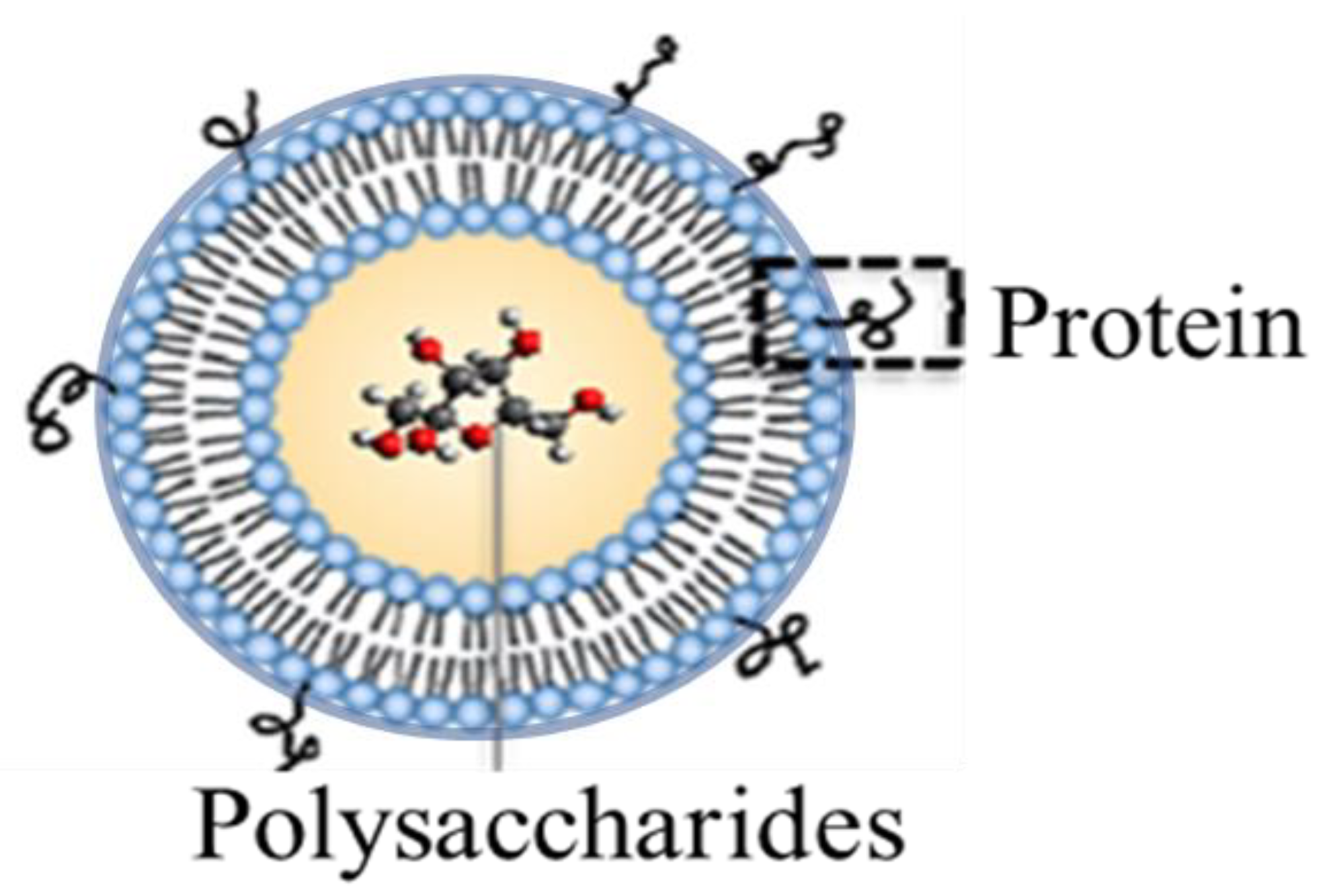
Design Variation of a Dual-Antigen Liposomal Vaccine Carrier System

 ,
, 

Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials and Reagents
2.2. Liposomal Preparation
2.3. Polysaccharide Encapsulation Analysis
2.4. Size and Zeta Potential Analysis
2.5. Liposomal Protein Surface Assessment
2.6. Experimental Repetition
3. Results
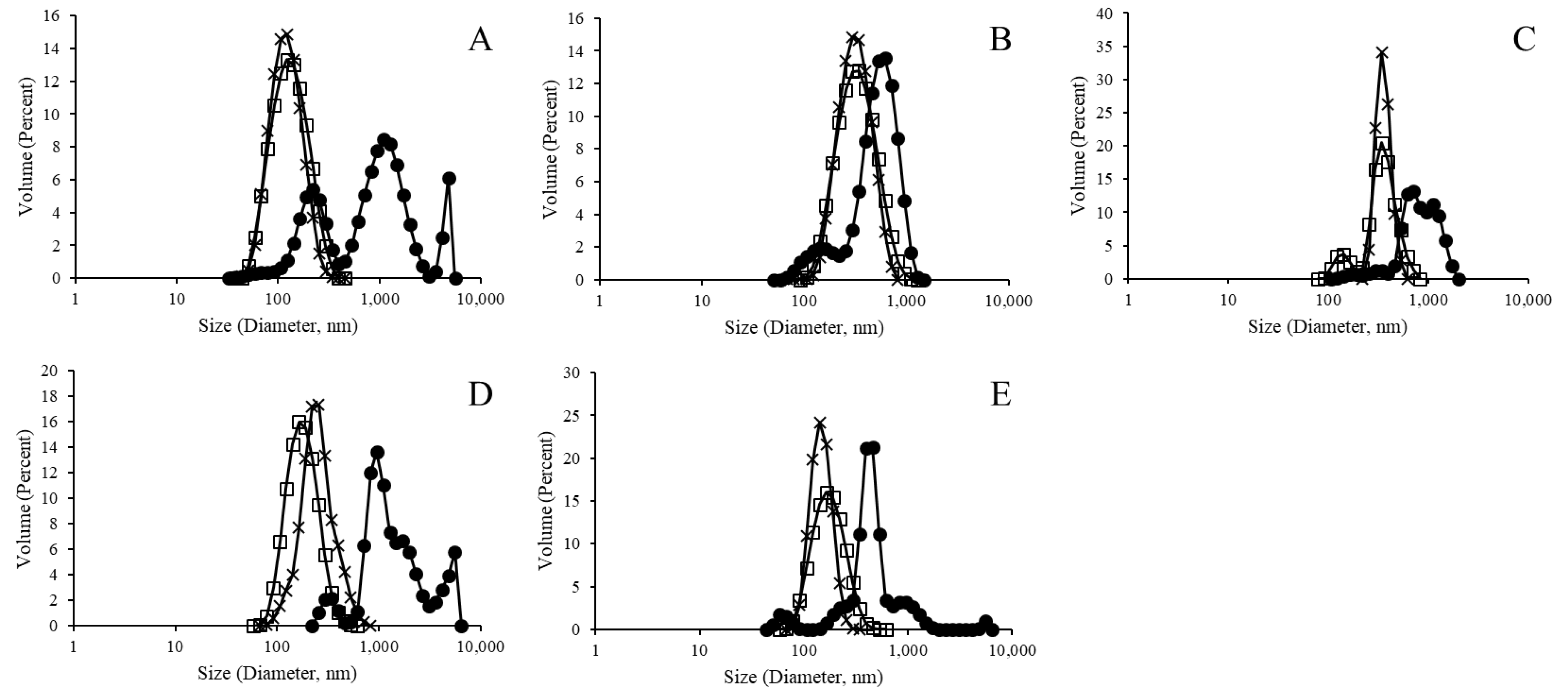
3.1. Liposomal Formulation without Polysaccharide Encapsulation and without Protein Surface Binding
3.2. Liposomal Purification Effect on Liposomal Formulation with Polysaccharide Encapsulation But without Protein Surface Binding
3.3. Complete LEPS Formulation Characterization after Protein Surface Binding
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Sadanand, S. Vaccination: The present and the future. Yale J. Biol. Med. 2011, 84, 353–359. [Google Scholar] [PubMed]
- Schmidt, C. Vaccines for pandemics. Nat. Biotechnol. 2013, 31, 957–960. [Google Scholar] [CrossRef] [PubMed]
- Siegrist, C. Vaccine Immunology (General Aspects of Vaccination); WHO: Geneva, Switzerland, 2008. [Google Scholar]
- Starke, J.R.; Connelly, K.K. Vaccines; Saunders: Philadelphia, PA, USA, 1994. [Google Scholar]
- Plotkin, S.A. Vaccines: Past, present and future. Nat. Med. 2005, 11, S5–S11. [Google Scholar] [CrossRef] [PubMed]
- Flingai, S.; Czerwonko, M.; Goodman, J.; Kudchodkar, S.B.; Muthumani, K.; Weiner, D.B. Synthetic DNA vaccines: Improved vaccine potency by electroporation and co-delivered genetic adjuvants. Front. Immunol. 2013, 4, 354. [Google Scholar] [CrossRef] [PubMed]
- Cohen, E.P. Cancer therapy with DNA-based vaccines. Immunol. Lett. 2000, 74, 59–65. [Google Scholar] [CrossRef]
- Beitelshees, M.; Li, Y.; Pfeifer, B.A. Enhancing vaccine effectiveness with delivery technology. Curr. Opin Biotechnol. 2016, 42, 24–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beitelshees, M.; Hill, A.; Jones, C.H.; Pfeifer, B.A. Phenotypic Variation during Biofilm Formation: Implications for Anti-Biofilm Therapeutic Design. Materials 2018, 11, 1086. [Google Scholar] [CrossRef] [PubMed]
- Chao, Y.; Bergenfelz, C.; Hakansson, A.P. In Vitro and In Vivo Biofilm Formation by Pathogenic Streptococci. Methods Mol. Biol. 2017, 1535, 285–299. [Google Scholar] [PubMed]
- Bogaert, D.; de Groot, R.; Hermans, P.W.M. Streptococcus pneumoniae colonisation: The key to pneumococcal disease. Lancet Infect. Dis. 2004, 4, 144–154. [Google Scholar] [CrossRef]
- Daniels, C.C.; Rogers, P.D.; Shelton, C.M. A Review of Pneumococcal Vaccines: Current Polysaccharide Vaccine Recommendations and Future Protein Antigens. J. Pediatr. Pharmacol. Ther. 2016, 21, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Feldman, C.; Anderson, R. Review: Current and new generation pneumococcal vaccines. J. Infect. 2014, 69, 309–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moffitt, K.L.; Malley, R. Next generation pneumococcal vaccines. Curr. Opin Immunol. 2011, 23, 407–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonten, M.J.; Huijts, S.M.; Bolkenbaas, M.; Webber, C.; Patterson, S.; Gault, S.; van Werkhoven, C.H.; van Deursen, A.M.; Sanders, E.A.; Verheij, T.J.; et al. Polysaccharide conjugate vaccine against pneumococcal pneumonia in adults. N. Engl. J. Med. 2015, 372, 1114–1125. [Google Scholar] [CrossRef] [PubMed]
- Malito, E.; Bursulaya, B.; Chen, C.; Lo Surdo, P.; Picchianti, M.; Balducci, E.; Biancucci, M.; Brock, A.; Berti, F.; Bottomley, M.J.; et al. Structural basis for lack of toxicity of the diphtheria toxin mutant CRM197. Proc. Natl. Acad. Sci. USA 2012, 109, 5229–5234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gruber, W.C.; Scott, D.A.; Emini, E.A. Development and clinical evaluation of Prevnar 13, a 13-valent pneumocococcal CRM197 conjugate vaccine. Ann. N. Y. Acad. Sci. 2012, 1263, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Hill, A.B.; Beitelshees, M.; Nayerhoda, R.; Pfeifer, B.A.; Jones, C.H. Engineering a Next-Generation Glycoconjugate-Like Streptococcus pneumoniae Vaccine. ACS Infect. Dis. 2018, 4, 1553–1563. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.H.; Zhang, G.; Nayerhoda, R.; Beitelshees, M.; Hill, A.; Rostami, P.; Li, Y.; Davidson, B.A.; Knight, P.; Pfeifer, B.A. Comprehensive vaccine design for commensal disease progression. Sci. Adv. 2017, 3, e1701797. [Google Scholar] [CrossRef] [PubMed]
- MacNair, J.E.; Desai, T.; Teyral, J.; Abeygunawardana, C.; Hennessey, J.P., Jr. Alignment of absolute and relative molecular size specifications for a polyvalent pneumococcal polysaccharide vaccine (PNEUMOVAX 23). Biologicals 2005, 33, 49–58. [Google Scholar] [CrossRef]
- Kim, J.S.; Laskowich, E.R.; Arumugham, R.G.; Kaiser, R.E.; MacMichael, G.J. Determination of saccharide content in pneumococcal polysaccharides and conjugate vaccines by GC-MSD. Anal. Biochem. 2005, 347, 262–274. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Binding Element | Lipids or Liposomal Component | ||||||
|---|---|---|---|---|---|---|---|
| DOPG | DOPC | DGS–NTA(Ni) | DGS–NTA | DSPE–PEG(2000) | CH | DSPE–PEG(2000)–Biotin | |
| NTA–Ni | 3 | 3 | 1 | 0 | 0.1 | 4 | 0 |
| NTA–Co | 3 | 3 | 0 | 1 | 0.1 | 4 | 0 |
| NTA–Zn | 3 | 3 | 0 | 1 | 0.1 | 4 | 0 |
| NTA | 3 | 3 | 0 | 1 | 0.1 | 4 | 0 |
| Biotinylation | 3 | 3 | 0 | 0 | 0 | 4 | 0.1 |
| Particle | Zeta Potential (mV) |
|---|---|
| Without metallic binding element pre extrusion | −34.23 |
| Without metallic binding element post extrusion | −33.73 |
| Ni pre extrusion | −46.2 |
| Ni post extrusion | −18.43 |
| Co pre extrusion | −3.42 |
| Co post extrusion | 0.01 |
| Zn pre extrusion | −4.52 |
| Zn post extrusion | −7.16 |
| Biotinylated pre extrusion | −36.93 |
| Biotinylated post extrusion | −10.52 |
| Particle | Zeta Potential (mV) |
|---|---|
| Without metallic binding element post extrusion | −3.50 |
| Without metallic binding element post purification | −41.16 |
| Ni post extrusion | −41.7 |
| Ni post purification | −29.6 |
| Co post extrusion | −6.04 |
| Co post purification | −22.93 |
| Zn post extrusion | −4.19 |
| Zn post purification | −3.27 |
| Biotinylated post extrusion | −29.7 |
| Biotinylated post purification | −35.06 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nayerhoda, R.; Hill, A.; Beitelshees, M.; Jones, C.; Pfeifer, B. Design Variation of a Dual-Antigen Liposomal Vaccine Carrier System. Materials 2019, 12, 2809. https://doi.org/10.3390/ma12172809
Nayerhoda R, Hill A, Beitelshees M, Jones C, Pfeifer B. Design Variation of a Dual-Antigen Liposomal Vaccine Carrier System. Materials. 2019; 12(17):2809. https://doi.org/10.3390/ma12172809
Chicago/Turabian StyleNayerhoda, Roozbeh, Andrew Hill, Marie Beitelshees, Charles Jones, and Blaine Pfeifer. 2019. "Design Variation of a Dual-Antigen Liposomal Vaccine Carrier System" Materials 12, no. 17: 2809. https://doi.org/10.3390/ma12172809
APA StyleNayerhoda, R., Hill, A., Beitelshees, M., Jones, C., & Pfeifer, B. (2019). Design Variation of a Dual-Antigen Liposomal Vaccine Carrier System. Materials, 12(17), 2809. https://doi.org/10.3390/ma12172809