosome [
40]. Regarding the post-treated cells,
Foxo3 remains upregulated and so does
Prkab1, while
Jun is downregulated, suggesting that the apoptotic pathway signaling in which
Foxo3 is involved is probably not followed.
Atm is a downstream gene of
Foxo3 [
41] and it is known to participate both in apoptosis and in DNA repair. Since apoptosis does not seem to be a followed pathway, it is reasonable to indicate DNA repair as the purpose of
Atm upregulation. These results point to the capacity of CBG to manage the oxidative stress status of the cells by improving the activity of antioxidant enzymes, reducing the apoptosis signals, regulating the intracellular calcium levels and the over-excitation of neurons. The use of CBG in targeted antioxidant therapies may enhance the survivability of motor neurons after a traumatic injury.
From the axonal regeneration point of view, the markers of neuroregeneration were inspected. Arp2/3 complex acts with N-WASP, encoded by the
Wasl gene, and they were shown to be active in Golgi polarization in cells close to the border of a wound of a culture of fibroblasts [
42]. Even though the Arp2/3 complex is important during neurodevelopment [
43], it has been shown that its inhibition, instead of overexpression, leads to axon elongation in primary neurons, suggesting that this complex is a growth cone negative regulator [
44,
45].
Arpc2,
Actr2, and
Actr3 are the genes of complex Arp2/3 found upregulated in scratched cells and in pre-treatment groups, as well as
Wasl.
Gap43 correlates with regeneration and it is often used as a classical marker for the growth cone and axon regeneration [
46], as well as
Prph, encoded for peripherin, whose expression varies during axonal regeneration [
47].
Gap43 and
Prph were downregulated in pre-treated cells.
Nrp1 codes for neuropilin-1, which was found downregulated in scratched cells, and its collateral pruning function seemed important in facilitating the recovery of motor neurons after a traumatic injury [
48].
Map1b knockdown does not affect the amount of regeneration per se, however it plays a role in the cytoskeleton organization in the new-formed branch. Both
Mapb1 and
Nrp1 are upregulated in the pre-treatment. Taken together, these results suggest that the cell might be managing its internal oxidative stress and could be on the path to balance it, because it seems to be preparing the signals to initiate the regeneration. On the other hand, other evidence suggests that CBG treatment after scratch injury is efficient for trigger regeneration signals.
Gap43 was found to be upregulated, along with
Prph.
Nrp1 and
Map1b upregulation points toward the idea that cytoskeleton remodeling is taking place.
Atf3, upregulated in this comparison, is known to be induced in dorsal root ganglion neurons after injury, but it also promotes the axonal regeneration [
49]. It was proposed as an interaction between
Atf3 and
Sox11, upregulated in post-treated cells, in the regeneration of peripheral injured nerves [
50]. Since cytoskeleton remodeling seems influenced by the administration of CBG, genes involved in the architecture of the cytoskeleton were inspected.
Nefl was upregulated after scratch, while the pre-treatment seems to reduce its expression. The stoichiometry between the different neurofilament chains (Neurofilament Light Chain, encoded by
Nefl gene; Neurofilament Medium Chain, encoded by
Nefm gene; Neurofilament Heavy Chain, encoded by
Nefh gene) is strictly regulated, at least in normal healthy neurons [
51]. The downregulation of
Nefl in pre-treated cells could be interpreted as an attempt of the cell to balance the broken stoichiometry. On the other hand, the expression of
Nefm is downregulated as well, again leading to the imbalance of the amount of neurofilament chains.
Tardbp, which encodes for the protein TDP-43, remained upregulated, as was already seen after the scratch injury. It has been suggested its capacity to bind to human NFL and form cytosolic aggregates and, in such a way, disturb the balance in neurofilaments stoichiometry [
52,
53]. The cytoplasmic accumulation of TDP-43, which gains toxic function, is known to deregulate the mRNA translation, so probably the decrease of
Nefm expression is correlated with the increase of TDP-43 [
54,
55]. Similar to the previous comparison,
Nefl is downregulated also in the post-treatment, but
Nefh was upregulated. Even though the heavy chain of neurofilaments could undergo the same fate as
Nefl in TDP-43 granule accumulation, it is worth noting that no upregulation of
Tardbp gene is taking place. Moreover, evidence suggests a possible role of neurofilament-heavy chain increase mediated by
Bdnf during regeneration, and this could be mediated also by HuD, encoded from the gene
Elavl4; in our experiment,
Nefh is upregulated and so is
Bdnf, while
Elavl4 is downregulated. The explanation of this apparently weird scenario is given by the timeline of upregulation observed in an experiment of HIV-neuropathy. After ddC injection, the first increase observed was of the BDNF protein level, one day after the injection, while HuD and NF-H followed after three days [
56]. It is reasonable to think that NF-H, involved in cytoskeleton organization, is already upregulated because of a regenerative process already started. This hypothesis is reinforced by Western Blot analyses; the protein level of
Nefl increases after scratch and decreases in the pretreated cells, as found in the transcriptomic analyses, but then it increases again in the post-treated cells, apparently in contrast with the transcriptomic data. It is important to note that, during axonal regeneration, the first upregulated genes are intein and peripherin, along with
Nefl, followed by
Nefm and
Nefh is the last to be overexpressed [
57]. This correlates very well with our transcriptomic data. Since
Nefl is the first neurofilament gene to be expressed, it is reasonable to think that its protein has already been synthesized when
Nefh started to be expressed, as in the case of post-treated cells. Indeed, the level of BDNF protein is also higher in post-treated cells compared to the scratched ones, connecting
Bdnf and neurofilaments with the path of axonal regeneration.
Since several signals point toward axonal regeneration, a deeper analysis on the mechanisms, pathways, and genes involved in this important process were done, to give a more complete scenario in the post-treated cells. As mentioned before,
Trp53, encoding for the protein p53, is upregulated. P53 is involved in a large variety of processes, ranging from apoptosis to DNA repair [
58], but also neuroregeneration.
Gap43 is a target gene of p53, which is acetylated by p300 [
59], encoding by
Ep300, upregulated in our experiment; it was also noticed that knocking down p53 has the potential to decrease the regeneration of nerve fibers in mice and its overexpression has the capacity to increase the size of the growth cone [
60], which could very well correlate with
Nefh upregulation. Other genes inducible by p53 and involved in axonal regeneration or guidance are
Slit2,
Slit3,
Neo1,
Unc5b and
Ephb3.
Unc5b is primarily known for its role in apoptosis, since its interaction with Caspase-3, which is not deregulated in this comparison, mediates cell death [
61]. An experiment involving peripheral nerve injury in mice evaluated the role of
Unc5b in regeneration, since it is the receptor of Netrin-1; the
Unc5b signal acts as a repulsive signal, so it will repel axon growth cones in order to help it to reach the right target. In the mentioned experiment, UNC5B protein level peaks 14 days after injury and the authors interpret this as the time when the regenerating axon passed over the analyzed nerve fragment [
62]. In our experimental comparison, the
Unc5b level is decreased. We harvest the cells long before 14 days, so it is reasonable to think that there was no need for repulsive signals since no axons had the time to reach the target.
Slit2 and
Slit3 are important components of SLIT/ROBO signaling, necessary for the control of the axon pathfinding. SLITs are the secreted protein and ROBOs are their receptors. An experiment of sciatic nerve crush in mice revealed the downregulation of
Slit2 after injury, but also its upregulation during axonal regeneration, which could be the case of our comparison where
Slit2 is slightly upregulated [
63]. The downregulation of the repellent signal from
Slit3 may be interpreted as the
Unc5b one, so the regeneration has just started and there is no point to repel an axon that is not already grown. Moreover, there is no change in
Robos genes expression, suggesting that probably no interaction between SLITs and ROBOs is taking place.
Neo1 is a transmembrane receptor, and it is involved in axon guidance and is downregulated in this comparison. RGMA and NET1 promote the formation of a complex known to mediate the collapse of the growth cone [
64], so the downregulation of
Neo1 could be interpreted as a positive signal of ongoing axonal regeneration.
Ephb3 is downregulated in post-treated cells as well. This belongs to another complex of signals because its ligand is Ephrin3B, encoded by
Efnb3, upregulated in our study, and their interaction can have different outcomes. They play a role in suppressing proliferation in neural stem cells and
Ephb3 can also induce cell death [
65]. It is important to notice that Ephrin3B presence seems to attenuate the cell death after adult TBI in mice [
66]. Taken together, this evidence points to an important role of p53 and
Bdnf in regenerating damaged motor neurons after CBG exposure, making them possible targets for future therapies. A careful management of these pathways through CBG administration could be useful to increase the regeneration of neurons after traumatic injuries.
The NSC-34 cell line does not express
Cnr2, which encodes for the Cannabinoid Receptor 2 [
67], while it does have
Cnr1, the gene for the Cannabinoid Receptor 1. CBG can normally exert its action on both with a lower affinity than CBD [
68], but the pre-treatment seems not to upregulate
Cnr1 gene expression. The treatment of CBG after the injury appears to increase the expression level of
Cnr1, while the scratch injury, without any other treatment, appears to decrease its expression compared to control cells. Another receptor with which CBG could interact is PPAR-γ, but neither the pre-treatment nor the treatment after injury upregulate
Pparg gene expression. CBG proved to be a potent agonist of α-2 adrenoceptor, encoded from the gene
Adra2a, upregulated in the post-injury treatment, and it is a mild antagonist of the 5HT
1A receptor, which is a serotoninergic receptor encoded from the gene
Htr1a [
69]. Even though no difference in this latter gene expression was found, it is worth mentioning that the pre-treatment strongly downregulates the
Htr3a gene, which encodes for the subunit A of the serotonin type 3 receptor. Even if there is no evidence in the literature connecting adrenoceptors and axonal regeneration in motor neurons, an experiment about optic nerve injury in rats elucidates the capacity of α-2 adrenoceptor agonists to promote axonal growth of retinal ganglion cells [
70]. Since, in our experimental set, post-injury CBG treatment seems to upregulate genes involved in the axonal regeneration, it is possible that an α-2 adrenoceptor action could trigger axonal regeneration in motor neurons. Further analyses could better clarify this association. Another experiment aimed to evaluate the mechanism behind the evidence that antagonists of the serotonin 3 receptor (5-HT
3R) have anti-apoptotic and anti-inflammatory effects. The proposed mechanism involved the inhibition of TNF-α pathway via antagonism of 5-HT
3R [
71] and this might be connected with our evidence that CBG pre-treatment reduced the expression of
Tnfaip1. This gene encodes for the TNF Alpha Induced Protein 1, identified as a protein which can be induced by TNF-α [
72]. The results of our experiment are reported in
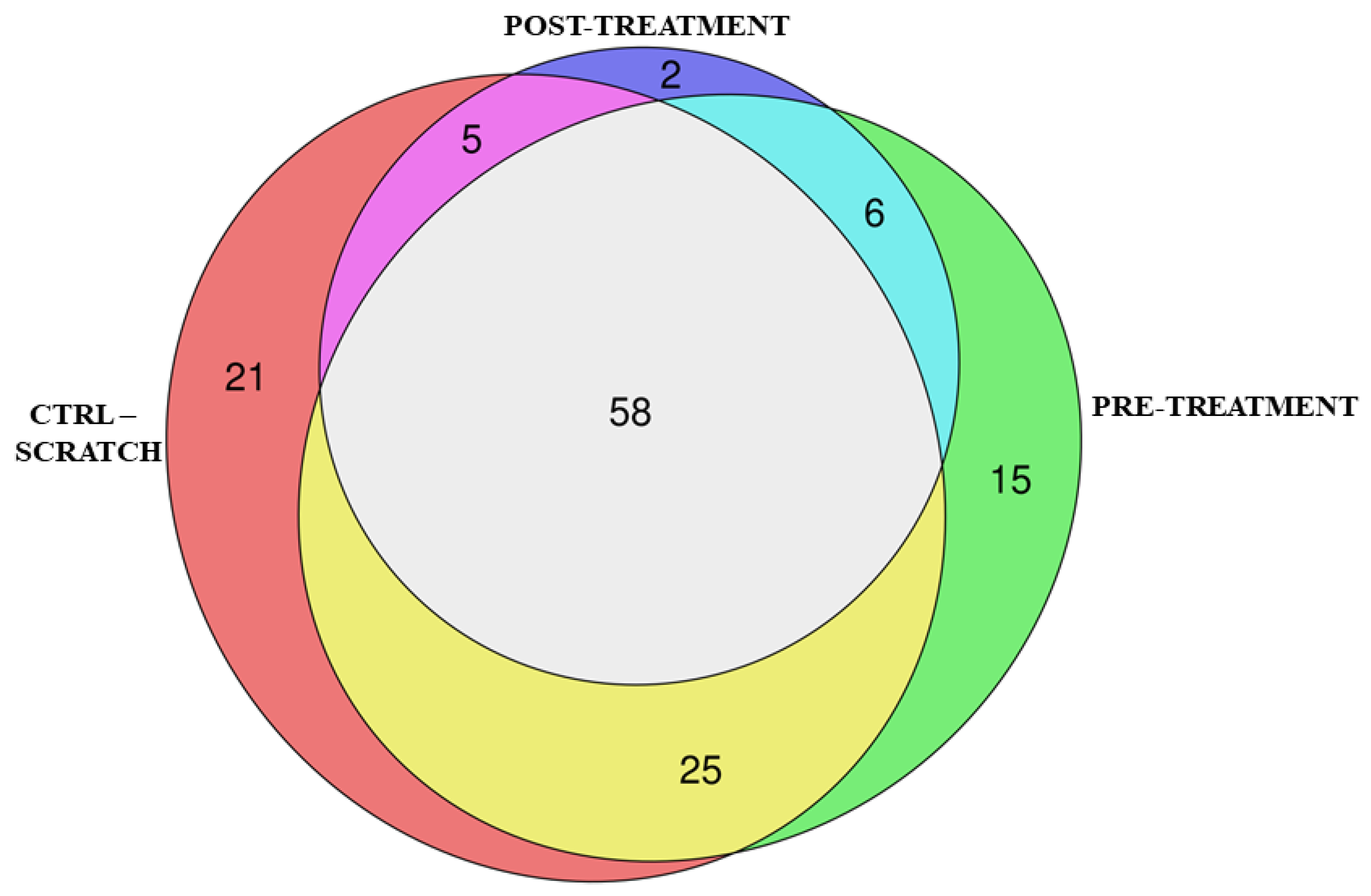
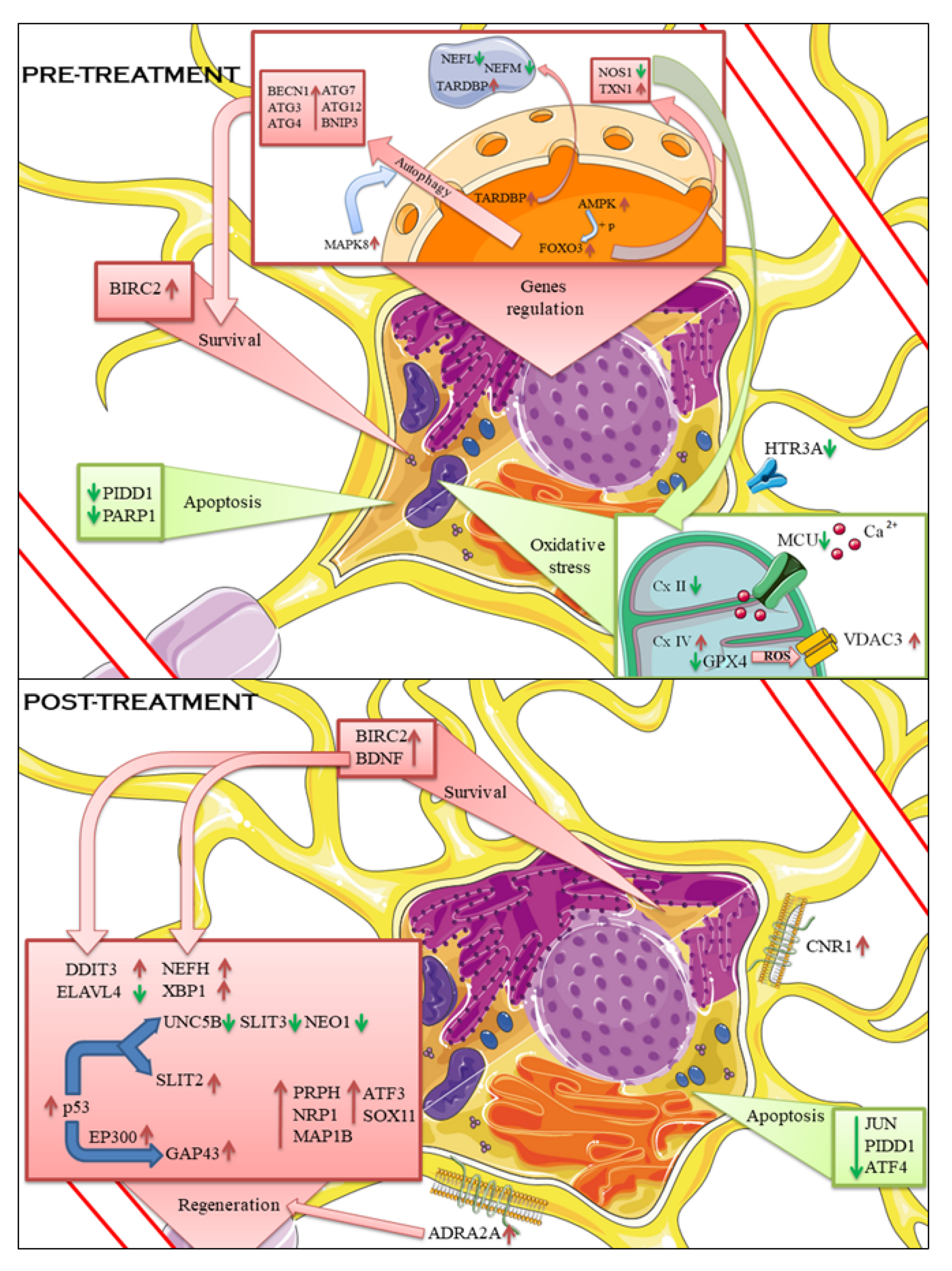
Figure 3.


 ,
, 



{kind=link}
{kind=link}
{kind=link}