
Expanding Roles of De Novo Lipogenesis in Breast Cancer

 ,
,  , , , , ,
, , , , ,  and
and 
{kind=link}
{kind=link}
Abstract
:1. Introduction
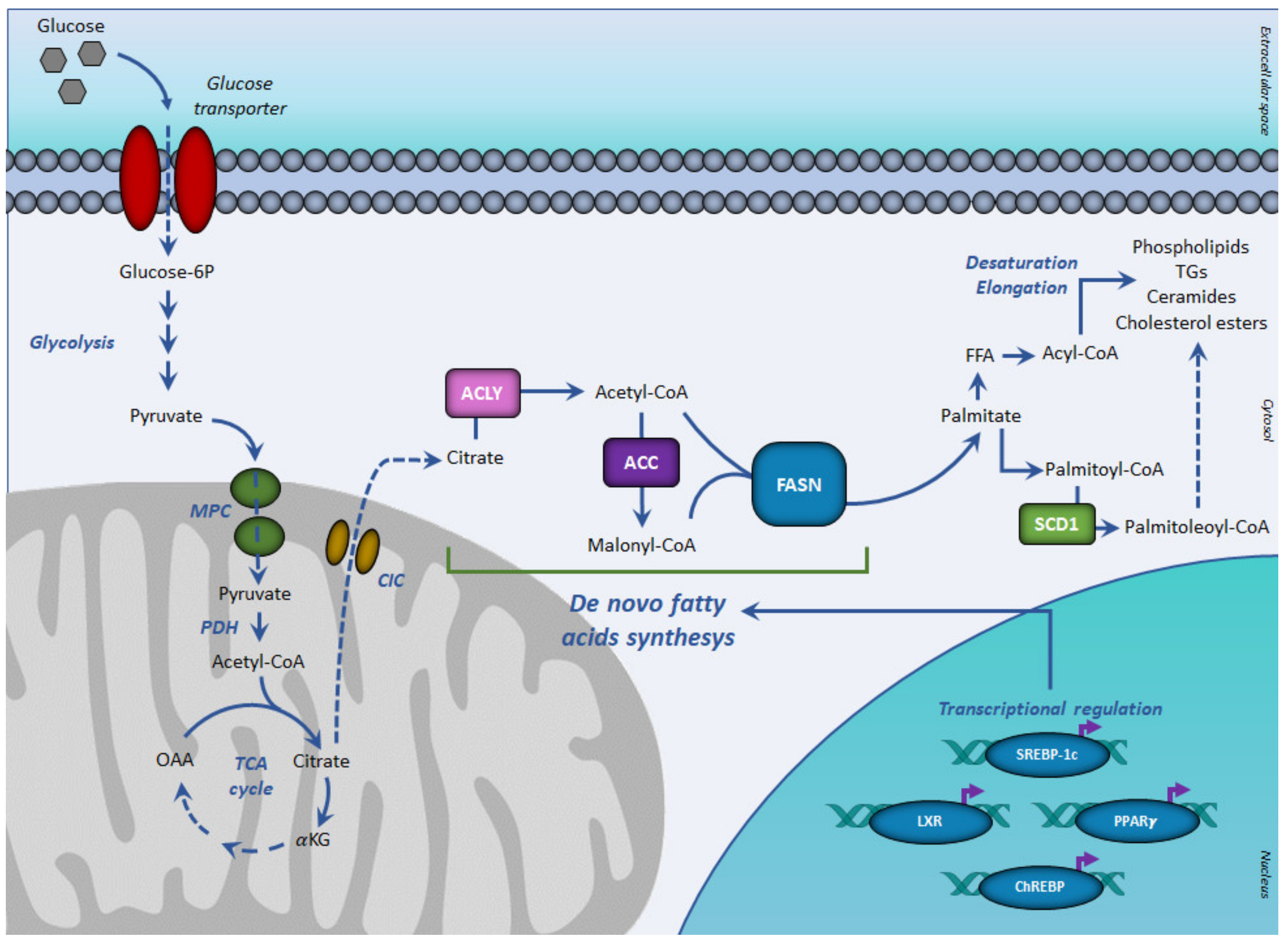
2. Lipogenesis in Cancer
3. Key Enzymes of De Novo Lipogenesis (DNL)
4. Transcription Factors Regulating De Novo Lipogenesis (DNL)
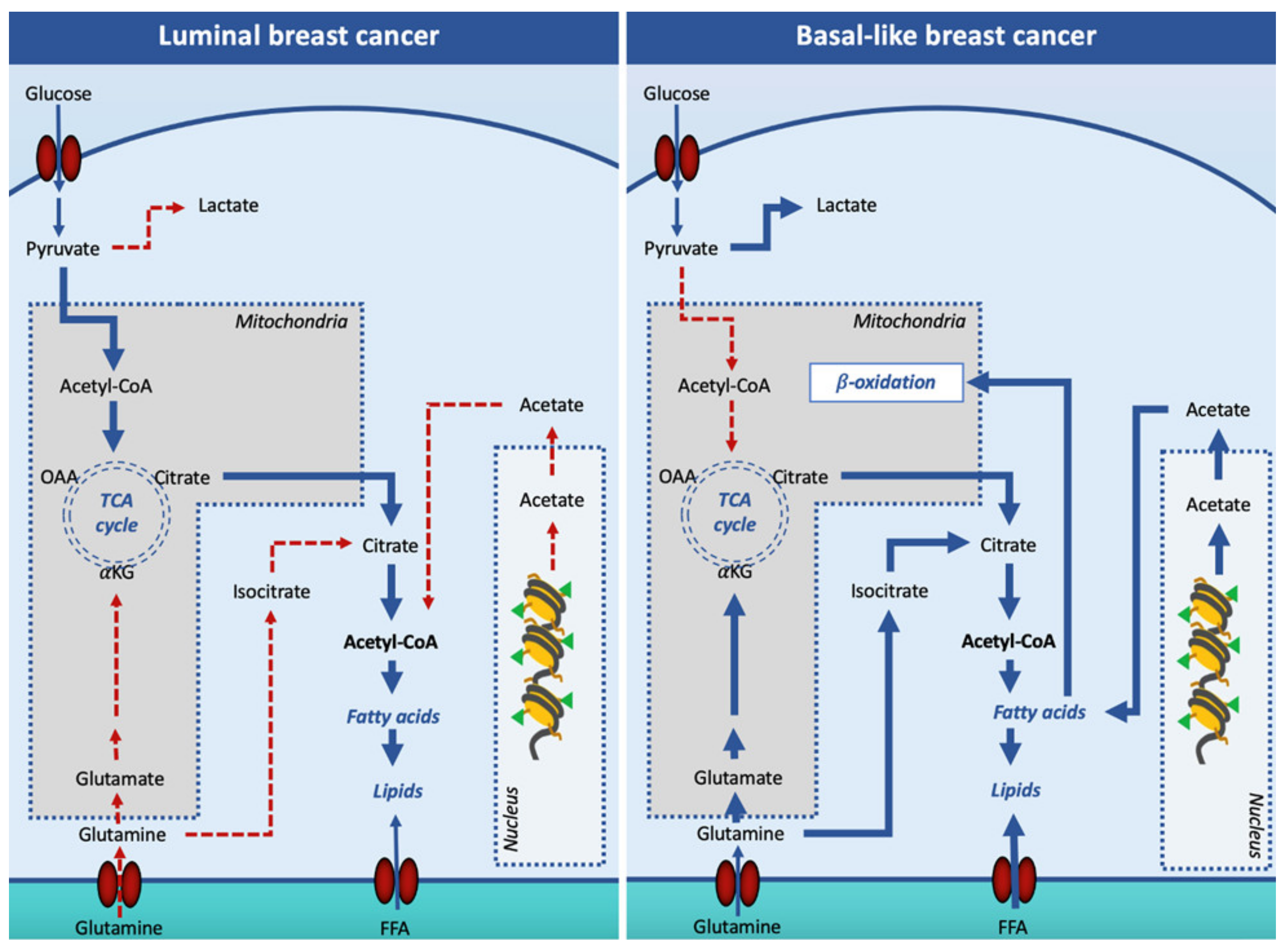
5. Role of DNL Enzymes in Breast Cancer
6. Role of DNL Transcription Factors in Breast Cancer
7. Tumor-Derived Extracellular Vesicles (EVs) Modulate Breast Cancer Metabolism
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Krug, K.; Jaehnig, E.J.; Satpathy, S.; Blumenberg, L.; Karpova, A.; Anurag, M.; Miles, G.; Mertins, P.; Geffen, Y.; Tang, L.C.; et al. Proteogenomic Landscape of Breast Cancer Tumorigenesis and Targeted Therapy. Cell 2020, 183, 1436–1456.e31. [Google Scholar] [CrossRef]
- Mertins, P.; Mani, D.R.; Ruggles, K.V.; Gillette, M.A.; Clauser, K.R.; Wang, P.; Wang, X.; Qiao, J.W.; Cao, S.; Petralia, F.; et al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 2016, 534, 55–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansson, H.J.; Socciarelli, F.; Vacanti, N.M.; Haugen, M.H.; Zhu, Y.; Siavelis, I.; Fernandez-Woodbridge, A.; Aure, M.R.; Sennblad, B.; Vesterlund, M.; et al. Breast cancer quantitative proteome and proteogenomic landscape. Nat. Commun. 2019, 10, 1600. [Google Scholar] [CrossRef]
- Koboldt, D.C.; Fulton, R.S.; McLellan, M.D.; Schmidt, H.; Kalicki-Veizer, J.; McMichael, J.F.; Fulton, L.L.; Dooling, D.J.; Ding, L.; Mardis, E.R.; et al. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Yersal, O.; Barutca, S. Biological subtypes of breast cancer: Prognostic and therapeutic implications. World J. Clin. Oncol. 2014, 5, 412–424. [Google Scholar] [CrossRef] [PubMed]
- Haukaas, T.H.; Euceda, L.R.; Giskeødegård, G.F.; Lamichhane, S.; Krohn, M.; Jernström, S.; Aure, M.R.; Lingjærde, O.C.; Schlichting, E.; Garred, Ø.; et al. Metabolic clusters of breast cancer in relation to gene- and protein expression subtypes. Cancer Metab. 2016, 4, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolfi, S.C.; Chan, L.L.-Y.; Qiu, J.; Tedeschi, P.M.; Bertino, J.R.; Hirshfield, K.M.; Oltvai, Z.N.; Vazquez, A. The metabolic demands of cancer cells are coupled to their size and protein synthesis rates. Cancer Metab. 2013, 1, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Z.; Ibekwe, E.; Chornenkyy, Y. Metabolic Alterations in Cancer Cells and the Emerging Role of Oncometabolites as Drivers of Neoplastic Change. Antioxidants 2018, 7, 16. [Google Scholar] [CrossRef] [Green Version]
- Naik, A.; Decock, J. Lactate Metabolism and Immune Modulation in Breast Cancer: A Focused Review on Triple Negative Breast Tumors. Front. Oncol. 2020, 10, 2668. [Google Scholar] [CrossRef]
- Feng, W.W.; Kurokawa, M. Lipid metabolic reprogramming as an emerging mechanism of resistance to kinase inhibitors in breast cancer. Cancer Drug Resist. 2020, 3, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Beloribi-Djefaflia, S.; Vasseur, S.; Guillaumond, F. Lipid metabolic reprogramming in cancer cells. Oncogenesis 2016, 5, e189. [Google Scholar] [CrossRef]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar] [CrossRef] [Green Version]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef] [Green Version]
- Jones, S.F.; Infante, J.R. Molecular Pathways: Fatty Acid Synthase. Clin. Cancer Res. 2015. clincanres.0126.2015. [Google Scholar] [CrossRef] [Green Version]
- Munir, R.; Lisec, J.; Swinnen, J.V.; Zaidi, N. Lipid metabolism in cancer cells under metabolic stress. Br. J. Cancer 2019, 120, 1090–1098. [Google Scholar] [CrossRef]
- Giudetti, A.M.; De Domenico, S.; Ragusa, A.; Lunetti, P.; Gaballo, A.; Franck, J.; Simeone, P.; Nicolardi, G.; De Nuccio, F.; Santino, A.; et al. A specific lipid metabolic profile is associated with the epithelial mesenchymal transition program. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2019, 1864, 344–357. [Google Scholar] [CrossRef]
- Chen, M.; Huang, J. The expanded role of fatty acid metabolism in cancer: New aspects and targets. Precis. Clin. Med. 2019, 2, 183–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.J.; Nieto, M.A. Epithelial-Mesenchymal Transitions in Development and Disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Stefania, D.D.; Vergara, D. The Many-Faced Program of Epithelial-Mesenchymal Transition: A System Biology-Based View. Front. Oncol. 2017, 7, 274. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Settleman, J. EMT, cancer stem cells and drug resistance: An emerging axis of evil in the war on cancer. Oncogene 2010, 29, 4741–4751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papandreou, I.; Cairns, R.A.; Fontana, L.; Lim, A.L.; Denko, N.C. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006, 3, 187–197. [Google Scholar] [CrossRef] [Green Version]
- Metallo, C.M.; Gameiro, P.A.; Bell, E.L.; Mattaini, K.R.; Yang, J.; Hiller, K.; Jewell, C.M.; Johnson, Z.R.; Irvine, D.J.; Guarente, L.; et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 2012, 481, 380–384. [Google Scholar] [CrossRef] [Green Version]
- Lampa, M.; Arlt, H.; He, T.; Ospina, B.; Reeves, J.; Zhang, B.; Murtie, J.; Deng, G.; Barberis, C.; Hoffmann, D.; et al. Glutaminase is essential for the growth of triple-negative breast cancer cells with a deregulated glutamine metabolism pathway and its suppression synergizes with mTOR inhibition. PLoS ONE 2017, 12, e0185092. [Google Scholar] [CrossRef]
- Kung, H.-N.; Marks, J.R.; Chi, J.-T. Glutamine synthetase is a genetic determinant of cell type-specific glutamine independence in breast epithelia. PLoS Genet. 2011, 7, e1002229. [Google Scholar] [CrossRef] [Green Version]
- Röhrig, F.; Schulze, A. The multifaceted roles of fatty acid synthesis in cancer. Nat. Rev. Cancer 2016, 16, 732–749. [Google Scholar] [CrossRef] [PubMed]
- Schug, Z.T.; Peck, B.; Jones, D.T.; Zhang, Q.; Grosskurth, S.; Alam, I.S.; Goodwin, L.M.; Smethurst, E.; Mason, S.; Blyth, K.; et al. Acetyl-CoA Synthetase 2 Promotes Acetate Utilization and Maintains Cancer Cell Growth under Metabolic Stress. Cancer Cell 2015, 27, 57–71. [Google Scholar] [CrossRef] [Green Version]
- Kamphorst, J.J.; Chung, M.K.; Fan, J.; Rabinowitz, J.D. Quantitative analysis of acetyl-CoA production in hypoxic cancer cells reveals substantial contribution from acetate. Cancer Metab. 2014, 2, 23. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.K.; Gebregiworgis, T.; Purohit, V.; Chaika, N.V.; Gunda, V.; Radhakrishnan, P.; Mehla, K.; Pipinos, I.I.; Powers, R.; Yu, F.; et al. Erratum to: Metabolic reprogramming induced by ketone bodies diminishes pancreatic cancer cachexia. Cancer Metab. 2014, 2, 23. [Google Scholar] [CrossRef] [Green Version]
- Yoshii, Y.; Furukawa, T.; Yoshii, H.; Mori, T.; Kiyono, Y.; Waki, A.; Kobayashi, M.; Tsujikawa, T.; Kudo, T.; Okazawa, H.; et al. Cytosolic acetyl-CoA synthetase affected tumor cell survival under hypoxia: The possible function in tumor acetyl-CoA/acetate metabolism. Cancer Sci. 2009, 100, 821–827. [Google Scholar] [CrossRef]
- Bensaad, K.; Favaro, E.; Lewis, C.A.; Peck, B.; Lord, S.; Collins, J.M.; Pinnick, K.E.; Wigfield, S.; Buffa, F.M.; Li, J.-L.; et al. Fatty acid uptake and lipid storage induced by HIF-1α contribute to cell growth and survival after hypoxia-reoxygenation. Cell Rep. 2014, 9, 349–365. [Google Scholar] [CrossRef] [Green Version]
- Mwaikambo, B.R.; Yang, C.; Chemtob, S.; Hardy, P. Hypoxia Up-regulates CD36 Expression and Function via Hypoxia-inducible Factor-1- and Phosphatidylinositol 3-Kinase-dependent Mechanisms*. J. Biol. Chem. 2009, 284, 26695–26707. [Google Scholar] [CrossRef] [Green Version]
- Chabowski, A.; Górski, J.; Calles-Escandon, J.; Tandon, N.N.; Bonen, A. Hypoxia-induced fatty acid transporter translocation increases fatty acid transport and contributes to lipid accumulation in the heart. FEBS Lett. 2006, 580, 3617–3623. [Google Scholar] [CrossRef] [Green Version]
- Lewis, C.A.; Brault, C.; Peck, B.; Bensaad, K.; Griffiths, B.; Mitter, R.; Chakravarty, P.; East, P.; Dankworth, B.; Alibhai, D.; et al. SREBP maintains lipid biosynthesis and viability of cancer cells under lipid- and oxygen-deprived conditions and defines a gene signature associated with poor survival in glioblastoma multiforme. Oncogene 2015, 34, 5128–5140. [Google Scholar] [CrossRef] [PubMed]
- Gharpure, K.M.; Pradeep, S.; Sans, M.; Rupaimoole, R.; Ivan, C.; Wu, S.Y.; Bayraktar, E.; Nagaraja, A.S.; Mangala, L.S.; Zhang, X.; et al. FABP4 as a key determinant of metastatic potential of ovarian cancer. Nat. Commun. 2018, 9, 2923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casciano, J.C.; Perry, C.; Cohen-Nowak, A.J.; Miller, K.D.; Vande Voorde, J.; Zhang, Q.; Chalmers, S.; Sandison, M.E.; Liu, Q.; Hedley, A.; et al. MYC regulates fatty acid metabolism through a multigenic program in claudin-low triple negative breast cancer. Br. J. Cancer 2020, 122, 868–884. [Google Scholar] [CrossRef] [PubMed]
- Sp, N.; Kang, D.Y.; Kim, D.H.; Park, J.H.; Lee, H.G.; Kim, H.J.; Darvin, P.; Park, Y.-M.; Yang, Y.M. Nobiletin Inhibits CD36-Dependent Tumor Angiogenesis, Migration, Invasion, and Sphere Formation Through the Cd36/Stat3/Nf-Κb Signaling Axis. Nutrients 2018, 10, 772. [Google Scholar] [CrossRef] [Green Version]
- Hellerstein, M.K. De novo lipogenesis in humans: Metabolic and regulatory aspects. Eur. J. Clin. Nutr. 1999, 53, s53–s65. [Google Scholar] [CrossRef] [Green Version]
- Abu-Elheiga, L.; Jayakumar, A.; Baldini, A.; Chirala, S.S.; Wakil, S.J. Human Acetyl-CoA Carboxylase: Characterization, Molecular Cloning, and Evidence for Two Isoforms. Proc. Natl. Acad. Sci. USA 1995, 92, 4011–4015. [Google Scholar] [CrossRef] [Green Version]
- Abu-Elheiga, L.; Almarza-Ortega, D.B.; Baldini, A.; Wakil, S.J. Human Acetyl-CoA Carboxylase 2: MOLECULAR CLONING, CHARACTERIZATION, CHROMOSOMAL MAPPING, AND EVIDENCE FOR TWO ISOFORMS. J. Biol. Chem. 1997, 272, 10669–10677. [Google Scholar] [CrossRef] [Green Version]
- Abu-Elheiga, L.; Brinkley, W.R.; Zhong, L.; Chirala, S.S.; Woldegiorgis, G.; Wakil, S.J. The subcellular localization of acetyl-CoA carboxylase 2. Proc. Natl. Acad. Sci. USA 2000, 97, 1444–1449. [Google Scholar] [CrossRef] [Green Version]
- Ntambi, J.M.; Miyazaki, M. Recent insights into stearoyl-CoA desaturase-1. Curr. Opin. Lipidol. 2003, 14, 255–261. [Google Scholar] [CrossRef]
- Angelucci, C.; D’Alessio, A.; Iacopino, F.; Proietti, G.; Di Leone, A.; Masetti, R.; Sica, G. Pivotal role of human stearoyl-CoA desaturases (SCD1 and 5) in breast cancer progression: Oleic acid-based effect of SCD1 on cell migration and a novel pro-cell survival role for SCD5. Oncotarget 2018, 9, 24364. [Google Scholar] [CrossRef]
- Shimomura, I.; Bashmakov, Y.; Ikemoto, S.; Horton, J.D.; Brown, M.S.; Goldstein, J.L. Insulin selectively increases SREBP-1c mRNA in the livers of rats with streptozotocin-induced diabetes. Proc. Natl. Acad. Sci. USA 1999, 96, 13656–13661. [Google Scholar] [CrossRef] [Green Version]
- Damiano, F.; Gnoni, G.V.; Siculella, L. Citrate carrier promoter is target of peroxisome proliferator-activated receptor alpha and gamma in hepatocytes and adipocytes. Int. J. Biochem. Cell Biol. 2012, 44, 659–668. [Google Scholar] [CrossRef]
- Cheng, C.; Geng, F.; Cheng, X.; Guo, D. Lipid metabolism reprogramming and its potential targets in cancer. Cancer Commun. 2018, 38, 27. [Google Scholar] [CrossRef]
- Guo, D.; Bell, E.H.; Mischel, P.; Chakravarti, A. Targeting SREBP-1-driven lipid metabolism to treat cancer. Curr. Pharm. Des. 2014, 20, 2619–2626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffin, M.J.; Wong, R.H.F.; Pandya, N.; Sul, H.S. Direct Interaction between USF and SREBP-1c Mediates Synergistic Activation of the Fatty-acid Synthase Promoter. J. Biol. Chem. 2007, 282, 5453–5467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishii, S.; IIzuka, K.; Miller, B.C.; Uyeda, K. Carbohydrate response element binding protein directly promotes lipogenic enzyme gene transcription. Proc. Natl. Acad. Sci. USA 2004, 101, 15597–15602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uyeda, K.; Yamashita, H.; Kawaguchi, T. Carbohydrate responsive element-binding protein (ChREBP): A key regulator of glucose metabolism and fat storage. Biochem. Pharmacol. 2002, 63, 2075–2080. [Google Scholar] [CrossRef]
- Dentin, R.; Girard, J.; Postic, C. Carbohydrate responsive element binding protein (ChREBP) and sterol regulatory element binding protein-1c (SREBP-1c): Two key regulators of glucose metabolism and lipid synthesis in liver. Biochimie 2005, 87, 81–86. [Google Scholar] [CrossRef]
- Airley, R.E.; McHugh, P.; Evans, A.R.; Harris, B.; Winchester, L.; Buffa, F.M.; Al-Tameemi, W.; Leek, R.; Harris, A.L. Role of carbohydrate response element-binding protein (ChREBP) in generating an aerobic metabolic phenotype and in breast cancer progression. Br. J. Cancer 2014, 110, 715–723. [Google Scholar] [CrossRef]
- Chawla, A.; Repa, J.J.; Evans, R.M.; Mangelsdorf, D.J. Nuclear Receptors and Lipid Physiology: Opening the X-Files. Science 2001, 294, 1866–1870. [Google Scholar] [CrossRef] [Green Version]
- Willy, P.J.; Umesono, K.; Ong, E.S.; Evans, R.M.; Heyman, R.A.; Mangelsdorf, D.J. LXR, a nuclear receptor that defines a distinct retinoid response pathway. Genes Dev. 1995, 9, 1033–1045. [Google Scholar] [CrossRef] [Green Version]
- Janowski, B.A.; Willy, P.J.; Devi, T.R.; Falck, J.R.; Mangelsdorf, D.J. An oxysterol signalling pathway mediated by the nuclear receptor LXRα. Nature 1996, 383, 728–731. [Google Scholar] [CrossRef]
- Joseph, S.B.; McKilligin, E.; Pei, L.; Watson, M.A.; Collins, A.R.; Laffitte, B.A.; Chen, M.; Noh, G.; Goodman, J.; Hagger, G.N.; et al. Synthetic LXR ligand inhibits the development of atherosclerosis in mice. Proc. Natl. Acad. Sci. USA 2002, 99, 7604–7609. [Google Scholar] [CrossRef] [Green Version]
- Sugden, M.C.; Zariwala, M.G.; Holness, M.J. PPARs and the orchestration of metabolic fuel selection. Pharmacol. Res. 2009, 60, 141–150. [Google Scholar] [CrossRef]
- Wang, Y.-X. PPARs: Diverse regulators in energy metabolism and metabolic diseases. Cell Res. 2010, 20, 124–137. [Google Scholar] [CrossRef] [PubMed]
- Chao, L.; Marcus-Samuels, B.; Mason, M.M.; Moitra, J.; Vinson, C.; Arioglu, E.; Gavrilova, O.; Reitman, M.L. Adipose tissue is required for the antidiabetic, but not for the hypolipidemic, effect of thiazolidinediones. J. Clin. Investig. 2000, 106, 1221–1228. [Google Scholar] [CrossRef] [Green Version]
- Kubota, N.; Terauchi, Y.; Miki, H.; Tamemoto, H.; Yamauchi, T.; Komeda, K.; Satoh, S.; Nakano, R.; Ishii, C.; Sugiyama, T.; et al. PPAR gamma mediates high-fat diet-induced adipocyte hypertrophy and insulin resistance. Mol. Cell 1999, 4, 597–609. [Google Scholar] [CrossRef]
- Miles, P.D.; Barak, Y.; He, W.; Evans, R.M.; Olefsky, J.M. Improved insulin-sensitivity in mice heterozygous for PPAR-gamma deficiency. J. Clin. Investig. 2000, 105, 287–292. [Google Scholar] [CrossRef] [Green Version]
- Phan, L.M.; Yeung, S.-C.J.; Lee, M.-H. Cancer metabolic reprogramming: Importance, main features, and potentials for precise targeted anti-cancer therapies. Cancer Biol. Med. 2014, 11, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Burak Ozkaya, A.; Ak, H.; Aydin, S.; Aydin, H.H. Targeting Mitochondrial Citrate Transport in Breast Cancer Cell Lines. Anticancer. Agents Med. Chem. 2015, 15, 374–381. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, N.; Swinnen, J.V.; Smans, K. ATP-Citrate Lyase: A Key Player in Cancer Metabolism. Cancer Res. 2012, 72, 3709–3714. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Yin, L.; Wei, J.; Yang, Z.; Jiang, G. ATP citrate lyase is increased in human breast cancer, depletion of which promotes apoptosis. Tumor Biol. 2017, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatzivassiliou, G.; Zhao, F.; Bauer, D.E.; Andreadis, C.; Shaw, A.N.; Dhanak, D.; Hingorani, S.R.; Tuveson, D.A.; Thompson, C.B. ATP citrate lyase inhibition can suppress tumor cell growth. Cancer Cell 2005, 8, 311–321. [Google Scholar] [CrossRef] [Green Version]
- Lucenay, K.S.; Doostan, I.; Karakas, C.; Bui, T.; Ding, Z.; Mills, G.B.; Hunt, K.K.; Keyomarsi, K. Cyclin E Associates with the Lipogenic Enzyme ATP-Citrate Lyase to Enable Malignant Growth of Breast Cancer Cells. Cancer Res. 2016, 76, 2406–2418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Jiang, Q.; Dong, C. Metabolic reprogramming in triple-negative breast cancer. Cancer Biol. Med. 2020, 17, 44–59. [Google Scholar] [CrossRef]
- Pandey, P.R.; Xing, F.; Sharma, S.; Watabe, M.; Pai, S.K.; Iiizumi-Gairani, M.; Fukuda, K.; Hirota, S.; Mo, Y.-Y.; Watabe, K. Elevated lipogenesis in epithelial stem-like cell confers survival advantage in ductal carcinoma in situ of breast cancer. Oncogene 2013, 32, 5111–5122. [Google Scholar] [CrossRef] [Green Version]
- Chajès, V.; Cambot, M.; Moreau, K.; Lenoir, G.M.; Joulin, V. Acetyl-CoA Carboxylase α Is Essential to Breast Cancer Cell Survival. Cancer Res. 2006, 66, 5287–5294. [Google Scholar] [CrossRef] [Green Version]
- Milgraum, L.Z.; Witters, L.A.; Pasternack, G.R.; Kuhajda, F.P. Enzymes of the fatty acid synthesis pathway are highly expressed in in situ breast carcinoma. Clin. Cancer Res. 1997, 3, 2115–2120. [Google Scholar] [PubMed]
- Menendez, J.A.; Vellon, L.; Mehmi, I.; Oza, B.P.; Ropero, S.; Colomer, R.; Lupu, R. Inhibition of fatty acid synthase (FAS) suppresses HER2/neu (erbB-2) oncogene overexpression in cancer cells. Proc. Natl. Acad. Sci. USA 2004, 101, 10715–10720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zielinska, H.A.; Holly, J.M.P.; Bahl, A.; Perks, C.M. Inhibition of FASN and ERα signalling during hyperglycaemia-induced matrix-specific EMT promotes breast cancer cell invasion via a caveolin-1-dependent mechanism. Cancer Lett. 2018, 419, 187–202. [Google Scholar] [CrossRef]
- Alwarawrah, Y.; Hughes, P.; Loiselle, D.; Carlson, D.A.; Darr, D.B.; Jordan, J.L.; Xiong, J.; Hunter, L.M.; Dubois, L.G.; Thompson, J.W.; et al. Fasnall, a Selective FASN Inhibitor, Shows Potent Anti-tumor Activity in the MMTV-Neu Model of HER2(+) Breast Cancer. Cell Chem. Biol. 2016, 23, 678–688. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Salinas, F.; Rojo, R.; Martinez-Amador, C.; Herrera-Gamboa, J.; Trevino, V. Transcriptomic and cellular analyses of CRISPR/Cas9-mediated edition of FASN show inhibition of aggressive characteristics in breast cancer cells. Biochem. Biophys. Res. Commun. 2020, 529, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Alo, P.L.; Visca, P.; Marci, A.; Mangoni, A.; Botti, C.; Di Tondo, U. Expression of fatty acid synthase (FAS) as a predictor of recurrence in stage I breast carcinoma patients. Cancer 1996, 77, 474–482. [Google Scholar] [CrossRef]
- Li, G.; Zhao, F.; Cui, Y. Proteomics Using Mammospheres as a Model System to Identify Proteins Deregulated in Breast Cancer Stem Cells. Curr. Mol. Med. 2013, 13, 459–463. [Google Scholar]
- Brunet, J.; Vazquez-Martin, A.; Colomer, R.; Graña-Suarez, B.; Martin-Castillo, B.; Menendez, J.A. BRCA1 and acetyl-CoA carboxylase: The metabolic syndrome of breast cancer. Mol. Carcinog. 2008, 47, 157–163. [Google Scholar] [CrossRef]
- Rochefort, H.; Chalbos, D. The Role of Sex Steroid Receptors on Lipogenesis in Breast and Prostate Carcinogenesis: A Viewpoint. Horm. Cancer 2010, 1, 63–70. [Google Scholar] [CrossRef] [Green Version]
- Yoon, S.; Lee, M.-Y.; Park, S.W.; Moon, J.-S.; Koh, Y.-K.; Ahn, Y.-H.; Park, B.-W.; Kim, K.-S. Up-regulation of Acetyl-CoA Carboxylase α and Fatty Acid Synthase by Human Epidermal Growth Factor Receptor 2 at the Translational Level in Breast Cancer Cells. J. Biol. Chem. 2007, 282, 26122–26131. [Google Scholar] [CrossRef] [Green Version]
- Corominas-Faja, B.; Cuyàs, E.; Gumuzio, J.; Bosch-Barrera, J.; Leis, O.; Martin, Á.G.; Menendez, J.A. Chemical inhibition of acetyl-CoA carboxylase suppresses self-renewal growth of cancer stem cells. Oncotarget 2014, 5, 8306–8316. [Google Scholar] [CrossRef]
- Jin, Q.; Yuan, L.X.; Boulbes, D.; Baek, J.M.; Wang, Y.N.; Gomez-Cabello, D.; Hawke, D.H.; Yeung, S.C.; Lee, M.H.; Hortobagyi, G.N.; et al. Fatty acid synthase phosphorylation: A novel therapeutic target in HER2-overexpressing breast cancer cells. Breast Cancer Res. 2010, 12, R96. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.H.; Kim, N.H.; Yun, J.S.; Cho, E.S.; Cha, Y.H.; Cho, S.B.; Lee, S.-H.; Cha, S.Y.; Kim, S.-Y.; Choi, J.; et al. Snail augments fatty acid oxidation by suppression of mitochondrial ACC2 during cancer progression. Life Sci. Alliance 2020, 3, e202000683. [Google Scholar] [CrossRef]
- Liu, Q.; Huo, H.; Ao, S.; Liu, T.; Yang, L.; Fei, Z.; Zhang, Z.; Ding, L.; Cui, Q.; Lin, J.; et al. TGF-β1-induced epithelial-mesenchymal transition increases fatty acid oxidation and OXPHOS activity via the p-AMPK pathway in breast cancer cells. Oncol Rep. 2020, 44, 1206–1215. [Google Scholar] [CrossRef]
- Rios Garcia, M.; Steinbauer, B.; Srivastava, K.; Singhal, M.; Mattijssen, F.; Maida, A.; Christian, S.; Hess-Stumpp, H.; Augustin, H.G.; Müller-Decker, K.; et al. Acetyl-CoA Carboxylase 1-Dependent Protein Acetylation Controls Breast Cancer Metastasis and Recurrence. Cell Metab. 2017, 26, 842–855.e5. [Google Scholar] [CrossRef] [Green Version]
- Igal, R.A. Roles of StearoylCoA Desaturase-1 in the Regulation of Cancer Cell Growth, Survival and Tumorigenesis. Cancers 2011, 3, 2462–2477. [Google Scholar] [CrossRef] [Green Version]
- Mauvoisin, D.; Charfi, C.; Lounis, A.M.; Rassart, E.; Mounier, C. Decreasing stearoyl-CoA desaturase-1 expression inhibits β-catenin signaling in breast cancer cells. Cancer Sci. 2013, 104, 36–42. [Google Scholar] [CrossRef]
- Luyimbazi, D.; Akcakanat, A.; McAuliffe, P.F.; Zhang, L.; Singh, G.; Gonzalez-Angulo, A.M.; Chen, H.; Do, K.-A.; Zheng, Y.; Hung, M.-C.; et al. Rapamycin regulates stearoyl CoA desaturase 1 expression in breast cancer. Mol. Cancer Ther. 2010, 9, 2770–2784. [Google Scholar] [CrossRef] [Green Version]
- Holder, A.M.; Gonzalez-Angulo, A.M.; Chen, H.; Akcakanat, A.; Do, K.-A.; Fraser Symmans, W.; Pusztai, L.; Hortobagyi, G.N.; Mills, G.B.; Meric-Bernstam, F. High stearoyl-CoA desaturase 1 expression is associated with shorter survival in breast cancer patients. Breast Cancer Res. Treat. 2013, 137, 319–327. [Google Scholar] [CrossRef] [Green Version]
- Monaco, M.E. Fatty acid metabolism in breast cancer subtypes. Oncotarget 2017, 8, 29487–29500. [Google Scholar] [CrossRef] [Green Version]
- Kumar-Sinha, C.; Ignatoski, K.W.; Lippman, M.E.; Ethier, S.P.; Chinnaiyan, A.M. Transcriptome Analysis of HER2 Reveals a Molecular Connection to Fatty Acid Synthesis. Cancer Res. 2003, 63, 132–139. [Google Scholar]
- Pitroda, S.P.; Khodarev, N.N.; Beckett, M.A.; Kufe, D.W.; Weichselbaum, R.R. MUC1-induced alterations in a lipid metabolic gene network predict response of human breast cancers to tamoxifen treatment. Proc. Natl. Acad. Sci. USA 2009, 106, 5837–5841. [Google Scholar] [CrossRef] [Green Version]
- Mason, P.; Liang, B.; Li, L.; Fremgen, T.; Murphy, E.; Quinn, A.; Madden, S.L.; Biemann, H.-P.; Wang, B.; Cohen, A.; et al. SCD1 inhibition causes cancer cell death by depleting mono-unsaturated fatty acids. PLoS ONE 2012, 7, e33823. [Google Scholar] [CrossRef] [Green Version]
- Minville-Walz, M.; Pierre, A.-S.; Pichon, L.; Bellenger, S.; Fèvre, C.; Bellenger, J.; Tessier, C.; Narce, M.; Rialland, M. Inhibition of stearoyl-CoA desaturase 1 expression induces CHOP-dependent cell death in human cancer cells. PLoS ONE 2010, 5, e14363. [Google Scholar] [CrossRef]
- Hess, D.; Chisholm, J.W.; Igal, R.A. Inhibition of stearoylCoA desaturase activity blocks cell cycle progression and induces programmed cell death in lung cancer cells. PLoS ONE 2010, 5, e11394. [Google Scholar] [CrossRef] [Green Version]
- Fritz, V.; Benfodda, Z.; Rodier, G.; Henriquet, C.; Iborra, F.; Avancès, C.; Allory, Y.; de la Taille, A.; Culine, S.; Blancou, H.; et al. Abrogation of de novo lipogenesis by stearoyl-CoA desaturase 1 inhibition interferes with oncogenic signaling and blocks prostate cancer progression in mice. Mol. Cancer Ther. 2010, 9, 1740–1754. [Google Scholar] [CrossRef] [Green Version]
- Scaglia, N.; Igal Ariel, R. Inhibition of Stearoyl-CoA Desaturase 1 expression in human lung adenocarcinoma cells impairs tumorigenesis. Int. J. Oncol. 2008, 33, 839–850. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Zhi, Z.; Wang, C.; Xing, H.; Song, G.; Yu, X.; Zhu, Y.; Wang, X.; Zhang, X.; Di, Y. Exogenous lipids promote the growth of breast cancer cells via CD36. Oncol. Rep. 2017, 38, 2105–2115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belkaid, A.; Duguay, S.R.; Ouellette, R.J.; Surette, M.E. 17β-estradiol induces stearoyl-CoA desaturase-1 expression in estrogen receptor-positive breast cancer cells. BMC Cancer 2015, 15, 440. [Google Scholar] [CrossRef] [Green Version]
- Ide, Y.; Waki, M.; Hayasaka, T.; Nishio, T.; Morita, Y.; Tanaka, H.; Sasaki, T.; Koizumi, K.; Matsunuma, R.; Hosokawa, Y.; et al. Human breast cancer tissues contain abundant phosphatidylcholine(36:1) with high stearoyl-CoA desaturase-1 expression. PLoS ONE 2013, 8, e61204. [Google Scholar] [CrossRef]
- Bao, J.; Zhu, L.; Zhu, Q.; Su, J.; Liu, M.; Huang, W. SREBP-1 is an independent prognostic marker and promotes invasion and migration in breast cancer. Oncol. Lett. 2016, 12, 2409–2416. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.-A.; Morin, P.J.; Han, W.F.; Chen, T.; Bornman, D.M.; Gabrielson, E.W.; Pizer, E.S. Regulation of fatty acid synthase expression in breast cancer by sterol regulatory element binding protein-1c. Exp. Cell Res. 2003, 282, 132–137. [Google Scholar] [CrossRef]
- Freed-Pastor, W.A.; Mizuno, H.; Zhao, X.; Langerød, A.; Moon, S.-H.; Rodriguez-Barrueco, R.; Barsotti, A.; Chicas, A.; Li, W.; Polotskaia, A.; et al. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell 2012, 148, 244–258. [Google Scholar] [CrossRef] [Green Version]
- Tong, X.; Zhao, F.; Mancuso, A.; Gruber, J.J.; Thompson, C.B. The glucose-responsive transcription factor ChREBP contributes to glucose-dependent anabolic synthesis and cell proliferation. Proc. Natl. Acad. Sci. USA 2009, 106, 21660–21665. [Google Scholar] [CrossRef] [Green Version]
- Kotta-Loizou, I.; Theocharis, C.G.; Stamatios, T. The Role of Peroxisome Proliferator-Activated Receptor-γ in Breast Cancer. Anticancer. Agents Med. Chem. 2012, 12, 1025–1044. [Google Scholar] [CrossRef]
- Papadaki, I.; Mylona, E.; Giannopoulou, I.; Markaki, S.; Keramopoulos, A.; Nakopoulou, L. PPARγ expression in breast cancer: Clinical value and correlation with ERβ. Histopathology 2005, 46, 37–42. [Google Scholar] [CrossRef]
- Zaytseva, Y.Y.; Wallis, N.K.; Southard, R.C.; Kilgore, M.W. The PPARγ Antagonist T0070907 Suppresses Breast Cancer Cell Proliferation and Motility via Both PPARγ-dependent and -independent Mechanisms. Anticancer Res. 2011, 31, 813–823. [Google Scholar] [PubMed]
- Stefan, E.; Bister, K. MYC and RAF: Key Effectors in Cellular Signaling and Major Drivers in Human Cancer. Curr. Top. Microbiol. Immunol. 2017, 407, 117–151. [Google Scholar] [CrossRef]
- Sengupta, S.; Biarnes, M.C.; Jordan, V.C. Cyclin dependent kinase-9 mediated transcriptional de-regulation of cMYC as a critical determinant of endocrine-therapy resistance in breast cancers. Breast Cancer Res. Treat. 2014, 143, 113–124. [Google Scholar] [CrossRef] [Green Version]
- Cheng, A.S.L.; Jin, V.X.; Fan, M.; Smith, L.T.; Liyanarachchi, S.; Yan, P.S.; Leu, Y.-W.; Chan, M.W.Y.; Plass, C.; Nephew, K.P.; et al. Combinatorial Analysis of Transcription Factor Partners Reveals Recruitment of c-MYC to Estrogen Receptor-α Responsive Promoters. Mol. Cell 2006, 21, 393–404. [Google Scholar] [CrossRef]
- Cericatto, R.; Pozzobon, A.; Morsch, D.M.; Menke, C.H.; Brum, I.S.; Spritzer, P.M. Estrogen receptor-α, bcl-2 and c-myc gene expression in fibroadenomas and adjacent normal breast: Association with nodule size, hormonal and reproductive features. Steroids 2005, 70, 153–160. [Google Scholar] [CrossRef]
- Wang, C.; Mayer, J.A.; Mazumdar, A.; Fertuck, K.; Kim, H.; Brown, M.; Brown, P.H. Estrogen induces c-myc gene expression via an upstream enhancer activated by the estrogen receptor and the AP-1 transcription factor. Mol. Endocrinol. 2011, 25, 1527–1538. [Google Scholar] [CrossRef] [Green Version]
- Vergara, D.; Stanca, E.; Guerra, F.; Priore, P.; Gaballo, A.; Franck, J.; Simeone, P.; Trerotola, M.; De Domenico, S.; Fournier, I.; et al. β-Catenin Knockdown Affects Mitochondrial Biogenesis and Lipid Metabolism in Breast Cancer Cells. Front. Physiol. 2017, 8, 544. [Google Scholar] [CrossRef] [Green Version]
- Fotovati, A.; Abu-Ali, S.; Kage, M.; Shirouzu, K.; Yamana, H.; Kuwano, M. N-myc Downstream-regulated Gene 1 (NDRG1) a Differentiation Marker of Human Breast Cancer. Pathol. Oncol. Res. 2011, 17, 525–533. [Google Scholar] [CrossRef]
- Sevinsky, C.J.; Khan, F.; Kokabee, L.; Darehshouri, A.; Maddipati, K.R.; Conklin, D.S. NDRG1 regulates neutral lipid metabolism in breast cancer cells. Breast Cancer Res. 2018, 20, 55. [Google Scholar] [CrossRef] [PubMed]
- Van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef]
- Simeone, P.; Bologna, G.; Lanuti, P.; Pierdomenico, L.; Guagnano, M.T.; Pieragostino, D.; Del Boccio, P.; Vergara, D.; Marchisio, M.; Miscia, S.; et al. Extracellular Vesicles as Signaling Mediators and Disease Biomarkers across Biological Barriers. Int. J. Mol. Sci. 2020, 21, 2514. [Google Scholar] [CrossRef] [Green Version]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [Green Version]
- Marchisio, M.; Simeone, P.; Bologna, G.; Ercolino, E.; Pierdomenico, L.; Pieragostino, D.; Ventrella, A.; Antonini, F.; Del Zotto, G.; Vergara, D.; et al. Flow Cytometry Analysis of Circulating Extracellular Vesicle Subtypes from Fresh Peripheral Blood Samples. Int. J. Mol. Sci. 2020, 22, 48. [Google Scholar] [CrossRef]
- Dini, L.; Tacconi, S.; Carata, E.; Tata, A.M.; Vergallo, C.; Panzarini, E. Microvesicles and exosomes in metabolic diseases and inflammation. Cytokine Growth Factor Rev. 2020, 51. [Google Scholar] [CrossRef]
- Lv, Y.; Tan, J.; Miao, Y.; Zhang, Q. The role of microvesicles and its active molecules in regulating cellular biology. J. Cell. Mol. Med. 2019, 23, 7894–7904. [Google Scholar] [CrossRef]
- Buca, D.; Bologna, G.; D’Amico, A.; Cugini, S.; Musca, F.; Febbo, M.; D’Arcangelo, D.; Buca, D.; Simeone, P.; Liberati, M.; et al. Extracellular Vesicles in Feto-Maternal Crosstalk and Pregnancy Disorders. Int. J. Mol. Sci. 2020, 21, 2120. [Google Scholar] [CrossRef] [Green Version]
- Panzarini, E.; Tacconi, S.; Carata, E.; Mariano, S.; Tata, A.M.; Dini, L. Molecular Characterization of Temozolomide-Treated and Non Temozolomide-Treated Glioblastoma Cells Released Extracellular Vesicles and Their Role in the Macrophage Response. Int. J. Mol. Sci. 2020, 21, 8353. [Google Scholar] [CrossRef] [PubMed]
- Lucchetti, D.; Ricciardi Tenore, C.; Colella, F.; Sgambato, A. Extracellular Vesicles and Cancer: A Focus on Metabolism, Cytokines, and Immunity. Cancers 2020, 12, 171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rontogianni, S.; Synadaki, E.; Li, B.; Liefaard, M.C.; Lips, E.H.; Wesseling, J.; Wu, W.; Altelaar, M. Proteomic profiling of extracellular vesicles allows for human breast cancer subtyping. Commun. Biol. 2019, 2, 325. [Google Scholar] [CrossRef] [Green Version]
- Griffiths, S.G.; Cormier, M.T.; Clayton, A.; Doucette, A.A. Differential Proteome Analysis of Extracellular Vesicles from Breast Cancer Cell Lines by Chaperone Affinity Enrichment. Proteomes 2017, 5, 25. [Google Scholar] [CrossRef] [PubMed]
- Santi, A.; Caselli, A.; Ranaldi, F.; Paoli, P.; Mugnaioni, C.; Michelucci, E.; Cirri, P. Cancer associated fibroblasts transfer lipids and proteins to cancer cells through cargo vesicles supporting tumor growth. Biochim. Biophys. Acta-Mol. Cell Res. 2015, 1853, 3211–3223. [Google Scholar] [CrossRef] [Green Version]
- Achreja, A.; Zhao, H.; Yang, L.; Yun, T.H.; Marini, J.; Nagrath, D. Exo-MFA-A 13C metabolic flux analysis framework to dissect tumor microenvironment-secreted exosome contributions towards cancer cell metabolism. Metab. Eng. 2017, 43, 156–172. [Google Scholar] [CrossRef]
- Sansone, P.; Savini, C.; Kurelac, I.; Chang, Q.; Amato, L.B.; Strillacci, A.; Stepanova, A.; Iommarini, L.; Mastroleo, C.; Daly, L.; et al. Packaging and transfer of mitochondrial DNA via exosomes regulate escape from dormancy in hormonal therapy-resistant breast cancer. Proc. Natl. Acad. Sci. USA 2017, 114, E9066–E9075. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Yang, L.; Baddour, J.; Achreja, A.; Bernard, V.; Moss, T.; Marini, J.C.; Tudawe, T.; Seviour, E.G.; San Lucas, F.A.; et al. Tumor microenvironment derived exosomes pleiotropically modulate cancer cell metabolism. Elife 2016, 5, e10250. [Google Scholar] [CrossRef]
- Flaherty, S.E., 3rd; Grijalva, A.; Xu, X.; Ables, E.; Nomani, A.; Ferrante, A.W., Jr. A lipase-independent pathway of lipid release and immune modulation by adipocytes. Science 2019, 363, 989–993. [Google Scholar] [CrossRef]
- Garcia, N.A.; González-King, H.; Grueso, E.; Sánchez, R.; Martinez-Romero, A.; Jávega, B.; O’Connor, J.E.; Simons, P.J.; Handberg, A.; Sepúlveda, P. Circulating exosomes deliver free fatty acids from the bloodstream to cardiac cells: Possible role of CD36. PLoS ONE 2019, 14, e0217546. [Google Scholar] [CrossRef] [PubMed]
- Record, M.; Carayon, K.; Poirot, M.; Silvente-Poirot, S. Exosomes as new vesicular lipid transporters involved in cell–cell communication and various pathophysiologies. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2014, 1841, 108–120. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhu, N.; Yan, T.; Shi, Y.-N.; Chen, J.; Zhang, C.-J.; Xie, X.-J.; Liao, D.-F.; Qin, L. The crosstalk: Exosomes and lipid metabolism. Cell Commun. Signal. 2020, 18, 119. [Google Scholar] [CrossRef] [PubMed]
- Nishida-Aoki, N.; Izumi, Y.; Takeda, H.; Takahashi, M.; Ochiya, T.; Bamba, T. Lipidomic Analysis of Cells and Extracellular Vesicles from High- and Low-Metastatic Triple-Negative Breast Cancer. Metabolites 2020, 10, 67. [Google Scholar] [CrossRef] [Green Version]
- Chu, M.; Zhao, Y.; Feng, Y.; Zhang, H.; Liu, J.; Cheng, M.; Li, L.; Shen, W.; Cao, H.; Li, Q.; et al. MicroRNA-126 participates in lipid metabolism in mammary epithelial cells. Mol. Cell. Endocrinol. 2017, 454, 77–86. [Google Scholar] [CrossRef]
- Kang, H.S.; Lee, S.C.; Park, Y.S.; Jeon, Y.E.; Lee, J.H.; Jung, S.-Y.; Park, I.H.; Jang, S.H.; Park, H.M.; Yoo, C.W.; et al. Protein and lipid MALDI profiles classify breast cancers according to the intrinsic subtype. BMC Cancer 2011, 11, 465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Tontonoz, P. Phospholipid Remodeling in Physiology and Disease. Annu. Rev. Physiol. 2019, 81, 165–188. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, A.M.; Colombo, I.; Montorfano, G.; Zava, S.; Corsetto, P.A. Exogenous Fatty Acids Modulate ER Lipid Composition and Metabolism in Breast Cancer Cells. Cells 2021, 10, 175. [Google Scholar] [CrossRef]
- Barrera, G. Oxidative stress and lipid peroxidation products in cancer progression and therapy. ISRN Oncol. 2012, 2012, 137289. [Google Scholar] [CrossRef] [Green Version]
- Rysman, E.; Brusselmans, K.; Scheys, K.; Timmermans, L.; Derua, R.; Munck, S.; Van Veldhoven, P.P.; Waltregny, D.; Daniëls, V.W.; Machiels, J.; et al. De novo Lipogenesis Protects Cancer Cells from Free Radicals and Chemotherapeutics by Promoting Membrane Lipid Saturation. Cancer Res. 2010, 70, 8117–8126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Orozco, R.; Navarro-Tito, N.; Soto-Guzman, A.; Castro-Sanchez, L.; Perez Salazar, E. Arachidonic acid promotes epithelial-to-mesenchymal-like transition in mammary epithelial cells MCF10A. Eur. J. Cell Biol. 2010, 89, 476–488. [Google Scholar] [CrossRef] [PubMed]
- Eghlimi, R.; Shi, X.; Hrovat, J.; Xi, B.; Gu, H. Triple Negative Breast Cancer Detection Using LC–MS/MS Lipidomic Profiling. J. Proteome Res. 2020, 19, 2367–2378. [Google Scholar] [CrossRef] [PubMed]
- Jarc, E.; Kump, A.; Malavašič, P.; Eichmann, T.O.; Zimmermann, R.; Petan, T. Lipid droplets induced by secreted phospholipase A2 and unsaturated fatty acids protect breast cancer cells from nutrient and lipotoxic stress. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2018, 1863, 247–265. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Simeone, P.; Tacconi, S.; Longo, S.; Lanuti, P.; Bravaccini, S.; Pirini, F.; Ravaioli, S.; Dini, L.; Giudetti, A.M. Expanding Roles of De Novo Lipogenesis in Breast Cancer. Int. J. Environ. Res. Public Health 2021, 18, 3575. https://doi.org/10.3390/ijerph18073575
Simeone P, Tacconi S, Longo S, Lanuti P, Bravaccini S, Pirini F, Ravaioli S, Dini L, Giudetti AM. Expanding Roles of De Novo Lipogenesis in Breast Cancer. International Journal of Environmental Research and Public Health. 2021; 18(7):3575. https://doi.org/10.3390/ijerph18073575
Chicago/Turabian StyleSimeone, Pasquale, Stefano Tacconi, Serena Longo, Paola Lanuti, Sara Bravaccini, Francesca Pirini, Sara Ravaioli, Luciana Dini, and Anna M. Giudetti. 2021. "Expanding Roles of De Novo Lipogenesis in Breast Cancer" International Journal of Environmental Research and Public Health 18, no. 7: 3575. https://doi.org/10.3390/ijerph18073575
APA StyleSimeone, P., Tacconi, S., Longo, S., Lanuti, P., Bravaccini, S., Pirini, F., Ravaioli, S., Dini, L., & Giudetti, A. M. (2021). Expanding Roles of De Novo Lipogenesis in Breast Cancer. International Journal of Environmental Research and Public Health, 18(7), 3575. https://doi.org/10.3390/ijerph18073575