
_Kim.png)
Cycle Threshold (Ct) Values of SARS-CoV-2 Detected with the GeneXpert® System and a Mutation Associated with Different Target Gene Failure
 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Participants
2.2. Nucleic Acid Detection Testing of SARS-CoV-2
2.3. DNA Sequencing of the SARS-CoV-2 Genome
2.4. Statistical Analysis
3. Results
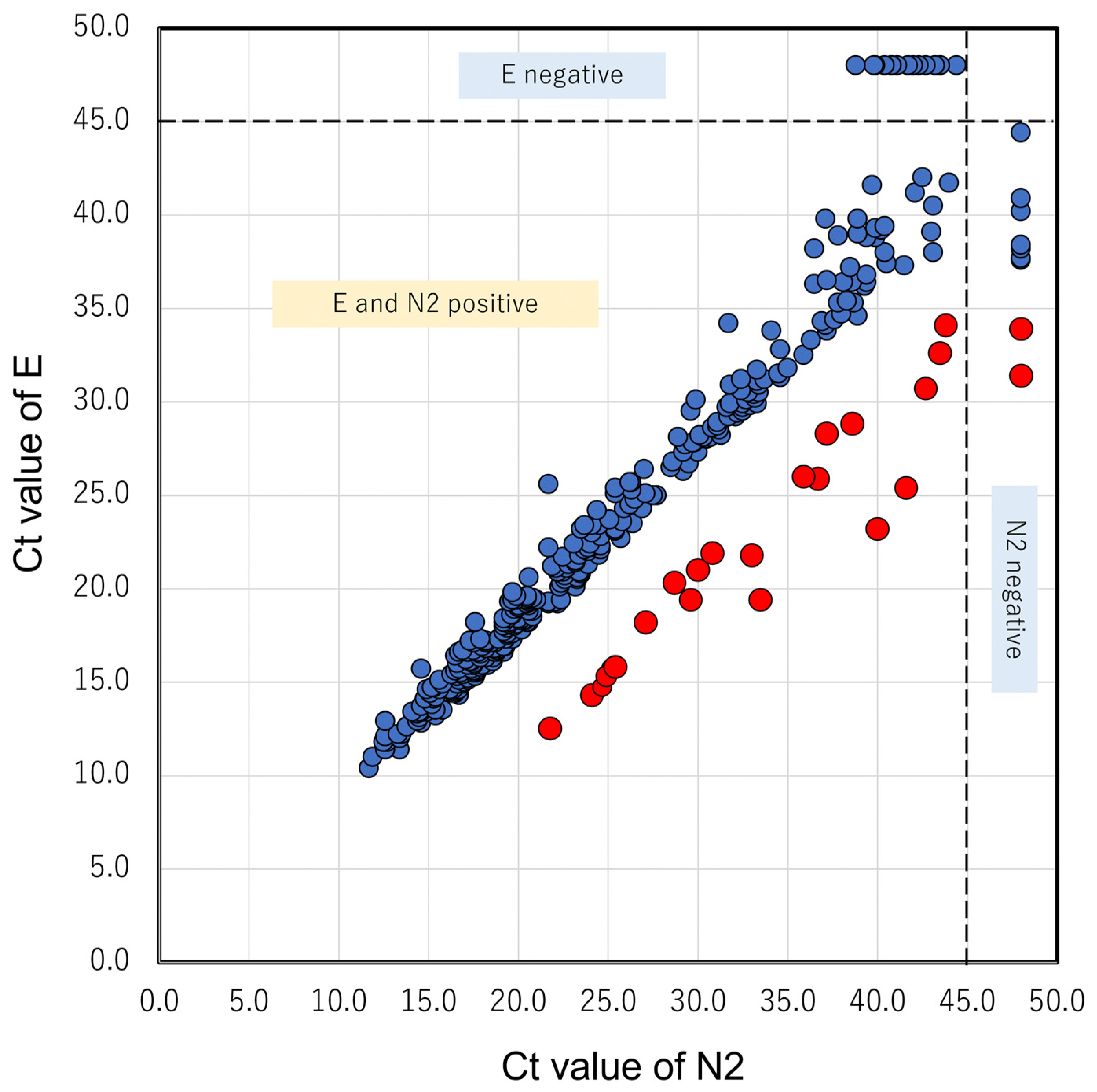
3.1. N2 Delay in GX
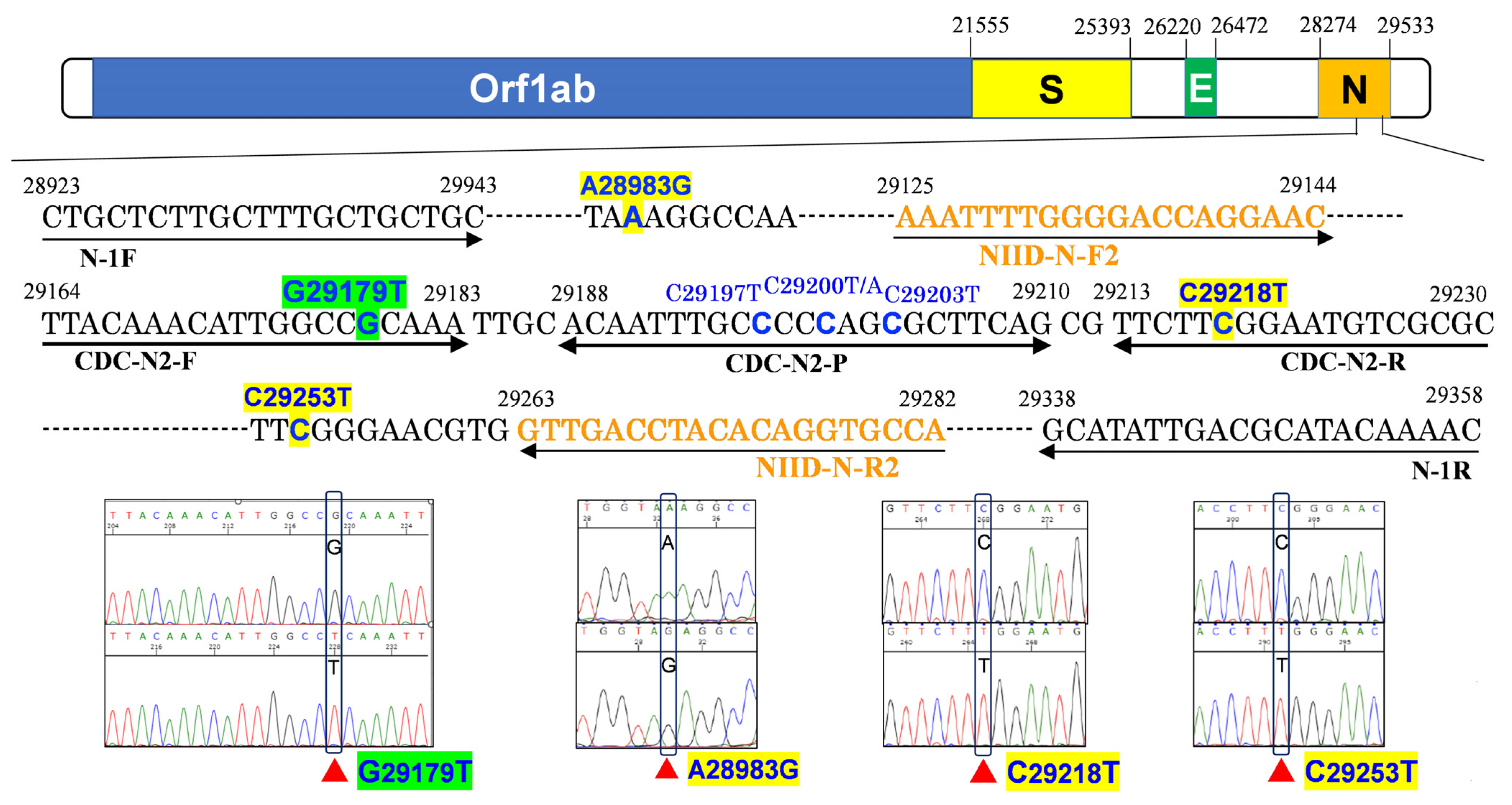
3.2. N2 Amplification Delay Is Linked to the G29179T Mutation
3.3. Past Cases Analyzed through GX
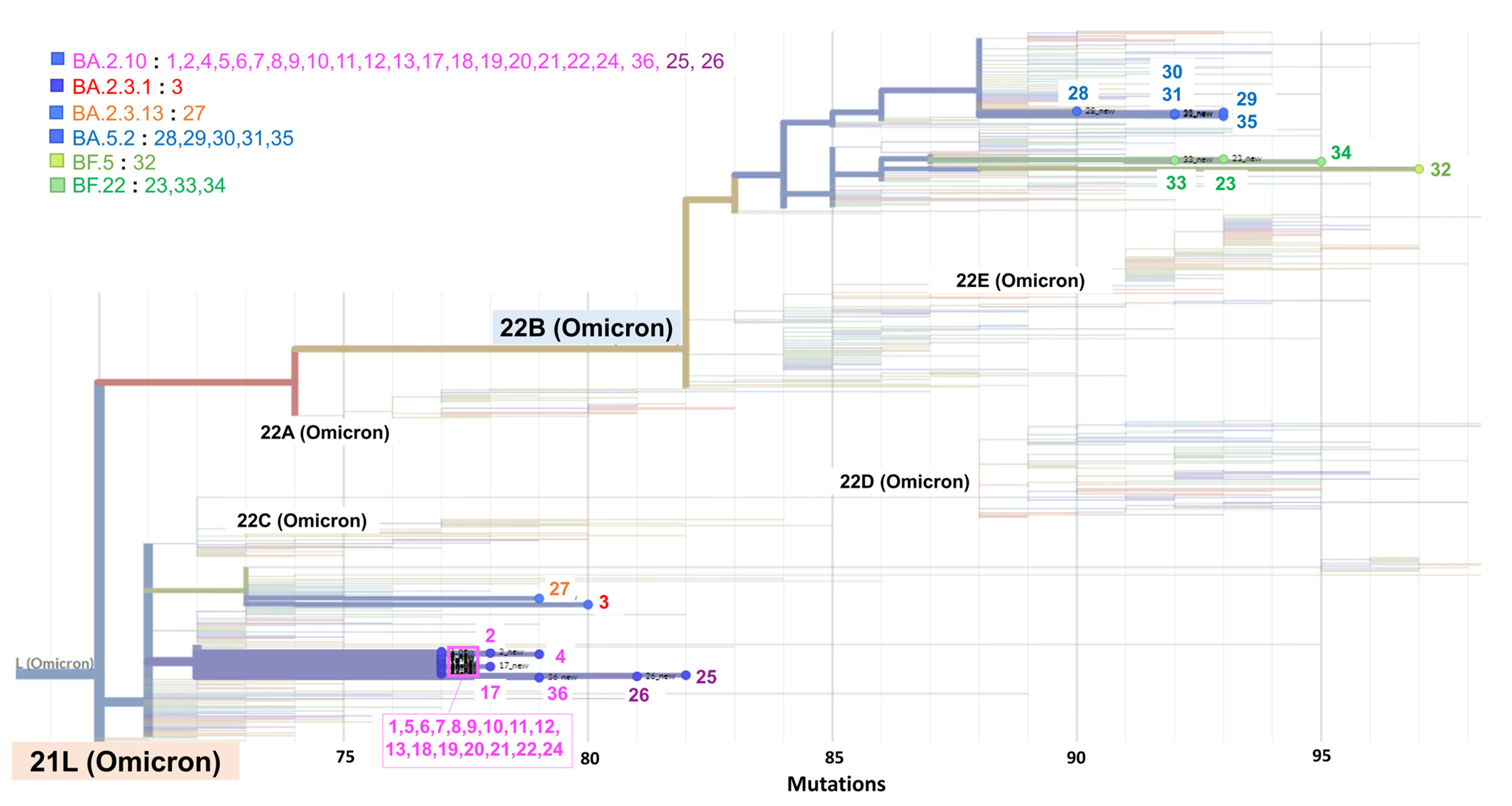
3.4. Phylogeny of SARS-CoV-2 Detected during the Cluster
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhu, Y.; Zhang, M.; Jie, Z.; Tao, S. Nucleic acid testing of SARS-CoV-2: A review of current methods, challenges, and prospects. Front. Microbiol. 2022, 13, 5004. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.M.; Kim, I.-H.; Kim, S. Nucleic acid testing of SARS-CoV-2. Int. J. Mol. Sci. 2021, 22, 6150. [Google Scholar] [CrossRef]
- Benda, A.; Zerajic, L.; Ankita, A.; Cleary, E.; Park, Y.; Pandey, S. COVID-19 testing and diagnostics: A review of commercialized technologies for cost, convenience and quality of tests. Sensors 2021, 21, 6581. [Google Scholar] [CrossRef] [PubMed]
- Loeffelholz, M.J.; Tang, Y.W. Detection of SARS-CoV-2 at the point of care. Bioanalysis 2021, 13, 1213–1223. [Google Scholar] [CrossRef]
- Cao, X.-J.; Fang, K.-Y.; Li, Y.-P.; Zhou, J.; Guo, X.-G. The diagnostic accuracy of Xpert Xpress to SARS-CoV-2: A systematic review. J. Virol. Methods 2022, 301, 114460. [Google Scholar] [CrossRef]
- Cepheid. Xpert® Xpress SARS-CoV-2 Instructions for Use; Cepheid: Sunnyvale, CA, USA, 2020; pp. 302–3562. [Google Scholar]
- Hong, K.H.; In, J.W.; Lee, J.; Kim, S.Y.; Lee, K.A.; Kim, S.; An, Y.; Lee, D.; Sung, H.; Kim, J.S.; et al. Prevalence of a single-nucleotide variant of SARS-CoV-2 in Korea and its impact on the diagnostic sensitivity of the Xpert Xpress SARS-CoV-2 assay. Ann. Lab. Med. 2022, 42, 96–99. [Google Scholar] [CrossRef]
- Foster, C.S.P.; Madden, M.; Chan, R.; Agapiou, D.; Bull, R.A.; Rawlinson, W.D.; Van Hal, S.J. SARS-CoV-2 N-gene mutation leading to Xpert Xpress SARS-CoV-2 assay instability. Pathology 2022, 54, 499–501. [Google Scholar] [CrossRef]
- Ziegler, K.; Steininger, P.; Ziegler, R.; Steinmann, J.; Korn, K.; Ensser, A. SARS-CoV-2 samples may escape detection because of a single point mutation in the N gene. Euro. Surveill. 2020, 25, 2001650. [Google Scholar] [CrossRef] [PubMed]
- Vanaerschot, M.; Mann, S.A.; Webber, J.T.; Kamm, J.; Bell, S.M.; Bell, J.; Hong, S.N.; Nguyen, M.P.; Chan, L.Y.; Bhatt, K.D.; et al. Identification of a polymorphism in the N Gene of SARS-CoV-2 that adversely impacts detection by reverse transcription-PCR. J. Clin. Microbiol. 2020, 59, e02369-20. [Google Scholar] [CrossRef]
- Hasan, M.R.; Sundararaju, S.; Manickam, C.; Mirza, F.; Al-Hail, H.; Lorenz, S.; Tang, P. A novel point mutation in the N Gene of SARS-CoV-2 may affect the detection of the virus by reverse transcription-quantitative PCR. J. Clin. Microbiol. 2021, 59, e03278-20. [Google Scholar] [CrossRef]
- Leelawong, M.; Mitchell, S.L.; Fowler, R.C.; Gonzalez, E.; Hughes, S.; Griffith, M.P.; Marsh, J.W.; Harrison, L.H.; Rakeman, J.L. SARS-CoV-2 N gene mutations impact detection by clinical molecular diagnostics: Reports in two cities in the United States. Diagn. Microbiol. Infect. Dis. 2021, 101, 115468. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.; Lee, T.; Merritt, A.; Pryce, T.; Levy, A.; Speers, D. Single-point mutations in the N Gene of SARS-CoV-2 adversely impact detection by a commercial dual target diagnostic assay. Microbiol. Spectr. 2021, 9, e01494-21. [Google Scholar] [CrossRef]
- Isabel, S.; Abdulnoor, M.; Boissinot, K.; Isabel, M.R.; de Borja, R.; Zuzarte, P.C.; Sjaarda, C.P.; Barker, K.R.; Sheth, P.M.; Matukas, L.M.; et al. Emergence of a mutation in the nucleocapsid gene of SARS-CoV-2 interferes with PCR detection in Canada. Sci. Rep. 2022, 12, 10867. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.; Hwang, M.; Lukey, J.; Jinadatha, C.; Navarathna, D.H. Presumptive positive with the Cepheid Xpert Xpress SARS-CoV-2 assay due to N mutations in the Delta variant. Diagn. Microbiol. Infect. Dis. 2022, 103, 115699. [Google Scholar] [CrossRef]
- Wang, H.; Jean, S.; Wilson, S.A.; Lucyshyn, J.M.; McGrath, S.; Wilson, R.K.; Magrini, V.; Leber, A.L. A deletion in the N gene of SARS-CoV-2 may reduce test sensitivity for detection of SARS-CoV-2. Diagn. Microbiol. Infect. Dis. 2022, 102, 115631. [Google Scholar] [CrossRef]
- Rhoads, D.D.; Plunkett, D.; Nakitandwe, J.; Dempsey, A.; Tu, Z.J.; Procop, G.W.; Bosler, D.; Rubin, B.P.; Loeffelholz, M.J.; Brock, J.E. Endemic SARS-CoV-2 polymorphisms can cause a higher diagnostic target failure rate than estimated by aggregate global sequencing data. J. Clin. Microbiol. 2021, 59, e00913-21. [Google Scholar] [CrossRef] [PubMed]
- Fox-Lewis, S.; Fox-Lewis, A.; Harrower, J.; Chen, R.; Wang, J.; de Ligt, J.; McAuliffe, G.; Taylor, S.; Smit, E. Lack of N2-gene amplification on the Cepheid Xpert Xpress SARS-CoV-2 assay and potential novel causative mutations: A case series from Auckland, New Zealand. IDCases 2021, 25, e01233. [Google Scholar] [CrossRef]
- Bozidis, P.; Tsaousi, E.T.; Kostoulas, C.; Sakaloglou, P.; Gouni, A.; Koumpouli, D.; Sakkas, H.; Georgiou, I.; Gartzonika, K. Unusual N gene dropout and Ct value shift in commercial multiplex PCR assays caused by mutated SARS-CoV-2 strain. Diagnostics 2022, 12, 973. [Google Scholar] [CrossRef]
- Artesi, M.; Bontems, S.; Göbbels, P.; Franckh, M.; Maes, P.; Boreux, R.; Meex, C.; Melin, P.; Hayette, M.P.; Bours, V.; et al. A recurrent mutation at position 26340 of SARS-CoV-2 is associated with failure of the E gene quantitative reverse transcription-PCR utilized in a commercial dual-target diagnostic assay. J. Clin. Microbiol. 2020, 58, e01598-20. [Google Scholar] [CrossRef]
- Hasan, R.; Hossain, M.E.; Miah, M.; Hasan, M.M.; Rahman, M.; Rahman, M.Z. Identification of novel mutations in the N gene of SARS-CoV-2 that adversely affect the detection of the virus by reverse transcription-quantitative PCR. Microbiol. Spectr. 2021, 9, e00545-21. [Google Scholar] [CrossRef]
- Lesbon, J.C.C.; Poleti, M.D.; de Mattos Oliveira, E.C.; Patané, J.S.L.; Clemente, L.G.; Viala, V.L.; Ribeiro, G.; Giovanetti, M.; de Alcantara, L.C.J.; Teixeira, O.; et al. Nucleocapsid (N) gene mutations of SARS-CoV-2 can affect real-time RT-PCR diagnostic and impact false-negative results. Viruses 2021, 13, 2474. [Google Scholar] [CrossRef] [PubMed]
- Shirato, K.; Nao, N.; Katano, H.; Takayama, I.; Saito, S.; Kato, F.; Katoh, H.; Sakata, M.; Nakatsu, Y.; Mori, Y.; et al. Development of genetic diagnostic methods for detection for novel coronavirus 2019(nCoV-2019) in Japan. Jpn. J. Infect. Dis. 2020, 73, 304–307. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention. Research Use Only 2019-Novel Coronavirus. nCoV Real-Time RT-PCR Primers and Probes. Available online: https://www.cdc.gov/coronavirus/2019-ncov/lab/rt-pcr-panel-primer-probes.html (accessed on 1 September 2022).
- Itokawa, K.; Sekizuka, T.; Hashino, M.; Tanaka, R.; Kuroda, M. Disentangling primer interactions improves SARS-CoV-2 genome sequencing by multiplex tiling PCR. PLoS ONE. 2020, 15, e0239403. [Google Scholar] [CrossRef]
- nCoV-2019 Sequencing Protocol for Illumina. V.4. Available online: https://www.protocols.io/view/ncov-2019-sequencing-protocol-for-illumina-eq2ly398mgx9/v4 (accessed on 16 February 2023).
- Grubaugh, N.D.; Gangavarapu, K.; Quick, J.; Matteson, N.L.; De Jesus, J.G.; Main, B.J.; Tan, A.L.; Paul, L.M.; Brackney, D.E.; Grewal, S.; et al. An amplicon-based sequencing framework for accurately measuring intrahost virus diversity using PrimalSeq and iVar. Genome Biol. 2019, 20, 8. [Google Scholar] [CrossRef]
- Kanda, Y. Investigation of the freely available easy-to-use software ‘EZR’ for medical statistics. Bone Marrow Transplant. 2013, 48, 452–458. [Google Scholar] [CrossRef]
- Alkharsah, K.R.; Rehman, S.; Alkhamis, F.; Alnimr, A.; Diab, A.; Al-Ali, A.K. Comparative and molecular analysis of MRSA isolates from infection sites and carrier colonization sites. Ann. Clin. Microbiol. Antimicrob. 2018, 17, 7. [Google Scholar] [CrossRef]
- Falasca, F.; Sciandra, I.; Di Carlo, D.; Gentile, M.; Deales, A.; Antonelli, G.; Turriziani, O. Detection of SARS-CoV N2 gene: Very low amounts of viral RNA or false positive? J. Clin. Virol. 2020, 133, 104660. [Google Scholar] [CrossRef]
- Khoshchehreh, M.; Wald-Dickler, N.; Holtom, P.; Butler-Wu, S.M. A needle in the haystack? Assessing the significance of envelope (E) gene-negative, nucleocapsid (N2) gene-positive SARS-CoV-2 detection by the Cepheid Xpert Xpress SARS-CoV-2 assay. J. Clin. Virol. 2020, 133, 104683. [Google Scholar] [CrossRef]
- Varadhan, H.; Ahuja, V.; Pitman, C.; Dwyer, D.E. Weak positive SARS-CoV-2 N2 gene results using the Xpress Xpert assay: The need for an alternate interpretative criterion in a low prevalence setting. Pathology 2022, 54, 116–120. [Google Scholar] [CrossRef] [PubMed]
- Navarathna, D.H.; Sharp, S.; Lukey, J.; Arenas, M.; Villas, H.; Wiley, L.; Englett, I.; Juan, M.R.S.; Jinadatha, C. Understanding false positives and the detection of SARS-CoV-2 using the Cepheid Xpert Xpress SARS-CoV-2 and BD MAX SARS-CoV-2 assays. Diagn. Microbiol. Infect. Dis. 2021, 100, 115334. [Google Scholar] [CrossRef]
- Leducq, V.; Jary, A.; Bridier-Nahmias, A.; Daniel, L.; Zafilaza, K.; Damond, F.; Goldstein, V.; Duval, A.; Blanquart, F.; Calvez, V.; et al. Nosocomial transmission clusters and lineage diversity characterized by SARS-CoV-2 genomes from two large hospitals in Paris, France, in 2020. Sci. Rep. 2022, 12, 1094. [Google Scholar] [CrossRef] [PubMed]
- Turakhia, Y.; Thornlow, B.; Hinrichs, A.; McBroome, J.; Ayala, N.; Ye, C.; Smith, K.; De Maio, N.; Haussler, D.; Lanfear, R.; et al. Pandemic-scale phylogenomics reveals the SARS-CoV-2 recombination landscape. Nature 2022, 609, 994–997. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| No. | Day | Sex | Age | Patient or Staff | Ct Value (GeneXpert) | Sanger Sequence of N2 | NGS | Pangolin Lineage | Nextclade Lineage | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| E | N2 | SPC | N2-E | c.29179 | Other Mutations | c.29179 | |||||||
| 1 | −36 | F | 31 | Outpatient | 15.7 | 25.2 | 29.2 | 9.5 | T | T | BA.2.10 | 21L | |
| 2 | −3 | F | 33 | Outpatient | 19.4 | 29.6 | 28.2 | 10.2 | T | T | BA.2.10 | 21L | |
| 3 | −1 | F | 78 | Outpatient | 20.1 | 22.3 | 27.9 | 2.2 | G | c.28983A > G c.29218C > T | G | BA.2.3.1 | 21L |
| 4 | 0 | F | 11 | Outpatient | 20.3 | 28.7 | 27.8 | 8.4 | T | T | BA.2.10 | 21L | |
| 5 | 0 | M | 65 | Medical engineer | 21.9 | 30.8 | 27.7 | 8.9 | T | T | BA.2.10 | 21L | |
| 6 | 0 | M | 24 | Medical engineer | 28.3 | 37.2 | 30.5 | 8.9 | T | T | BA.2.10 | 21L | |
| 7 | 0 | M | 41 | Medical engineer | 21.8 | 33.0 | 28.3 | 11.2 | T | T | BA.2.10 | 21L | |
| 8 | 0 | F | 57 | Nurse | 25.4 | 41.6 | 28.2 | 16.2 | T | T | BA.2.10 | 21L | |
| 9 | 0 | M | 41 | Inpatient | 14.3 | 24.1 | 27.6 | 9.8 | T | T | BA.2.10 | 21L | |
| 10 | 0 | F | 41 | Nurse | 30.7 | 42.7 | 28.0 | 12.0 | T | T | BA.2.10 | 21L | |
| 11 | 1 | M | 27 | Doctor | 15.8 | 25.4 | 28.7 | 9.6 | T | T | BA.2.10 | 21L | |
| 12 | 1 | F | 59 | Nursing assistant | 25.9 | 36.7 | 28.9 | 10.8 | T | T | BA.2.10 | 21L | |
| 13 | 1 | F | 26 | Nurse | 23.2 | 40.0 | 28.9 | 16.8 | T | T | BA.2.10 | 21L | |
| 14 | 1 | F | 25 | Nurse | 34.1 | 43.8 | 28.5 | 9.7 | Failed | T | Failed | Failed | |
| 15 | 2 | F | 26 | Nurse | 32.6 | 43.5 | 27.9 | 10.9 | Failed | T | Failed | Failed | |
| 16 | 2 | F | 47 | Nurse | 33.8 | 0.0 | 28.9 | −33.8 | Failed | T | Failed | Failed | |
| 17 | 2 | F | 26 | Medical engineer | 31.4 | 0.0 | 28.9 | −31.4 | T | T | BA.2.10 | 21L | |
| 18 | 4 | F | 53 | Inpatient | 12.5 | 21.8 | 27.7 | 9.3 | T | T | BA.2.10 | 21L | |
| 19 | 4 | F | 82 | Family of No. 18 | 28.8 | 38.6 | 28.6 | 9.8 | T | T | BA.2.10 | 21L | |
| 20 | 4 | M | 82 | Inpatient | 14.7 | 24.7 | 28.5 | 10.0 | T | T | BA.2.10 | 21L | |
| 21 | 6 | F | 24 | Nurse | 19.4 | 33.5 | 28.2 | 14.1 | T | T | BA.2.10 | 21L | |
| 22 | 8 | F | 27 | Nurse | 26.0 | 35.9 | 28.4 | 9.9 | T | T | BA.2.10 | 21L | |
| 23 | 10 | F | 25 | Medical engineer | 16.7 | 16.9 | 0.0 | 0.2 | G | G | BF.22 | 22B | |
| 24 | 11 | F | 79 | Family of No. 20 | 18.2 | 27.1 | 29.2 | 8.9 | T | T | BA.2.10 | 21L | |
| 25 | 13 | F | 26 | Nurse | 14.7 | 15.3 | 0.0 | 0.6 | G | G | BA.2.10 | 21L | |
| 26 | 14 | F | 26 | Nurse | 19.5 | 20.5 | 0.0 | 1 | G | G | BA.2.10 | 21L | |
| 27 | 14 | M | 77 | Outpatient | 16.6 | 18.7 | 0.0 | 2.1 | G | c.29253C > T | G | BA.2.3.13 | 21L |
| 28 | 19 | F | 24 | Nurse | 25.4 | 25.4 | 30.4 | 0 | Not tested | G | BA.5.2 | 22B | |
| 29 | 19 | F | 26 | Physical therapist | 16.4 | 16.5 | 0.0 | 0.1 | Not tested | G | BA.5.2 | 22B | |
| 30 | 22 | M | 79 | Inpatient | 16.1 | 18.6 | 0.0 | 2.5 | Not tested | G | BA.5.2 | 22B | |
| 31 | 23 | F | 23 | Nurse | 20.7 | 22.6 | 0.0 | 1.9 | Not tested | G | BA.5.2 | 22B | |
| 32 | 23 | M | 29 | Doctor | 18.0 | 19.2 | 0.0 | 1.2 | Not tested | G | BF.5 | 22B | |
| 33 | 24 | F | 37 | Nurse | 19.6 | 20.5 | 0.0 | 0.9 | Not tested | G | BF.22 | 22B | |
| 34 | 25 | F | 25 | Nurse | 19.4 | 19.9 | 0.0 | 0.5 | Not tested | G | BF.22 | 22B | |
| 35 | 33 | F | 27 | Nurse | 18.1 | 19.5 | 0.0 | 1.4 | G | G | BA.5.2 | 22B | |
| 36 | 36 | F | 79 | Inpatient | 21.0 | 30.0 | 28.9 | 9.0 | T | T | BA.2.10 | 21L | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yamashita, K.; Taniguchi, T.; Niizeki, N.; Nagao, Y.; Suzuki, A.; Toguchi, A.; Takebayashi, S.; Ishikawa, J.; Nagura, O.; Furuhashi, K.; et al. Cycle Threshold (Ct) Values of SARS-CoV-2 Detected with the GeneXpert® System and a Mutation Associated with Different Target Gene Failure. Curr. Issues Mol. Biol. 2023, 45, 4124-4134. https://doi.org/10.3390/cimb45050262
Yamashita K, Taniguchi T, Niizeki N, Nagao Y, Suzuki A, Toguchi A, Takebayashi S, Ishikawa J, Nagura O, Furuhashi K, et al. Cycle Threshold (Ct) Values of SARS-CoV-2 Detected with the GeneXpert® System and a Mutation Associated with Different Target Gene Failure. Current Issues in Molecular Biology. 2023; 45(5):4124-4134. https://doi.org/10.3390/cimb45050262
Chicago/Turabian StyleYamashita, Keita, Terumi Taniguchi, Noriyasu Niizeki, Yuki Nagao, Akira Suzuki, Akihiro Toguchi, Shiori Takebayashi, Jinko Ishikawa, Osanori Nagura, Kazuki Furuhashi, and et al. 2023. "Cycle Threshold (Ct) Values of SARS-CoV-2 Detected with the GeneXpert® System and a Mutation Associated with Different Target Gene Failure" Current Issues in Molecular Biology 45, no. 5: 4124-4134. https://doi.org/10.3390/cimb45050262
APA StyleYamashita, K., Taniguchi, T., Niizeki, N., Nagao, Y., Suzuki, A., Toguchi, A., Takebayashi, S., Ishikawa, J., Nagura, O., Furuhashi, K., Iwaizumi, M., & Maekawa, M. (2023). Cycle Threshold (Ct) Values of SARS-CoV-2 Detected with the GeneXpert® System and a Mutation Associated with Different Target Gene Failure. Current Issues in Molecular Biology, 45(5), 4124-4134. https://doi.org/10.3390/cimb45050262