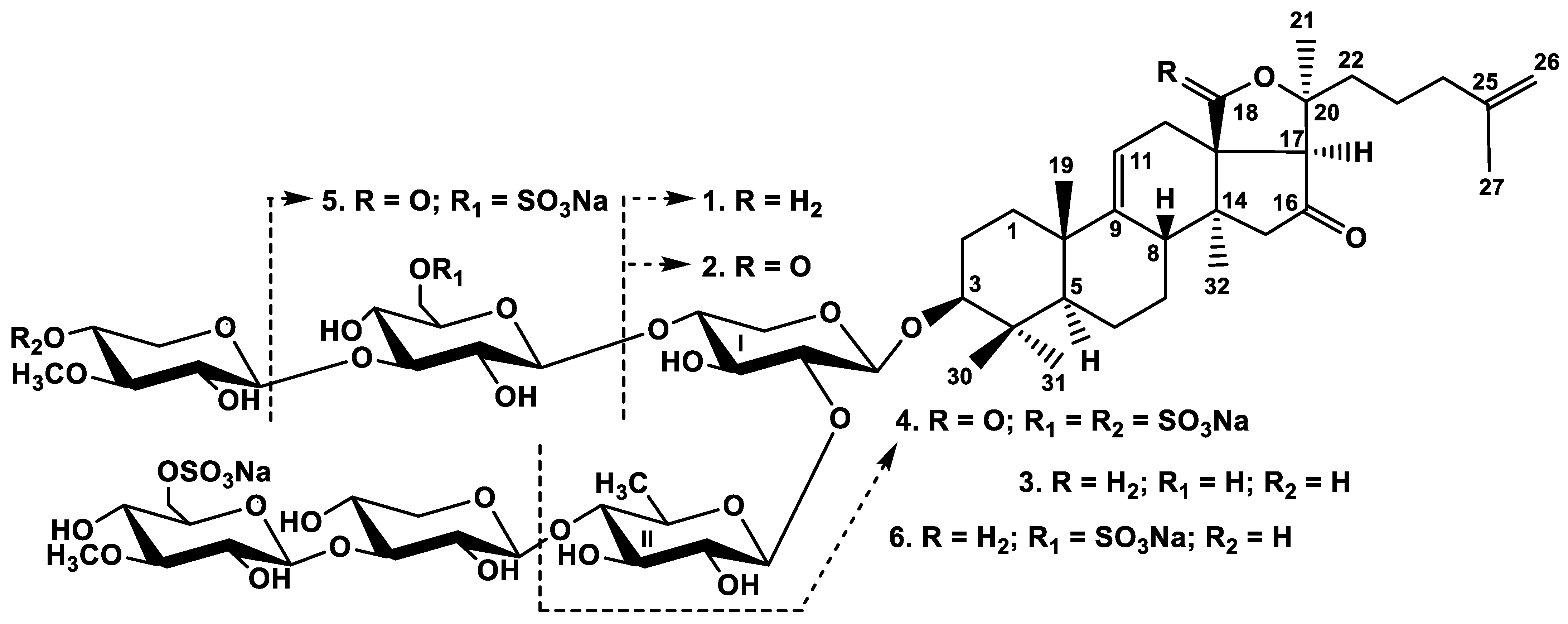
2.1. Structural Elucidation of the Glycosides
The concentrated ethanolic extract of the sea cucumber
Psolus chitonoides was chromatographed on a Polychrom-1 column (powdered Teflon, Biolar, Latvia). The glycosides were eluted after using water as a mobile phase with 50% EtOH and separated by repeated chromatography on Si gel columns with the eluents CHCl
3/EtOH/H
2O (100:75:10), (100:100:17), and (100:125:25) to give three fractions (I–III). The individual compounds
1–
6 (
Figure 1) were isolated by HPLC of the obtained fractions on silica-based column Supelcosil LC-Si (4.6 × 150 mm) and reversed-phase semipreparative columns Phenomenex Synergi Fusion RP (10 × 250 mm) or Supelco Ascentis RP-Amide (10 × 250 mm).
All the configurations of monosaccharides in all glycosides were assigned to D-series along with biogenetical analogies with all known sea cucumber triterpene glycosides.
The molecular formula of chitonoidoside A (
1) was determined to be C
53H
83O
23SNa from the [M
Na−Na]
− ion peak at
m/z 1119.5049 (calc. 1119.5051) in the (−)HR-ESI-MS and [M
Na+Na]
+ ion peak at
m/z 1165.4816 (calc. 1165.4836) in the (+)HR-ESI-MS. The
1H and
13C NMR spectra of the carbohydrate chain of chitonoidoside A (
1) (
Table 1,
Figures S1–S6) demonstrated four characteristic doublets at δ
H 4.82–5.30 (
J = 7.2–8.6 Hz) and, corresponding to them, signals of anomeric carbons at δ
C 104.2–105.5 indicating the presence of four monosaccharide residues, bonded to each other by
β-glycosidic linkages. Analysis of the
1H,
1H-COSY, HSQC, and 1D TOCSY spectra showed that two xylose (Xyl1 and Xyl3), one quinovose (Qui2), and one 3-
O-methylglucose (MeGlc4) residues are present in the carbohydrate chain of chitonoidoside A (
1). The signal of C-6 MeGlc4 was observed at δ
C 66.6, due to α-shifting effect of a sulfate group at this position. The signal of C-4 Xyl1 (δ
C 70.7) showed the presence of a free hydroxy group at this carbon atom in
1. The positions of interglycosidic linkages were established by the ROESY and HMBC spectra (
Table 1). Thus, despite the typical sugar composition for the sea cucumber glycosides, the oligosaccharide chain of
1 turned out to be new.
Analysis of the NMR spectra of the aglycone part of
1 (
Table 2,
Figures S1–S6) indicated the presence of C-30 triterpene nucleus with the 9(11)-double bond (characteristic signals δ
C 151.0 (C-9) and 114.7 (C-11)) and the C-16 keto-group (δ
C 215.9 (C-16)). These positions were confirmed by the HMBC correlations H-8, H
2-12, H
3-19/C-9, H
2-12/C-11, and H
2-15, H-17/C-16. The signal of C-18 at δ
C~178–180 characteristic of 18(20)-lactone in holostane aglycones was absent in the
13C NMR spectrum of compound
1.
The singlet signal of H-17 at δH 2.38 indicated its bonding to three quaternary carbons C-13, C-16, and C-20. The signal of C-20 at δC 86.2 was deduced by the characteristic HMBC correlation between the signal of methyl group H3-21 and C-20. Whereas H2-12 (δC 2.37 (m) and 2.27 (dd, J = 5.4; 16.9 Hz)) and H-17 were correlated in the HMBC spectrum with the signal at δC 73.8 assigned to the methylene group, having the proton signals at δH 4.08 (d, H-18a) and 3.71 (d, H-18b), the position of this group was established as C-18. The cross-peaks H-8β, H-12a,b, and H-15β/H-18 in the ROESY spectrum confirmed this. Based on the deshielded signals of C-18 and C-20, it was supposed that they linked by ether bonds. The configurations of all the asymmetric centers were the same as in holostane aglycones, which was confirmed by the ROESY spectrum. Thus, a new type of non-holostane aglycone lacking a lactone was discovered in the glycoside 1 of P. chitonoides.
The (−)ESI-MS/MS of 1 demonstrated the fragmentation of [MNa−Na]− ion at m/z 1119.5 resulting in the appearance of the fragment ion-peaks at m/z 665.1 [MNa−Na–C30H46O3 (Agl)]−, corresponding to the aglycone loss, 533.1 [MNa−Na–C30H46O3 (Agl)−C5H8O4 (Xyl)]−, 387.1 [MNa−Na–C30H46O3 (Agl)−C5H8O4 (Xyl)−C6H10O4 (Qui)]−, 255.0 [MNa−Na–C30H46O3 (Agl)−C5H8O4 (Xyl)−C6H10O4 (Qui)−C5H8O4 (Xyl)]−. The (+)ESI-MS/MS of 1 demonstrated the fragmentation of [MNa+Na]+ ion at m/z 1165.5. The peak of fragment ion had m/z 1045.5 [MNa+Na−NaSO4−H]+, corroborating the presence of a sulfate group. Other fragment ion peaks illustrated the same patterns that were observed in (−)ESI-MS/MS (see Materials and Methods) corroborating both of the aglycone and carbohydrate chain structures in chitonoidoside A (1).
All of these data indicate that chitonoidoside A (1) is 3β-O-[6-O-sodium sulfate-3-O-methyl-β-d-glucopyranosyl-(1→3)-β-d-xylopyranosyl-(1→4)-β-d-quinovopyranosyl-(1→2)-β-d-xylopyranosyl]-16-oxo-18(20)-epoxylanosta-9(11),25(26)-diene.
The molecular formula of chitonoidoside A
1 (
2) was determined to be C
53H
81O
24SNa from the [M
Na−Na]
− ion peak at
m/z 1133.4856 (calc. 1133.4844) in the (−)HR-ESI-MS and [M
Na+Na]
+ ion peak at
m/z 1179.4620 (calc. 1179.4628) in the (+)HR-ESI-MS. The NMR spectra corresponding to the carbohydrate part of
2 were close to those of
1, indicating the identity of their sugar chains (
Table S1,
Figures S8–S13). Analysis of the
1H and
13C NMR spectra of the aglycone part of
2 indicated the presence of earlier known and broadly distributed in the sea cucumber glycosides holostane aglycone holotoxinogenin, first found in
Apostichopus japonicus [
18] (
Table 3,
Figures S8–S13), which differed from the aglycone of
1 by the presence of 18(20)-lactone. The same aglycone was identified in chitonoidosides C (
4) and D (
5) (
Tables S3, S4, Figures S24–S29 and S32–S37).
The (−)ESI-MS/MS of 2 demonstrated the fragmentation of [MNa−Na]− ion with m/z 1133.5 The peak of fragment ion, observed at m/z 665.1 [MNa−Na–C30H44O4 (Agl)]−, corresponded to the aglycone loss. The subsequent fragmentation led to the appearance of the same fragmentary ion-peaks at m/z 533.1, 387.1 and 255.0 that were observed in the MS/MS of 1, corroborating the identity of the carbohydrate chains of chitonoidosides A1 (2) and A (1).
All these data indicate that chitonoidoside A1 (2) is 3β-O-[6-O-sodium sulfate-3-O-methyl-β-d-glucopyranosyl-(1→3)-β-d-xylopyranosyl-(1→4)-β-d-quinovopyranosyl-(1→2)-β-d-xylopyranosyl]-16-oxo-holosta-9(11),25(26)-diene.
The molecular formula of chitonoidoside B (
3) was determined to be C
65H
103O
32SNa from the [M
Na−Na]
− ion peak at
m/z 1427.6157 (calc. 1427.6159) and [M
Na+Na]
+ ion-peak at
m/z 1473.5925 (calc. 1473.5943) in the (−) and (+)HR-ESI-MS, correspondingly. The
1H and
13C NMR spectra of the carbohydrate part of chitonoidoside B (
3) showed six characteristic doublets at δ
H 4.67–5.12 (
J = 7.6–8.7 Hz), correlated by the HSQC spectrum with corresponding anomeric carbons at δ
C 102.2–105.3. These signals were indicative of a hexasaccharide chain and
β-configurations of glycosidic bonds (
Table 4,
Figures S15–S22). The signals of each monosaccharide unit were deduced as an isolated spin system based on the
1H,
1H-COSY and 1D TOCSY spectra. Further analysis of the HSQC and ROESY spectra of
3 resulted in the assigning of the monosaccharide residues as two xylose (Xyl1 and Xyl3), one quinovose (Qui2), one glucose (Glc5), one 3-
O-methylglucose (MeGlc4), and one 3-
O-methylxylose (MeXyl6) residues. The positions of interglycosidic linkages, established by the ROESY and HMBC spectra of
3, were characteristic of the majority of hexaosides from the sea cucumbers (
Table 4).
The signal of C-6 MeGlc4 in the
13C NMR spectrum of
3 was deshielded to δ
C 67.1 due to α-shifting effect of the sulfate group at this position. 3-
O-methylxylose residue in the upper semi-chain of
3 is not often found in the sea cucumber glycosides and was discovered earlier in some representatives of such taxa as
Synallactes nozawai (order Synallactida) [
19] and four species of the order Dendrochirotida:
Eupentacta fraudatrix [
5] and
E. pseudoquinquesemita [
20] (family Sclerodactylidae),
Pentamera calcigera [
21,
22], and
Thyone aurea [
23] (family Phyllophoridae). None of the previously investigated species of genus
Psolus contained this residue in the glycosides, so its finding was a surprise.
The NMR data of the aglycone part of
3 indicated the identity of the aglycone to that of chitonoidoside A (
1) (
Table S2, Figures S15–S20).
The (−)ESI-MS/MS of 3 demonstrated the fragmentation of [MNa−Na]− ion with m/z 1427.6. The peaks of fragment ions were observed at m/z 1281.5 [MNa−Na–C6H11O4 (MeXyl)+H]−, 1119.5 [MNa−Na–C6H11O4 (MeXyl)−C6H10O5 (Glc)+H]−. The (+)ESI-MS/MS of 3 demonstrated the fragmentation of [MNa+Na]+ ion with m/z 1473.5 resulting in the appearance of the peaks of fragment ions at m/z 1354.5 [MNa+Na−NaHSO4]+, 1327.5 [MNa+Na−C6H11O4 (MeXyl)+H]+, 1194.5 [MNa+Na−C7H11O8SNa (MeGlcSO3Na)]+, 1165.5 [MNa+Na−C6H11O4 (MeXyl)−C6H10O5 (Glc)+H]+, 1062.5 [MNa+Na−C7H11O8SNa (MeGlcSO3Na)−C5H8O4 (Xyl)]+, 917.5 [MNa+Na−C7H11O8SNa (MeGlcSO3Na)−C5H8O4 (Xyl)−C6H10O4 (Qui)]+, confirming the sequence of monosaccharides in the carbohydrate chain of 3.
All these data indicate that chitonoidoside B (3) is 3β-O-{6-O-sodium sulfate-3-O-methyl-β-d-glucopyranosyl-(1→3)-β-d-xylopyranosyl-(1→4)-β-d-quinovopyranosyl-(1→2)-[3-O-methyl-β-d-xylopyranosyl-(1→3)-β-d-glucopyranosyl-(1→4)]-β-d-xylopyranosyl}-16-oxo-18(20)-epoxylanosta-9(11),25(26)-diene.
The molecular formula of chitonoidoside C (
4) was determined to be C
53H
80O
27S
2Na
2 from the [M
2Na−Na]
− ion peak at
m/z 1235.4256 (calc. 1235.4232) and [M
2Na−2Na]
2− ion peak at
m/z 606.2183 (calc. 606.2170) in the (−)HR-ESI-MS. The
1H and
13C NMR spectra corresponding to the carbohydrate chain of chitonoidoside C (
4) (
Table 5,
Figures S24–S30) demonstrated four signals of anomeric protons at δ
H 4.68–5.08 (d,
J = 6.6–8.2 Hz) and corresponding to them signals of anomeric carbons at δ
C 102.2–104.9 deduced by the HSQC spectrum. These signals indicated the presence of tetrasaccharide moiety with
β-glycosidic bonds. The signals of each sugar residue were assigned by the analysis of the
1H,
1H-COSY, 1D TOCSY, ROESY, and HSQC spectra, enabling identification of the monosaccharide units in the chain of
4 as xylose (Xyl1), quinovose (Qui2), glucose (Glc3), and 3-
O-methylxylose (MeXyl4). Analysis of the glycosidic bond positions by the ROESY and HMBC spectra showed that quinovose residue was terminal in the bottom semi-chain because only the correlation between the anomeric proton H-1 Qui2 (δ
H 5.06, d,
J = 7.5 Hz) and H-2 (C-2) Xyl1 took place. Actually, the glycosylation effects were not observed for C-3–C-5 Qui2 in the
13C NMR spectrum of
4. In fact, the signal C-4 Qui2 was shielded to δ
C 76.2 when compared with the same signal in the spectrum of
3 (δ
C 85.6), and the signals of C-3 and C-5 Qui2 were observed at δ
C 76.8 and 72.5 instead of δ
C 74.9 and 71.4 in the spectrum of
3, correspondingly. Therefore, the remaining monosaccharide residues formed the upper semi-chain. The cross-peaks were observed between H-1 Glc3 and H-4 (C-4) Xyl1 and between H-1 MeXyl4 and H-3 (C-3) Glc3 in the ROESY and HMBC spectra, correspondingly. Therefore, the architecture of carbohydrate chain of
4 was the same as in kurilosides of the group C from
Thyonidium kurilensis [
24]. The comparison of the
13C NMR spectra of sugar chains of chitonoidoside C (
4) and kuriloside C
1 showed the coincidence of the signals of three monosachharide units with the exception of the signals of (fourth) terminal sugar. The comparison of the signals of 3-
O-methylxylose, attached to C-3 of Glc in the upper semi-chain, in the
13C NMR spectra of
3 and
4 revealed their differences due the presence of the sulfate group attached to C-4 of 3-
O-methylxylose in
4. Actually, the corresponding signal was deshielded (δ
C 75.5) and the signals of C-3 and C-5 MeXyl4 were shielded (δ
C 84.1 and 64.5) due to α- and β-effects of the sulfate group. The sulfated 3-
O-methylxylose residue was for the first time found in the glycosides from Holothuroidea. Additionally, C-4 in terminal sugars is not a common position of the sulfation in the glycosides in contrast to C-6 of terminal 3-
O-methylglucose or glucose residues. The glycosides characterized by the sulfate group at C-4 of a glucose residue were so far recently found only in
Psolus fabricii [
14].
The second sulfate group in the sugar chain of 4 was attached to C-6 Glc3 (δC 67.2). Thus, the new carbohydrate chain of chitonoidoside C (4) combined the features of the glycosides from T. kurilensis and P. fabricii as a result of parallel chemical evolution.
The aglycone part of chitonoidoside C (
4) was identical to that of chitonoidoside A
1 (
2). This was supposed based on the coincidence of their NMR spectra (
Table S3, Figures S24–S29).
The (−)ESI-MS/MS of chitonoidoside C (4) demonstrated the fragmentation of [M2Na−Na]− ion at m/z 1235.4. The peaks of fragment ions were observed at m/z 1115.5 [M2Na−Na–NaHSO4]−, 1089.4 [M2Na−Na−C6H10O4 (Qui)]−, 969.4 [M2Na−Na−NaHSO4–C6H10O4 (Qui)]−, 841.4 [M2Na−Na−C6H10O7SNa (MeXylOSO3)−C6H10O4 (Qui)+H]−, 517.1 [M2Na−Na–C30H44O4 (Agl)−C6H10O7SNa (MeXylOSO3)−H]−, corroborating the structure established based on NMR analyses.
All these data indicate that chitonoidoside C (4) is 3β-O-{β-d-quinovopyranosyl-(1→2)-[4-O-sodium sulfate-3-O-methyl-β-d-xylopyranosyl-(1→3)-6-O-sodium sulfate-β-d-glucopyranosyl-(1→4)]-β-d-xylopyranosyl}-16-oxo-holosta-9(11),25(26)-diene.
The molecular formula of chitonoidoside D (
5) was determined to be C
59H
90O
32S
2Na
2 from the [M
2Na−Na]
− ion peak at
m/z 1397.4706 (calc. 1397.4760), [M
2Na−2Na]
2− ion peak at
m/z 687.2412 (calc. 687.2434) in the (−)HR-ESI-MS. The presence of two-charged ions in the MS spectra indicated that chitonoidoside D (
5) contains two sulfate groups. The
1H and
13C NMR spectra of the carbohydrate part of chitonoidoside D (
5) (
Table 6,
Figures S32–S38) demonstrated five characteristic doublets at δ
H 4.67–5.11 (d,
J = 7.0–8.1 Hz) and, corresponding to them, signals of anomeric carbons at δ
C 102.7–104.8 deduced by the HSQC spectrum, that indicated the presence of five monosaccharide residues in
5. The monosaccharide composition of
5 was determined to include two xylose (Xyl1 and Xyl3), quinovose (Qui2), glucose (Glc5), and 3-
O-methylglucose (MeGlc4) residues. The positions of glycosidic bonds established by the ROESY and HMBC spectra were typical and showed bottom semi-chains composed of three monosaccharide units and the upper semi-chain—from two monosaccharide units, forming the chain with the common architecture. The comparison of the
13C NMR spectrum of the carbohydrate chain of cladolosides of the group I, having the same sugar composition and architecture [
25], with the spectrum of chitonoidoside D (
5) showed they differed in the signals C-5 and C-6 Glc5 only, shifted in the spectrum of
5 due to the presence of the second sulfate group (δ
C 75.8 (C-5 Glc5) and 67.2 (C-6 Glc5)) (
Table 6). Thus, chitonoidoside D (
5) contained a new branched disulfated pentasaccharide carbohydrate chain.
The aglycone part of chitonoidoside D (
5) (
Table S4, Figures S32–S37) was identical to that of chitonoidosides A
1 (
2) and C (
4), which was confirmed by the closeness of their NMR spectra.
The (−)ESI-MS/MS of chitonoidoside D (5) demonstrated the fragmentation of [M2Na−Na]− ion at m/z 1397.5. The peaks of fragment ions were observed at m/z 1277.5 [M2Na−Na–NaHSO4]−, 1119.5 [M2Na−Na–C7H11O8SNa (MeGlcOSO3)]−, 987.4 [M2Na−Na–C7H11O8SNa (MeGlcOSO3)−C5H8O4 (Xyl)]−, 841.3 [M2Na−Na–C7H11O8SNa (MeGlcOSO3)−C5H8O4 (Xyl)–C6H10O4 (Qui)]−, corroborating the structure of the oligosaccharide chain of 5.
All these data indicate that chitonoidoside D (5) is 3β-O-{6-O-sodium sulfate-3-O-methyl-β-d-glucopyranosyl-(1→3)-β-D-xylopyranosyl-(1→4)-β-d-quinovopyranosyl-(1→2)-[6-O-sodium sulfate-β-d-glucopyranosyl-(1→4)]-β-d-xylopyranosyl}-16-oxo-holosta-9(11),25(26)-diene.
The molecular formula of chitonoidoside E (
6) was determined to be C
65H
102O
35S
2Na
2 from the [M
2Na−Na]
− ion peak at
m/z 1529.5567 (calc. 1529.5546) and [M
2Na−2Na]
2− ion peak at
m/z 753.2836 (calc. 753.2827) in the (−)HR-ESI-MS. The
1H and
13C NMR spectra, corresponding to the carbohydrate chain of
6 (
Table 7,
Figures S40–S47), demonstrated six characteristic doublets at δ
H 4.67–5.12 (
J = 6.9–7.8 Hz) correlated with corresponding anomeric carbons at δ
C 102.2–105.2 by the HSQC spectrum, indicating the presence of a hexasaccharide moiety and
β-configurations of glycosidic bonds. The analysis of the
1H,
1H-COSY, 1D TOCSY, HSQC, and ROESY spectra of
6 revealed the same monosaccharide composition and positions of glycosidic bonds as in the carbohydrate chain of chitonoidoside B (
3). Actually, their
13C NMR spectra were almost coincident differing by the signals C-5 and C-6 of Glc5. The second sulfate group, attached to C-6 of Glc5 in
6, caused the shifting of the signals C-5 Glc5 to δ
C 75.5 and C-6 Glc5 to δ
C 67.1. The difference in the MS spectra of
3 and
6 in 102
amu corroborated this.
The NMR data of the aglycone part of
6 indicated the identity of the aglycone to that of chitonoidoside A (
1) and B (
3) (
Table S5, Figures S40–S45).
The (−)ESI-MS/MS of 6 demonstrated the fragmentation of [M2Na−Na]− ion at m/z 1529.5. The peaks of fragment ions were observed at m/z: 1409.5 [M2Na−Na–NaHSO4]−, 1383.5 [M2Na−Na–C6H11O4 (MeXyl)+H]−, 1263.5 [M2Na−Na–C6H11O4 (MeXyl) –NaHSO4]−, 1251.5 [M2Na−Na–C7H11O8SNa (MeGlcOSO3)]−, 1119.5 [M2Na−Na–C7H11O8SNa (MeGlcOSO3)−C5H8O4 (Xyl)]−, 973.4 [M2Na−Na–C7H11O8SNa (MeGlcOSO3)−C5H8O4 (Xyl)–C6H10O4 (Qui)]−, 827.4 [M2Na−Na–C7H11O8SNa (MeGlcOSO3)−C5H8O4 (Xyl)–C6H10O4 (Qui) –C6H11O4 (MeXyl)+H]−, confirming the sequence of the monosaccharide residues in the chain of 6.
All these data indicate that chitonoidoside E (6) is 3β-O-{6-O-sodium sulfate-3-O-methyl-β-d-glucopyranosyl-(1→3)-β-d-xylopyranosyl-(1→4)-β-d-quinovopyranosyl-(1→2)-[3-O-methyl-β-d-xylopyranosyl-(1→3)-6-O-sodium sulfate-β-d-glucopyranosyl-(1→4)]-β-d-xylopyranosyl}-16-oxo-18(20)-epoxylanosta-9(11),25(26)-diene.



 ,
, 



{kind=link}
{kind=link}