

Genome-Wide Identification of Candidate Genes Associated with Heat Stress in Mulberry (Morus alba L.)

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. RNA-Sequencing and Data Analysis
2.2. DEGs Confirmation Using RT-qPCR
3. Results
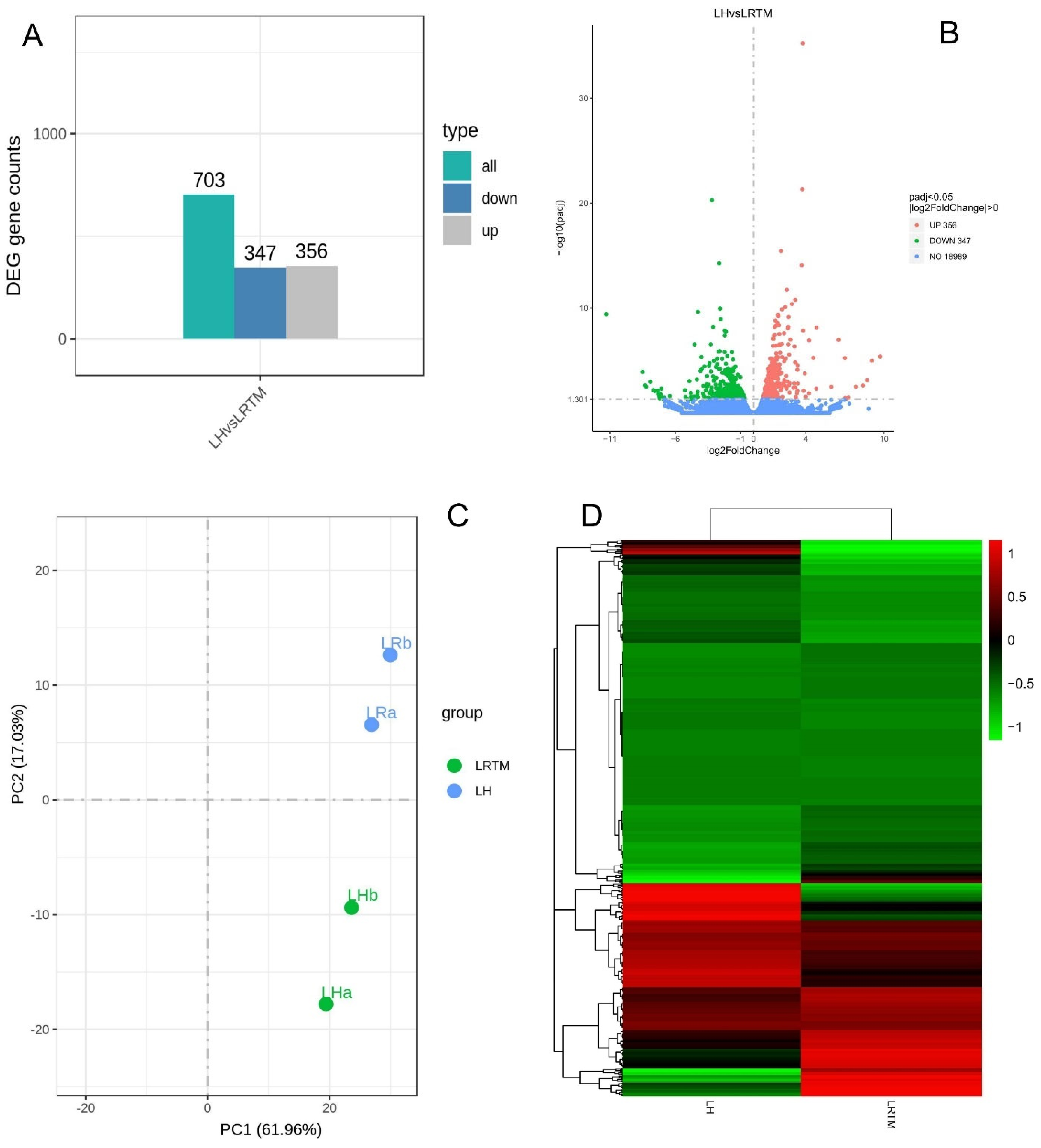
3.1. RNA-Seq-Based Transcriptomic Profiles of Mulberry (M. alba)
3.2. Differential Expressed Genes (DEGs) of Mulberry Responsive to Heat Stress
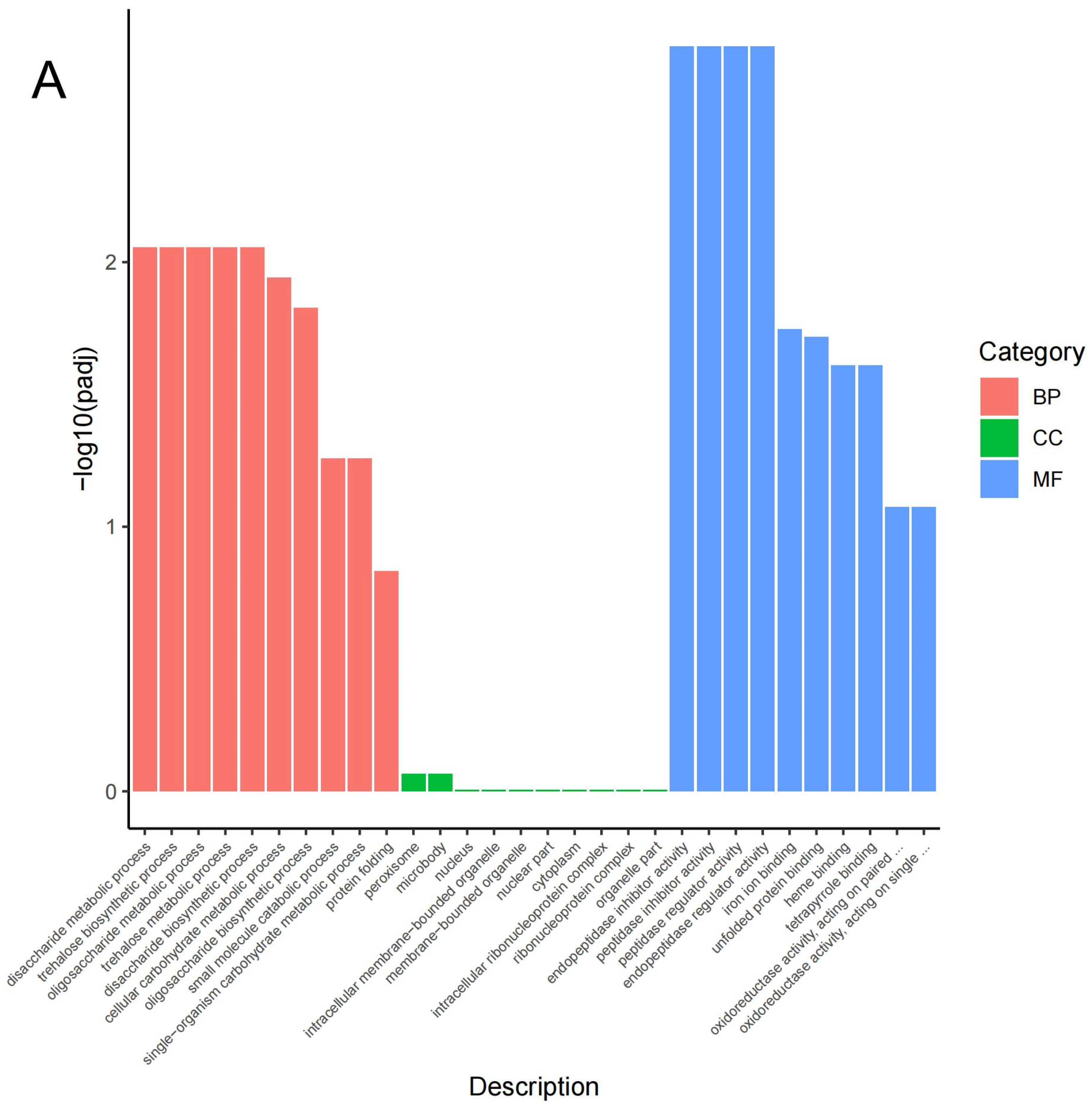
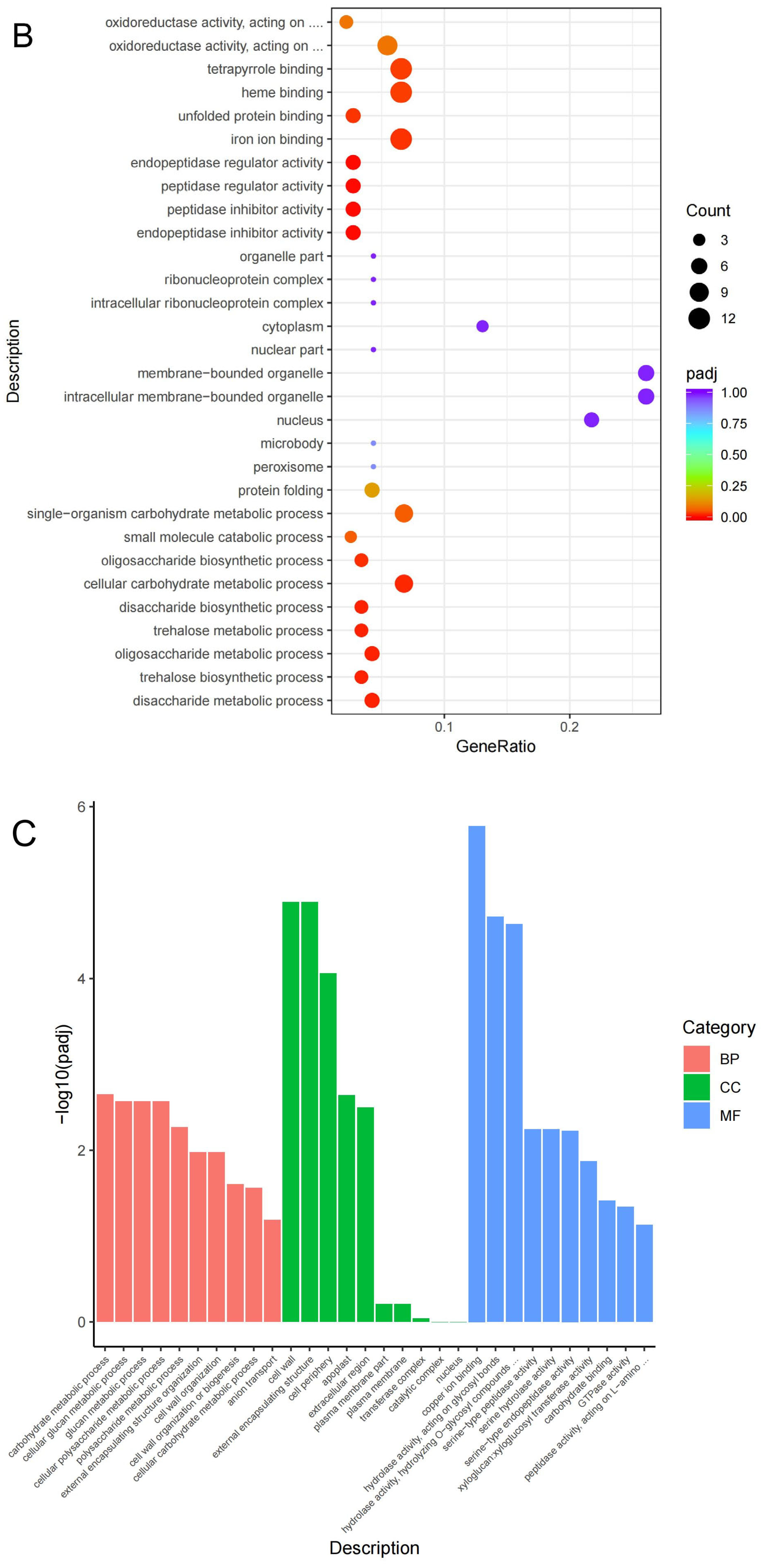
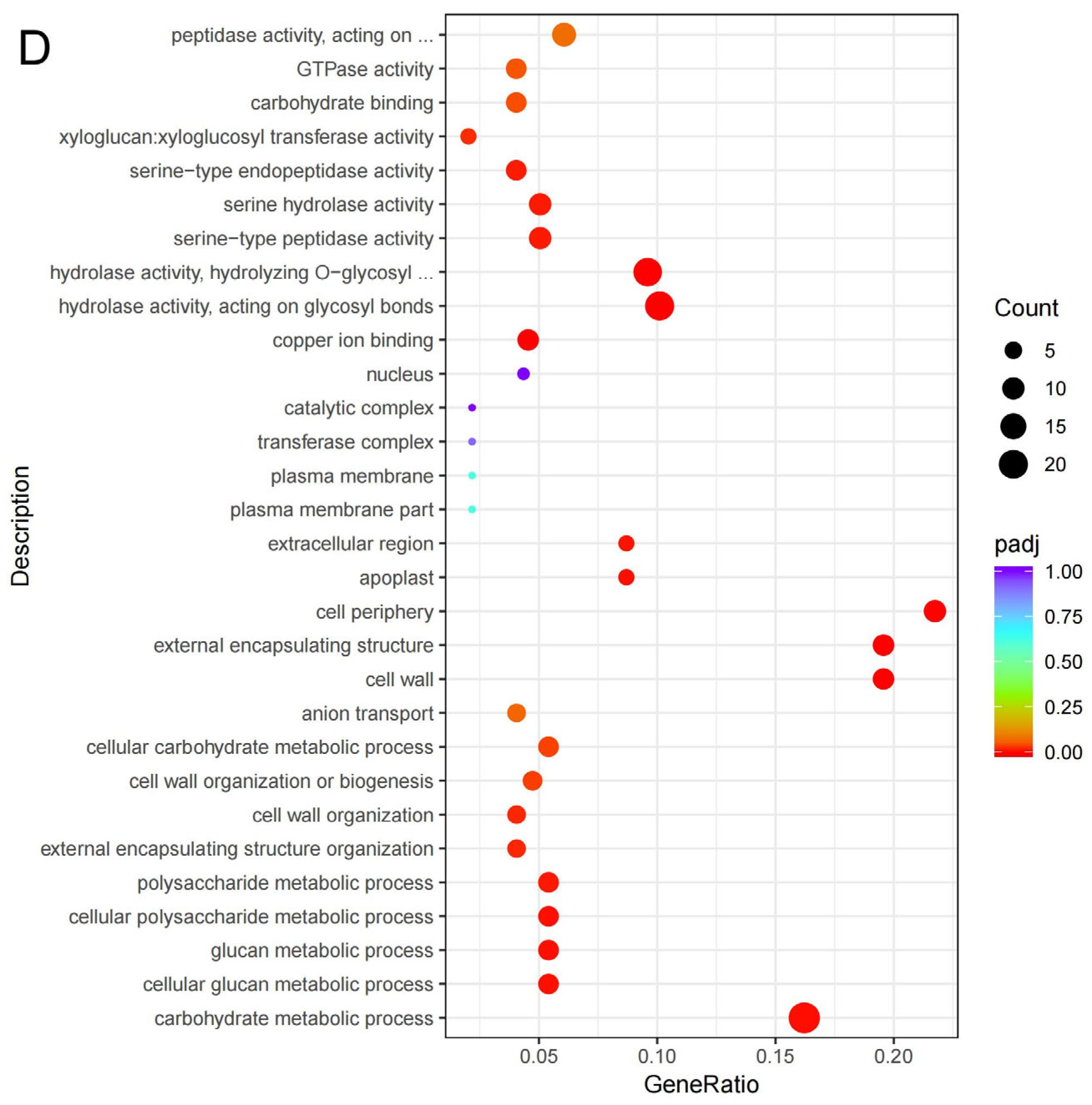
3.3. GO and KEGG Classification of the DEGs
3.4. Heat-Responsive Transcription Factors
3.5. Validation of DEGs Using Real-Time RT-PCR
4. Discussion
4.1. Transcriptomic Changes in M. alba under High Temperature
4.2. Heat Stress Response in Mulberry Induced Up-Regulated Genes Related to Valine, Leucine and Isoleucine Degradation
4.3. TFs Responsive to High Temperature
4.4. Validation of the RNA-Seq Results by qRT-PCR
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Qin, D.; Wang, G.; Dong, Z.; Xia, Q.; Zhao, P. Comparative Fecal Metabolomes of Silkworms Being Fed Mulberry Leaf and Artificial Diet. Insects 2020, 11, 851. [Google Scholar] [CrossRef]
- Sarkar, T.; Mogili, T.; Sivaprasad, V. Improvement of abiotic stress adaptive traits in mulberry (Morus spp.): An update on biotechnological interventions. 3 Biotech 2017, 7, 214. [Google Scholar] [CrossRef]
- Sharma, A.; Kumar, V.; Shahzad, B.; Ramakrishnan, M.; Sidhu, G.P.S.; Bali, A.S.; Handa, N.; Kapoor, D.; Yadav, P.; Khanna, K.; et al. Photosynthetic Response of Plants Under Different Abiotic Stresses: A Review. J. Plant Growth Regul. 2020, 39, 509–531. [Google Scholar] [CrossRef]
- Zhang, B.; Dong, S.; Hu, C.; Wang, K. Research progress on heat stress and adaptation of maize. J. Weifang Univ. 2006, 6, 90–94. [Google Scholar]
- Sohrabi, S.S.; Ismaili, A.; Nazarian-Firouzabadi, F.; Fallahi, H.; Hosseini, S.Z. Identification of key genes and molecular mechanisms associated with temperature stress in lentil. Gene 2021, 807, 145952. [Google Scholar] [CrossRef]
- Ackah, M.; Guo, L.; Li, S.; Jin, X.; Asakiya, C.; Tawiah Aboagye, E.; Yuan, F.; Wu, M.; Gyllye Essoh, L.; Adjibolosoo, D.; et al. DNA Methylation Changes and Its Associated Genes in Mulberry (Morus alba L.) Yu-711 Response to Drought Stress Using MethylRAD Sequencing. Plants 2022, 11, 190. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.R.; Goswami, S.; Sharma, S.K.; Singh, K.; Gadpayle, K.A.; Singh, S.D.; Pathak, H.; Rai, R.D. Differential expression of heat shock protein and alteration in osmolyte accumulation under heat stress in wheat. J. Plant Biochem. Biotechnol. 2012, 22, 16–26. [Google Scholar] [CrossRef]
- Guy, C. Molecular responses of plants to cold shock and cold acclimation. J. Mol. Microbiol. Biotechnol. 1999, 1, 231–242. [Google Scholar]
- Schleussner, C.-F.; Lissner, T.K.; Fischer, E.M.; Wohland, J.; Perrette, M.; Golly, A.; Rogelj, J.; Childers, K.; Schewe, J.; Frieler, K.; et al. Differential climate impacts for policy-relevant limits to global warming: The case of 1.5 C and 2 C. Earth Syst. Dyn. 2016, 7, 327–351. [Google Scholar] [CrossRef]
- Mousavi, S.; Alisoltani, A.; Shiran, B.; Fallahi, H.; Ebrahimie, E.; Imani, A.; Houshmand, S. De Novo Transcriptome Assembly and Comparative Analysis of Differentially Expressed Genes in Prunus dulcis Mill. in Response to Freezing Stress. PLoS ONE 2014, 9, e104541. [Google Scholar] [CrossRef]
- Kugler, K.G.; Siegwart, G.; Nussbaumer, T.; Ametz, C.; Spannagl, M.; Steiner, B.; Lemmens, M.; Mayer, K.F.; Buerstmayr, H.; Schweiger, W. Quantitative trait loci-dependent analysis of a gene co-expression network associated with Fusarium head blight resistance in bread wheat (Triticum aestivum L.). BMC Genom. 2013, 14, 728. [Google Scholar] [CrossRef] [PubMed]
- González-Schain, N.; Dreni, L.; Lawas, L.M.; Galbiati, M.; Colombo, L.; Heuer, S.; Jagadish, K.S.V.; Kater, M.M. Genome-Wide Transcriptome Analysis During Anthesis Reveals New Insights into the Molecular Basis of Heat Stress Responses in Tolerant and Sensitive Rice Varieties. Plant Cell Physiol. 2015, 57, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Frey, F.P.; Urbany, C.; Hüttel, B.; Reinhardt, R.; Stich, B. Genome-wide expression profiling and phenotypic evaluation of European maize inbreds at seedling stage in response to heat stress. BMC Genom. 2015, 16, 123. [Google Scholar] [CrossRef] [PubMed]
- Aulakh, S.S.; Veilleux, R.E.; Dickerman, A.W.; Tang, G.; Flinn, B.S. Characterization and RNA-seq analysis of underperformer, an activation-tagged potato mutant. Plant Mol. Biol. 2013, 84, 635–658. [Google Scholar] [CrossRef]
- Park, S.J.; Kim, K.-Y.; Baik, M.-Y.; Koh, Y.H. Sericulture and the edible-insect industry can help humanity survive: Insects are more than just bugs, food, or feed. Food Sci. Biotechnol. 2022, 31, 657–668. [Google Scholar] [CrossRef]
- Adolf, A.; Liu, L.; Ackah, M.; Li, Y.; Du, Q.; Zheng, D.; Guo, P.; Shi, Y.; Lin, Q.; Qiu, C.; et al. Transcriptome profiling reveals candidate genes associated with cold stress in mulberry. Braz. J. Bot. 2021, 44, 125–137. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.-C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Kotak, S.; Larkindale, J.; Lee, U.; von Koskull-Döring, P.; Vierling, E.; Scharf, K.-D. Complexity of the heat stress response in plants. Curr. Opin. Plant Biol. 2007, 10, 310–316. [Google Scholar] [CrossRef]
- Van Sandt, V.S.T.; Suslov, D.; Verbelen, J.-P.; Vissenberg, K. Xyloglucan Endotransglucosylase Activity Loosens a Plant Cell Wall. Ann. Bot. 2007, 100, 1467–1473. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Purugganan, M.M.; Polisensky, D.H.; Antosiewicz, D.M.; Fry, S.C.; Braam, J. Arabidopsis TCH4, regulated by hormones and the environment, encodes a xyloglucan endotransglycosylase. Plant Cell 1995, 7, 1555–1567. [Google Scholar]
- Iurlaro, A.; De Caroli, M.; Sabella, E.; De Pascali, M.; Rampino, P.; De Bellis, L.; Perrotta, C.; Dalessandro, G.; Piro, G.; Fry, S.C.; et al. Drought and Heat Differentially Affect XTH Expression and XET Activity and Action in 3-Day-Old Seedlings of Durum Wheat Cultivars with Different Stress Susceptibility. Front. Plant Sci. 2016, 7, 1686. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Wang, C.C.; Guo, W.D.; Li, X.B.; Lu, M.; Yu, C.L. Differential expression of cell wall related genes in the elongation zone of rice roots under water deficit. Russ. J. Plant Physiol. 2006, 53, 390–395. [Google Scholar] [CrossRef]
- Sihag, P.; Kumar, U.; Sagwal, V.; Kapoor, P.; Singh, Y.; Mehla, S.; Balyan, P.; Mir, R.R.; Varshney, R.K.; Singh, K.P.; et al. Effect of terminal heat stress on osmolyte accumulation and gene expression during grain filling in bread wheat (Triticum aestivum L.). Plant Genome 2023, e20307. [Google Scholar] [CrossRef]
- Attfield, P.V. Trehalose accumulates in Saccharomyces cerevisiae during exposure to agents that induce heat shock response. FEBS Lett. 1987, 225, 259–263. [Google Scholar] [CrossRef]
- Li, H.-W.; Zang, B.-S.; Deng, X.-W.; Wang, X.-P. Overexpression of the trehalose-6-phosphate synthase gene OsTPS1 enhances abiotic stress tolerance in rice. Planta 2011, 234, 1007–1018. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Fu, L.; Qin, P.; Sun, Y.; Liu, J.; Wang, X. Overexpression of the wheat trehalose 6-phosphate synthase 11 gene enhances cold tolerance in Arabidopsis thaliana. Gene 2019, 710, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Xiang, L.; Le Roy, K.; Moghaddam, M.R.B.; Vanhaecke, M.; Lammens, W.; Rolland, F.; Ende, W.V.D. Exploring the neutral invertase–oxidative stress defence connection in Arabidopsis thaliana. J. Exp. Bot. 2011, 62, 3849–3862. [Google Scholar] [CrossRef]
- Murayama, S.; Handa, H. Genes for alkaline/neutral invertase in rice: Alkaline/neutral invertases are located in plant mitochondria and also in plastids. Planta 2006, 225, 1193–1203. [Google Scholar] [CrossRef]
- Dahro, B.; Wang, F.; Peng, T.; Liu, J.-H. PtrA/NINV, an alkaline/neutral invertase gene of Poncirus trifoliata, confers enhanced tolerance to multiple abiotic stresses by modulating ROS levels and maintaining photosynthetic efficiency. BMC Plant Biol. 2016, 16, 76. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Chen, Z.; Wang, F.; Jia, W.; Xu, Z. Combined transcriptomic and metabolomic analyses uncover rearranged gene expression and metabolite metabolism in tobacco during cold acclimation. Sci. Rep. 2020, 10, 5242. [Google Scholar] [CrossRef] [PubMed]
- Ke, X.; Wang, H.; Li, Y.; Zhu, B.; Zang, Y.; He, Y.; Cao, J.; Zhu, Z.; Yu, Y. Genome-Wide Identification and Analysis of Polygalacturonase Genes in Solanum lycopersicum. Int. J. Mol. Sci. 2018, 19, 2290. [Google Scholar] [CrossRef]
- Sitrit, Y.; Hadfield, K.A.; Bennett, A.B.; Bradford, K.J.; Downie, A.B. Expression of a Polygalacturonase Associated with Tomato Seed Germination. Plant Physiol. 1999, 121, 419–428. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Ma, Y.; Chen, N.; Guo, S.; Liu, H.; Guo, X.; Chong, K.; Xu, Y. Overexpression of stress-inducible OsBURP16, the β subunit of polygalacturonase 1, decreases pectin content and cell adhesion and increases abiotic stress sensitivity in rice. Plant Cell Environ. 2014, 37, 1144–1158. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.-M.; Ge, X.-Y.; Zhang, W.-G. Improvement of polygalacturonase production at high temperature by mixed culture of Aspergillus niger and Saccharomyces cerevisiae. Bioresour. Technol. 2011, 102, 10085–10088. [Google Scholar] [CrossRef] [PubMed]
- Hildebrandt, T.M.; Nesi, A.N.; Araújo, W.L.; Braun, H.-P. Amino Acid Catabolism in Plants. Mol. Plant 2015, 8, 1563–1579. [Google Scholar] [CrossRef]
- Joshi, V.; Joung, J.-G.; Fei, Z.; Jander, G. Interdependence of threonine, methionine and isoleucine metabolism in plants: Accumulation and transcriptional regulation under abiotic stress. Amino Acids 2010, 39, 933–947. [Google Scholar] [CrossRef]
- McAllister, C.H.; Facette, M.; Holt, A.; Good, A.G. Analysis of the Enzymatic Properties of a Broad Family of Alanine Aminotransferases. PLoS ONE 2013, 8, e55032. [Google Scholar] [CrossRef]
- Rocha, M.; Licausi, F.; Araújo, W.L.; Nunes-Nesi, A.; Sodek, L.; Fernie, A.R.; van Dongen, J.T. Glycolysis and the Tricarboxylic Acid Cycle Are Linked by Alanine Aminotransferase during Hypoxia Induced by Waterlogging of Lotus japonicus. Plant Physiol. 2010, 152, 1501–1513. [Google Scholar] [CrossRef] [PubMed]
- Schertl, P.; Danne, L.; Braun, H.-P. 3-Hydroxyisobutyrate dehydrogenase is involved in both, valine and isoleucine degradation in Arabidopsis thaliana. Plant Physiol. 2017, 175, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Li, J.; Zhang, R.; Lin, Y.; Xiong, A.; Tan, G.; Luo, Y.; Zhang, Y.; Chen, Q.; Wang, Y.; et al. Combined Analysis of the Metabolome and Transcriptome to Explore Heat Stress Responses and Adaptation Mechanisms in Celery (Apium graveolens L.). Int. J. Mol. Sci. 2022, 23, 3367. [Google Scholar] [CrossRef] [PubMed]
- Ohama, N.; Sato, H.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Transcriptional Regulatory Network of Plant Heat Stress Response. Trends Plant Sci. 2018, 22, 53–65. [Google Scholar] [CrossRef]
- Li, G.-L.; Chang, H.; Li, B.; Zhou, W.; Sun, D.-Y.; Zhou, R.-G. The roles of the atDjA2 and atDjA3 molecular chaperone proteins in improving thermotolerance of Arabidopsis thaliana seedlings. Plant Sci. 2007, 173, 408–416. [Google Scholar] [CrossRef]
- Ma, C.; Haslbeck, M.; Babujee, L.; Jahn, O.; Reumann, S. Identification and Characterization of a Stress-Inducible and a Constitutive Small Heat-Shock Protein Targeted to the Matrix of Plant Peroxisomes. Plant Physiol. 2006, 141, 47–60. [Google Scholar] [CrossRef]
- Haq, S.U.; Khan, A.; Ali, M.; Khattak, A.M.; Gai, W.-X.; Zhang, H.-X.; Wei, A.-M.; Gong, Z.-H. Heat Shock Proteins: Dynamic Biomolecules to Counter Plant Biotic and Abiotic Stresses. Int. J. Mol. Sci. 2019, 20, 5321. [Google Scholar] [CrossRef]
- Zhuang, L.; Cao, W.; Wang, J.; Yu, J.; Yang, Z.; Huang, B. Characterization and Functional Analysis of FaHsfC1b from Festuca arundinacea Conferring Heat Tolerance in Arabidopsis. Int. J. Mol. Sci. 2018, 19, 2702. [Google Scholar] [CrossRef]
- Yang, K.-Z.; Xia, C.; Liu, X.-L.; Dou, X.-Y.; Wang, W.; Chen, L.-Q.; Zhang, X.-Q.; Xie, L.-F.; He, L.; Ma, X.; et al. A mutation in THERMOSENSITIVE MALE STERILE 1, encoding a heat shock protein with DnaJ and PDI domains, leads to thermosensitive gametophytic male sterility in Arabidopsis. Plant J. 2009, 57, 870–882. [Google Scholar] [CrossRef]
- Zhou, W.; Zhou, T.L.; Li, M.-X.; Zhao, C.-L.; Jia, N.; Wang, X.-X.; Sun, Y.-Z.; Li, G.-L.; Xu, M.; Zhou, R.-G.; et al. The Arabidopsis J-protein AtDjB1 facilitates thermotolerance by protecting cells against heat-induced oxidative damage. New Phytolog. 2012, 194, 364–378. [Google Scholar] [CrossRef]
- Seni, S.; Kaur, S.; Malik, P.; Yadav, I.S.; Sirohi, P.; Chauhan, H.; Kaur, A.; Chhuneja, P. Transcriptome based identification and validation of heat stress transcription factors in wheat progenitor species Aegilops speltoides. Sci. Rep. 2021, 11, 22049. [Google Scholar] [CrossRef] [PubMed]
- Chandler, J.W. Class VIIIb APETALA2 Ethylene Response Factors in Plant Development. Trends Plant Sci. 2018, 23, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Seo, P.J.; Xiang, F.; Qiao, M.; Park, J.-Y.; Na Lee, Y.; Kim, S.-G.; Lee, Y.-H.; Park, W.J.; Park, C.-M. The MYB96 Transcription Factor Mediates Abscisic Acid Signaling during Drought Stress Response in Arabidopsis. Plant Physiol. 2009, 151, 275–289. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.-C.; Liao, P.-M.; Kuo, W.-W.; Lin, T.-P. The Arabidopsis ETHYLENE RESPONSE FACTOR1 Regulates Abiotic Stress-Responsive Gene Expression by Binding to Different cis-Acting Elements in Response to Different Stress Signals. Plant Physiol. 2013, 162, 1566–1582. [Google Scholar] [CrossRef] [PubMed]
- Katiyar, A.; Smita, S.; Lenka, S.K.; Rajwanshi, R.; Chinnusamy, V.; Bansal, K.C. Genome-wide classification and expression analysis of MYB transcription factor families in rice and Arabidopsis. BMC Genom. 2012, 13, 544. [Google Scholar] [CrossRef]
- Seok, H.-Y.; Woo, D.-H.; Nguyen, L.V.; Tran, H.T.; Tarte, V.N.; Mehdi, S.M.M.; Lee, S.-Y.; Moon, Y.-H. Arabidopsis AtNAP functions as a negative regulator via repression of AREB1 in salt stress response. Planta 2016, 245, 329–341. [Google Scholar] [CrossRef]
- Cabello, J.V.; Arce, A.L.; Chan, R.L. The homologous HD-Zip I transcription factors HaHB1 and AtHB13 confer cold tolerance via the induction of pathogenesis-related and glucanase proteins. Plant J. 2011, 69, 141–153. [Google Scholar] [CrossRef]
- Phukan, U.J.; Jeena, G.S.; Shukla, R.K. WRKY Transcription Factors: Molecular Regulation and Stress Responses in Plants. Front. Plant Sci. 2016, 7, 760. [Google Scholar] [CrossRef]
- Li, S.; Fu, Q.; Chen, L.; Huang, W.; Yu, D. Arabidopsis thaliana WRKY25, WRKY26, and WRKY33 coordinate induction of plant thermotolerance. Planta 2011, 233, 1237–1252. [Google Scholar] [CrossRef]
- Zhao, J.; Missihoun, T.D.; Bartels, D. The role of Arabidopsis aldehyde dehydrogenase genes in response to high temperature and stress combinations. J. Exp. Bot. 2017, 68, 4295–4308. [Google Scholar] [CrossRef]
- de Abreu-Neto, J.B.; Turchetto-Zolet, A.C.; de Oliveira, L.F.V.; Zanettini, M.H.B.; Margis-Pinheiro, M. Heavy metal-associated isoprenylated plant protein (HIPP): Characterization of a family of proteins exclusive to plants. FEBS J. 2013, 280, 1604–1616. [Google Scholar] [CrossRef] [PubMed]
- Barth, O.; Vogt, S.; Uhlemann, R.; Zschiesche, W.; Humbeck, K. Stress induced and nuclear localized HIPP26 from Arabidopsis thaliana interacts via its heavy metal associated domain with the drought stress related zinc finger transcription factor ATHB29. Plant Mol. Biol. 2008, 69, 213–226. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, A.; Monteiro, F.; Sebastiana, M. Subtilisin-like proteases in plant–pathogen recognition and immune priming: A perspective. Front. Plant Sci. 2014, 5, 739. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | Raw Reads | Clean Reads | Clean Bases | Error Rate | Q30 | GC% |
|---|---|---|---|---|---|---|
| LRa | 57,243,672 | 56,159,618 | 8.4 G | 0.3 | 92.67 | 44.29 |
| LRb | 52,085,380 | 51,122,510 | 7.67 G | 0.3 | 92.5 | 44.63 |
| LHa | 66,765,656 | 65,546,612 | 9.83 G | 0.3 | 92.89 | 44.65 |
| LHb | 61,560,478 | 60,252,602 | 9.04 G | 0.3 | 92.84 | 44.43 |
| TF Family | Gene ID | Up/Down |
|---|---|---|
| Dnaj | 21394896, 21401054, 21395794, 21393813, 21398174, 21405132, 112091504 | Up |
| AP2 | 21407265, 21406253, 21398364, 21392224, 21387978 | Up |
| MYB | 21406818, 112092520, 21399045, 21394542 21394127, 21398688, 112093121 | Up Down |
| HLH | 21404443, 21404618, 21402833 | Up Down |
| NAC | 21389501 21397328 | Up Down |
| Bzip | 112092685 | Up |
| WRKY | 21404214 | Up |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jin, X.; Ackah, M.; Acheampong, A.; Zhang, Q.; Wang, L.; Lin, Q.; Qiu, C.; Zhao, W. Genome-Wide Identification of Candidate Genes Associated with Heat Stress in Mulberry (Morus alba L.). Curr. Issues Mol. Biol. 2023, 45, 4151-4167. https://doi.org/10.3390/cimb45050264
Jin X, Ackah M, Acheampong A, Zhang Q, Wang L, Lin Q, Qiu C, Zhao W. Genome-Wide Identification of Candidate Genes Associated with Heat Stress in Mulberry (Morus alba L.). Current Issues in Molecular Biology. 2023; 45(5):4151-4167. https://doi.org/10.3390/cimb45050264
Chicago/Turabian StyleJin, Xin, Michael Ackah, Adolf Acheampong, Qiaonan Zhang, Lei Wang, Qiang Lin, Changyu Qiu, and Weiguo Zhao. 2023. "Genome-Wide Identification of Candidate Genes Associated with Heat Stress in Mulberry (Morus alba L.)" Current Issues in Molecular Biology 45, no. 5: 4151-4167. https://doi.org/10.3390/cimb45050264
APA StyleJin, X., Ackah, M., Acheampong, A., Zhang, Q., Wang, L., Lin, Q., Qiu, C., & Zhao, W. (2023). Genome-Wide Identification of Candidate Genes Associated with Heat Stress in Mulberry (Morus alba L.). Current Issues in Molecular Biology, 45(5), 4151-4167. https://doi.org/10.3390/cimb45050264