
Unveiling the Origin of Alkali Metal (Na, K, Rb, and Cs) Promotion in CO2 Dissociation over Mo2C Catalysts

Abstract
:1. Introduction
2. Computation Detail and Models
3. Results and Discussion
3.1. Optimized Structure of Alkali-Metal-Modified β-Mo2C (001)


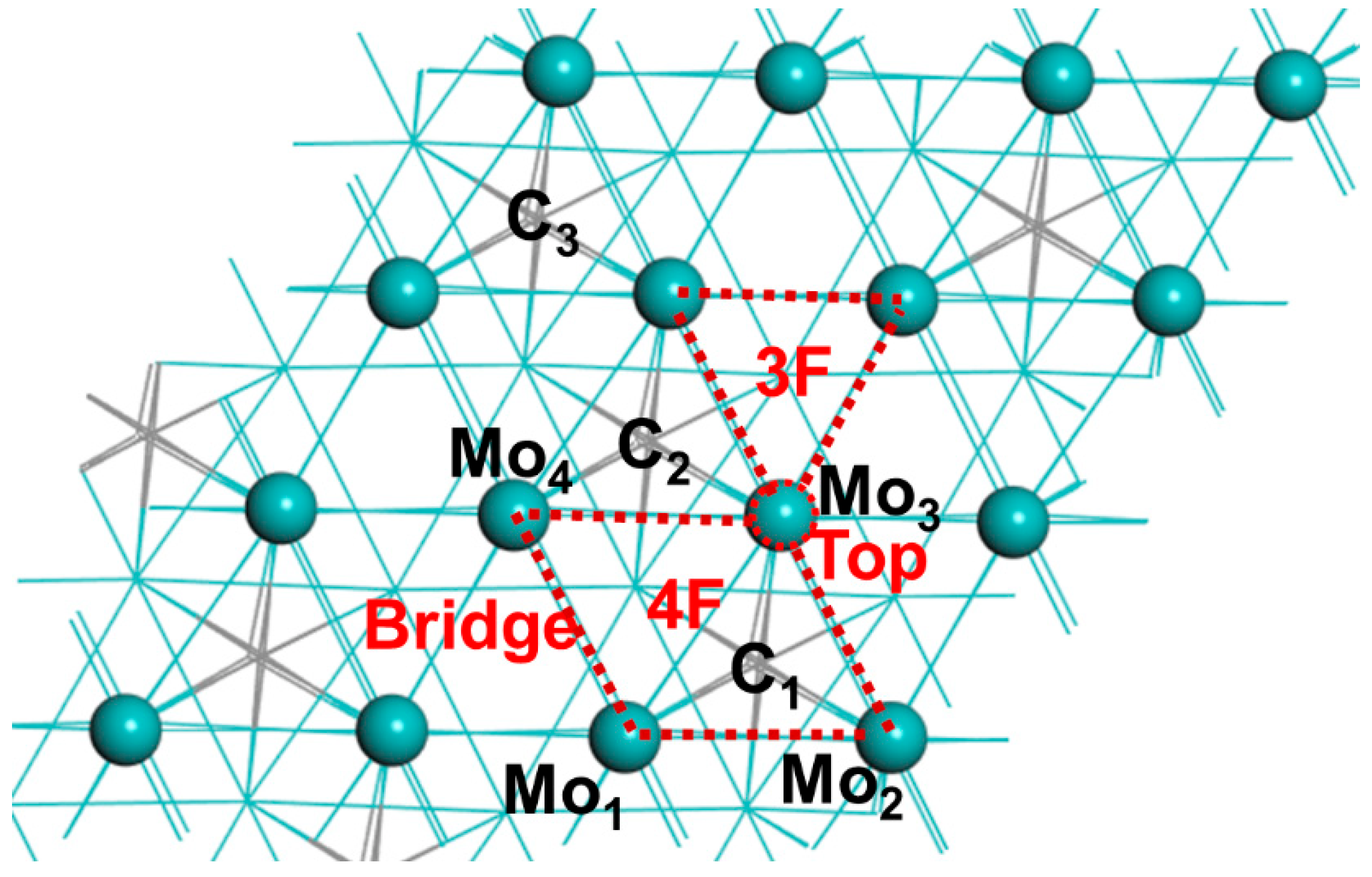{kind=link}
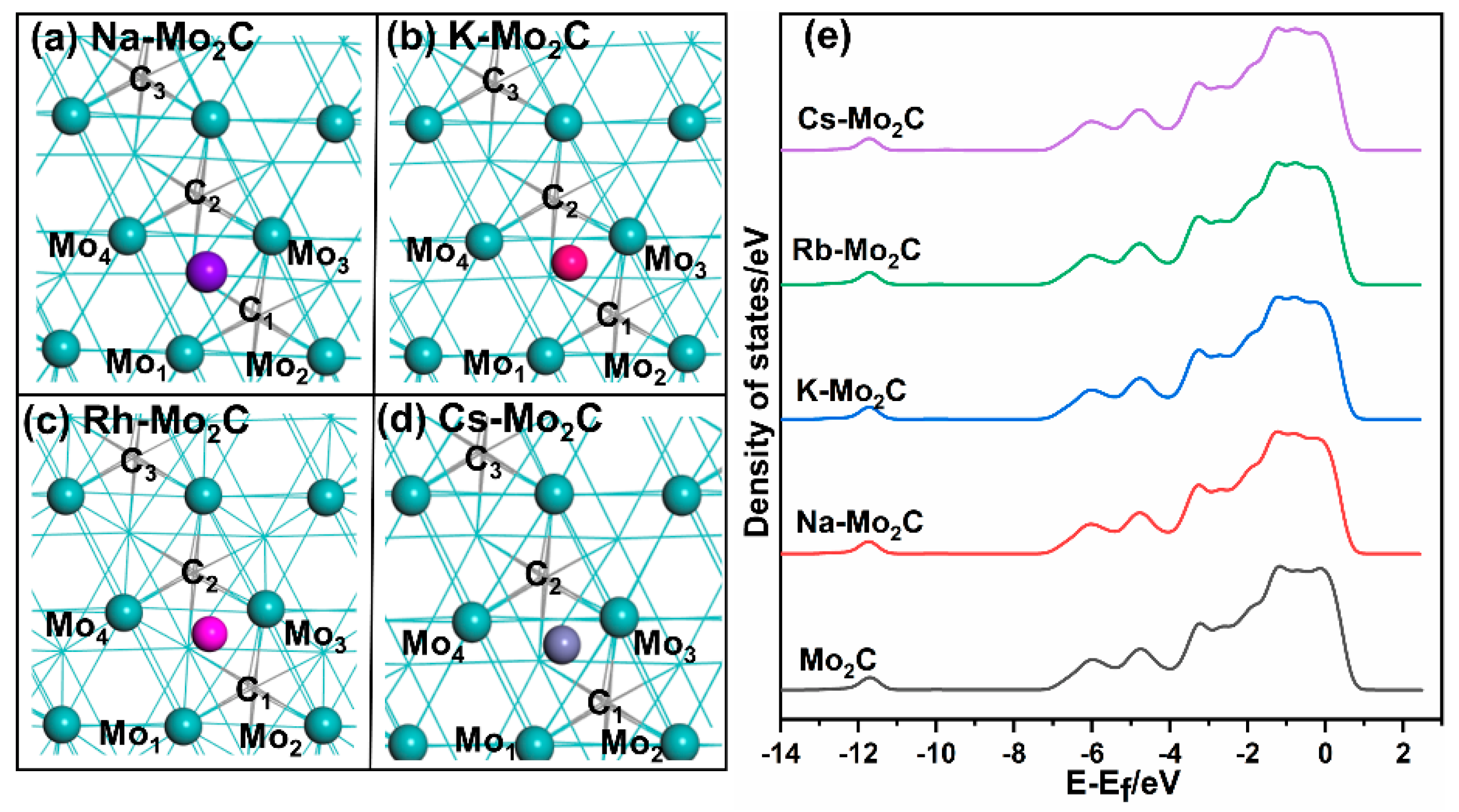{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalysts | Mulliken Charge (e) | ||||||
|---|---|---|---|---|---|---|---|
| X | Mo1 | Mo3 | Mo4 | C1 | C2 | C3 | |
| Mo2C | / | 0.05 | 0.05 | 0.13 | −0.50 | −0.50 | −0.50 |
| Na-Mo2C | 0.55 | −0.08 | −0.09 | 0.02 | −0.47 | −0.46 | −0.45 |
| K-Mo2C | 0.72 | −0.10 | −0.09 | 0.03 | −0.52 | −0.51 | −0.49 |
| Rb-Mo2C | 0.65 | −0.06 | −0.05 | 0.10 | −0.61 | −0.60 | −0.60 |
| Cs-Mo2C | 0.66 | −0.07 | −0.06 | 0.09 | −0.54 | −0.53 | −0.53 |
3.2. Adsorption of Intermediate Species on Mo2C and X-Mo2C Surfaces
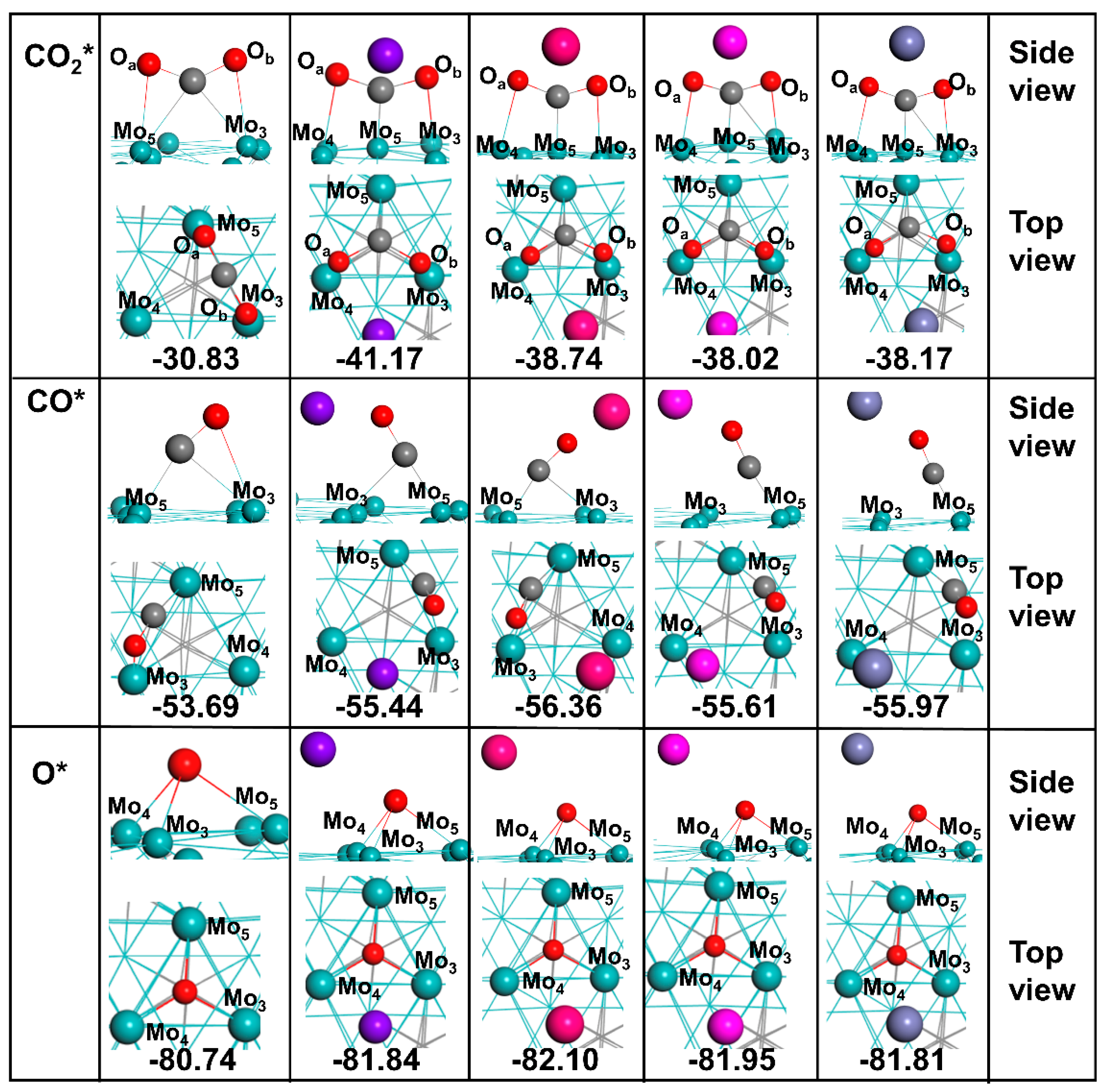
3.2.1. Adsorption of CO2*
3.2.2. Adsorption of CO*
3.2.3. Adsorption of O*
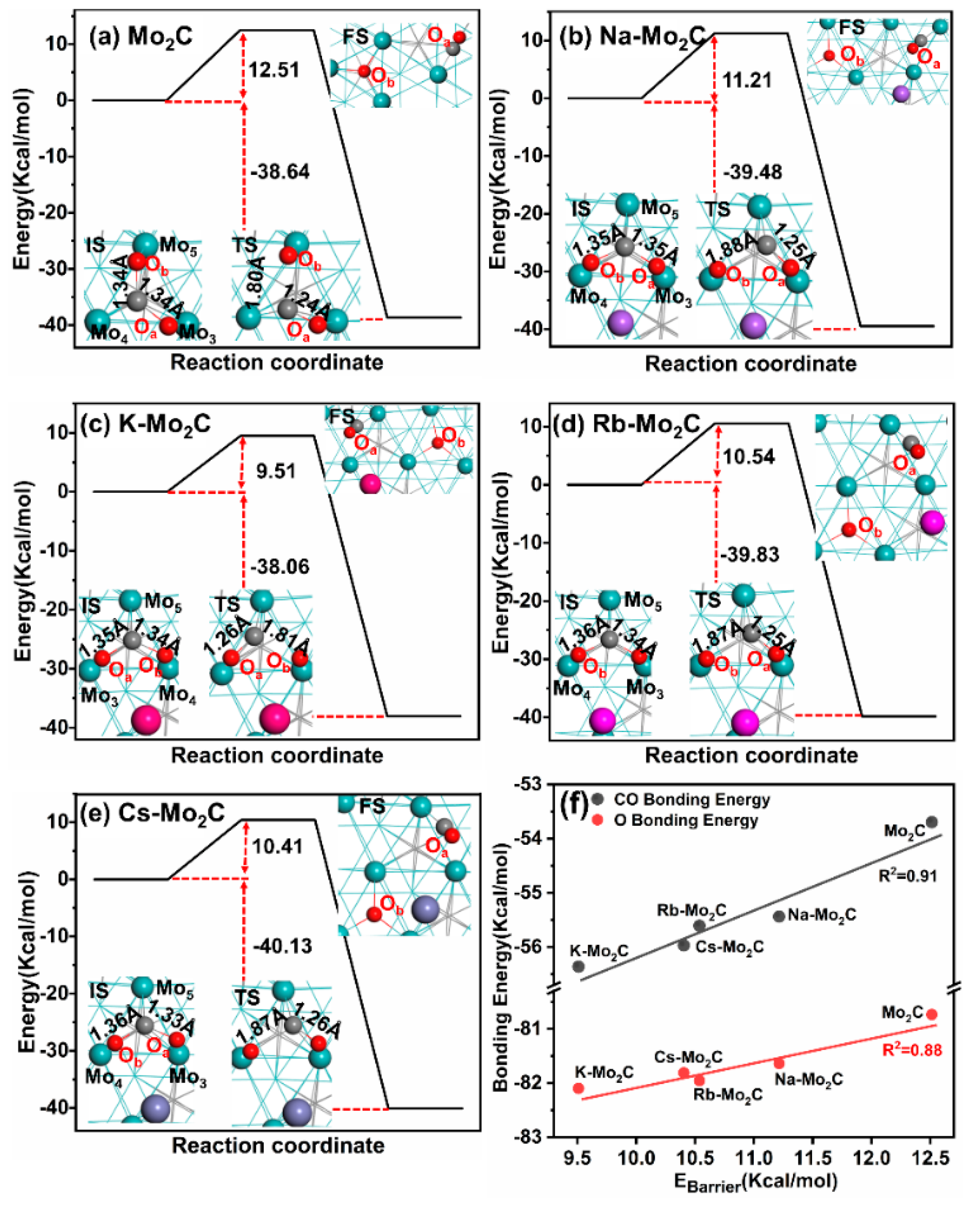
3.3. Energy Barriers for CO2 Dissociation on Mo2C and Alkali-Metal-Modified Mo2C Surfaces
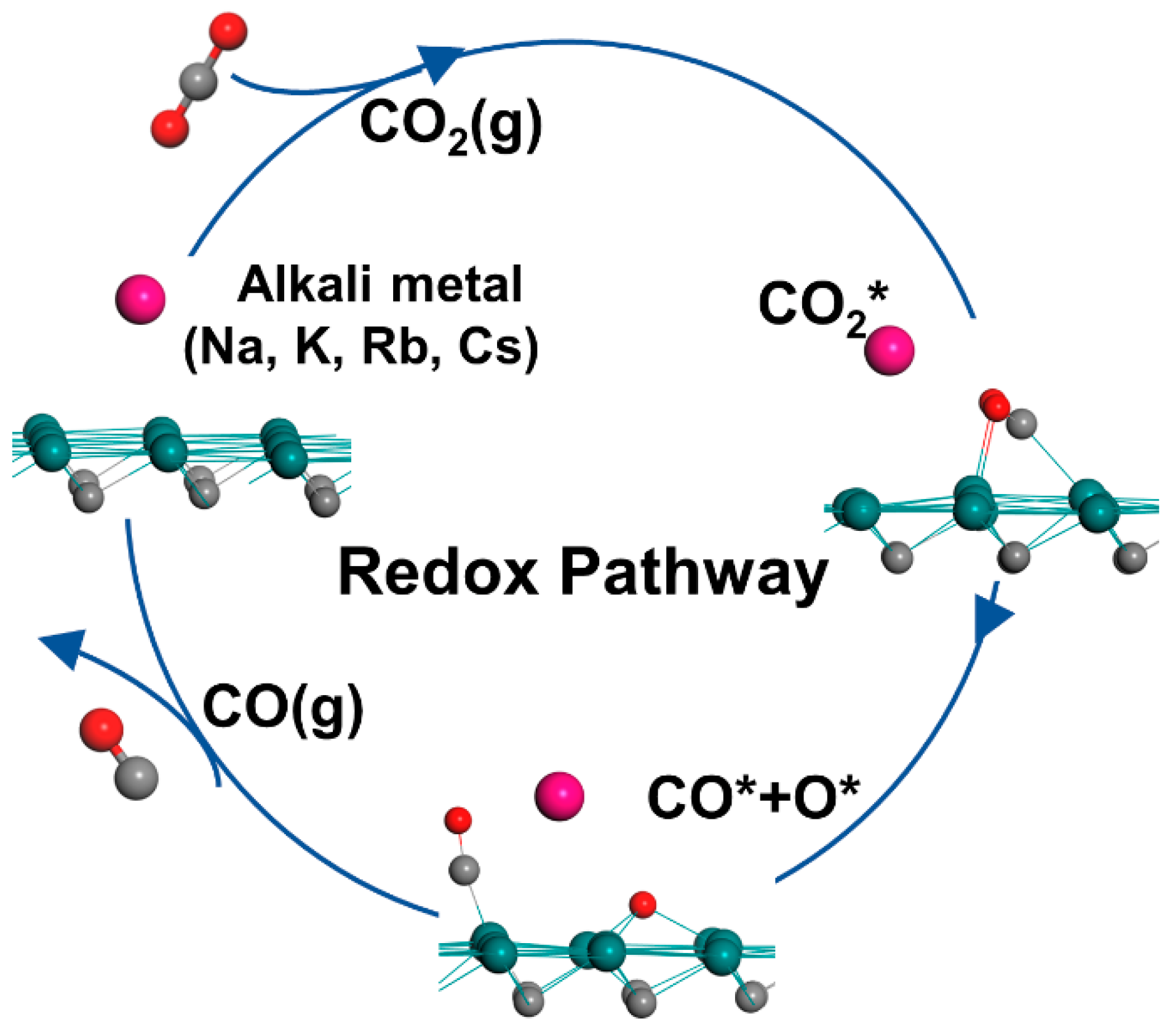
3.4. Energetic Analysis

| Mo | CO2 | |||||
|---|---|---|---|---|---|---|
| Mo1 a | Mo2 b | Oa | C | Ob | Alkali Metal | |
| Mo2C | 0.19 | 0.30 | −0.30 | 0.15 | −0.50 | |
| Na-Mo2C | 0.12 | 0.17 | −0.41 | 0.19 | −0.61 | 0.74 |
| K-Mo2C | 0.06 | 0.23 | −0.42 | 0.16 | −0.60 | 0.83 |
| Rb-Mo2C | 0.20 | 0.19 | −0.39 | 0.13 | −0.58 | 0.77 |
| Cs-Mo2C | 0.18 | 0.18 | −0.40 | 0.18 | −0.59 | 0.76 |
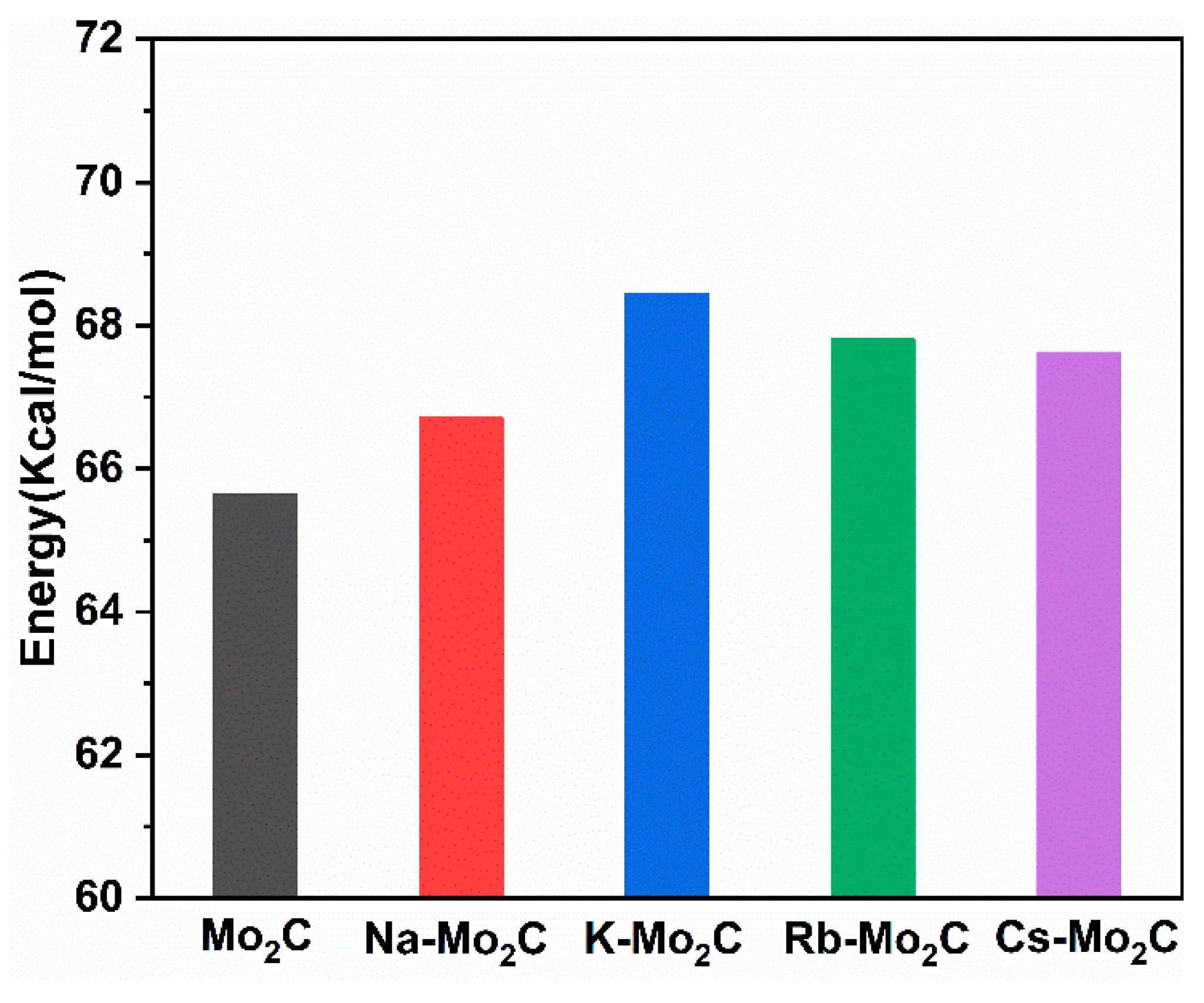
3.5. CO Desorption on Mo2C and Alkali-Metal-Modified Mo2C Surfaces
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pan, Y.-X.; Liu, C.-J.; Ge, Q. Effect of surface hydroxyls on selective CO2 hydrogenation over Ni4/γ-Al2O3: A density functional theory study. J. Catal. 2010, 272, 227–234. [Google Scholar] [CrossRef]
- Liu, C.; Liu, P. Mechanistic study of methanol synthesis from CO2 and H2 on a modified model Mo6S8 cluster. ACS Catal. 2015, 5, 1004–1012. [Google Scholar] [CrossRef]
- Wang, J.; Liu, C.-Y.; Senftle, T.P.; Zhu, J.; Zhang, G.; Guo, X.; Song, C. Variation in the In2O3 crystal phase alters catalytic performance toward the reverse water gas shift reaction. ACS Catal. 2019, 10, 3264–3273. [Google Scholar] [CrossRef]
- Aresta, M.; Dibenedetto, A.; Angelini, A. Catalysis for the Valorization of Exhaust Carbon: From CO2 to Chemicals, Materials, and Fuels. Technological Use of CO2. Chem. Rev. 2014, 114, 1709–1742. [Google Scholar] [CrossRef]
- Daza, Y.A.; Kuhn, J.N. CO2 conversion by reverse water gas shift catalysis: Comparison of catalysts, mechanisms and their consequences for CO2 conversion to liquid fuels. RSC Adv. 2016, 6, 49675–49691. [Google Scholar] [CrossRef]
- Zhao, Z.; Wang, M.; Ma, P.; Zheng, Y.; Chen, J.; Li, H.; Zhang, X.; Zheng, K.; Kuang, Q.; Xie, Z.-X. Atomically dispersed Pt/CeO2 catalyst with superior CO selectivity in reverse water gas shift reaction. Appl. Catal. B Environ. 2021, 291, 120101. [Google Scholar] [CrossRef]
- Kattel, S.; Yan, B.; Chen, J.G.; Liu, P. CO2 hydrogenation on Pt, Pt/SiO2 and Pt/TiO2: Importance of synergy between Pt and oxide support. J. Catal. 2016, 343, 115–126. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Asokan, C.; Xu, M.; Graham, G.W.; Pan, X.; Christopher, P.; Li, J.; Sautet, P. Rh single atoms on TiO2 dynamically respond to reaction conditions by adapting their site. Nat. Commun. 2019, 10, 4488. [Google Scholar] [CrossRef] [Green Version]
- Heyl, D.; Rodemerck, U.; Bentrup, U. Mechanistic study of low-temperature CO2 hydrogenation over modified Rh/Al2O3 catalysts. ACS Catal. 2016, 6, 6275–6284. [Google Scholar] [CrossRef]
- Aitbekova, A.; Wu, L.; Wrasman, C.J.; Boubnov, A.; Hoffman, A.S.; Goodman, E.D.; Bare, S.R.; Cargnello, M. Low-temperature restructuring of CeO2-supported Ru nanoparticles determines selectivity in CO2 catalytic reduction. J. Am. Chem. Soc. 2018, 140, 13736–13745. [Google Scholar] [CrossRef]
- Gao, J.; Wu, Y.; Jia, C.; Zhong, Z.; Gao, F.; Yang, Y.; Liu, B. Controllable synthesis of α-MoC1-x and β-Mo2C nanowires for highly selective CO2 reduction to CO. Catal. Commun. 2016, 84, 147–150. [Google Scholar] [CrossRef]
- Zhang, X.; Zhu, X.; Lin, L.; Yao, S.; Zhang, M.; Liu, X.; Wang, X.; Li, Y.-W.; Shi, C.; Ma, D. Highly dispersed copper over β-Mo2C as an efficient and stable catalyst for the reverse water gas shift (RWGS) reaction. ACS Catal. 2016, 7, 912–918. [Google Scholar] [CrossRef]
- Morse, J.R.; Juneau, M.; Baldwin, J.W.; Porosoff, M.D.; Willauer, H.D. Alkali promoted tungsten carbide as a selective catalyst for the reverse water gas shift reaction. J. CO2 Util. 2020, 35, 38–46. [Google Scholar] [CrossRef]
- Rodriguez, J.A.; Evans, J.; Feria, L.; Vidal, A.B.; Liu, P.; Nakamura, K.; Illas, F. CO2 hydrogenation on Au/TiC, Cu/TiC, and Ni/TiC catalysts: Production of CO, methanol, and methane. J. Catal. 2013, 307, 162–169. [Google Scholar] [CrossRef]
- Liu, X.; Kunkel, C.; Ramírez de la Piscina, P.; Homs, N.; Viñes, F.; Illas, F. Effective and highly selective CO generation from CO2 using a polycrystalline α-Mo2C catalyst. ACS Catal. 2017, 7, 4323–4335. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Ramírez, P.J.; Stacchiola, D.; Brito, J.L.; Rodriguez, J.A. The Carburization of Transition Metal Molybdates (MxMoO4, M=Cu, Ni or Co) and the Generation of Highly Active Metal/Carbide Catalysts for CO2 Hydrogenation. Catal. Lett. 2015, 145, 1365–1373. [Google Scholar] [CrossRef]
- Posada-Pérez, S.; Ramírez, P.J.; Gutiérrez, R.A.; Stacchiola, D.J.; Viñes, F.; Liu, P.; Illas, F.; Rodriguez, J.A. The conversion of CO2 to methanol on orthorhombic β-Mo2C and Cu/β-Mo2C catalysts: Mechanism for admetal induced change in the selectivity and activity. Catal. Sci. Technol. 2016, 6, 6766–6777. [Google Scholar] [CrossRef]
- Li, N.; Chen, X.; Ong, W.-J.; MacFarlane, D.R.; Zhao, X.; Cheetham, A.K.; Sun, C. Understanding of Electrochemical Mechanisms for CO2 Capture and Conversion into Hydrocarbon Fuels in Transition-Metal Carbides (MXenes). ACS Nano 2017, 11, 10825–10833. [Google Scholar] [CrossRef]
- Shi, Z.; Yang, H.; Gao, P.; Chen, X.; Liu, H.; Zhong, L.; Wang, H.; Wei, W.; Sun, Y. Effect of alkali metals on the performance of CoCu/TiO2 catalysts for CO2 hydrogenation to long-chain hydrocarbons. Chin. J. Catal. 2018, 39, 1294–1302. [Google Scholar] [CrossRef]
- Posada-Pérez, S.; Viñes, F.; Ramirez, P.J.; Vidal, A.B.; Rodriguez, J.A.; Illas, F. The bending machine: CO2 activation and hydrogenation on δ-MoC(001) and β-Mo2C(001) surfaces. Phys. Chem. Chem. Phys. 2014, 16, 14912–14921. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Su, X.; Chen, X.; Duan, H.; Liang, B.; Liu, Q.; Liu, X.; Ren, Y.; Huang, Y.; Zhang, T. Promotion effects of potassium on the activity and selectivity of Pt/zeolite catalysts for reverse water gas shift reaction. Appl. Catal. B Environ. 2017, 216, 95–105. [Google Scholar] [CrossRef]
- Wang, Y.-X.; Wang, G.-C. A systematic theoretical study of water gas shift reaction on Cu(111) and Cu(110): Potassium effect. ACS Catal. 2019, 9, 2261–2274. [Google Scholar] [CrossRef]
- Wang, Y.-X.; Zhang, H.-L.; An, P.; Wu, H.-S.; Jia, J.-F. Effect of Potassium on Methanol Steam Reforming on the Cu(111) and Cu(110) Surfaces: A DFT Study. J. Phys. Chem. C 2021, 125, 20905–20918. [Google Scholar] [CrossRef]
- An, W.; Xu, F.; Stacchiola, D.; Liu, P. Potassium-Induced Effect on the Structure and Chemical Activity of the CuxO/Cu(1 1 1) (x ≤ 2) Surface: A Combined Scanning Tunneling Microscopy and Density Functional Theory Study. ChemCatChem 2015, 7, 3865–3872. [Google Scholar] [CrossRef]
- Feng, Z.; Su, G.; Ding, H.; Ma, Y.; Li, Y.; Tang, Y.; Dai, X. Atomic alkali metal anchoring on graphdiyne as single-atom catalysts for capture and conversion of CO2 to HCOOH. Mol. Catal. 2020, 494, 111142. [Google Scholar] [CrossRef]
- Gao, M.; Zhang, J.; Zhu, P.; Liu, X.; Zheng, Z. Unveiling the origin of alkali metal promotion in CO2 methanation over Ru/ZrO2. Appl. Catal. B Environ. 2022, 314, 121476. [Google Scholar] [CrossRef]
- Juneau, M.; Vonglis, M.; Hartvigsen, J.; Frost, L.; Bayerl, D.; Dixit, M.; Mpourmpakis, G.; Morse, J.R.; Baldwin, J.W.; Willauer, H.D.; et al. Assessing the viability of K-Mo2C for reverse water–gas shift scale-up: Molecular to laboratory to pilot scale. Energy Environ. Sci. 2020, 13, 2524–2539. [Google Scholar] [CrossRef]
- Zhang, Q.; Pastor-Pérez, L.; Jin, W.; Gu, S.; Reina, T.R. Understanding the promoter effect of Cu and Cs over highly effective β-Mo2C catalysts for the reverse water-gas shift reaction. Appl. Catal. B Environ. 2019, 244, 889–898. [Google Scholar] [CrossRef]
- Bugyi, L.; Oszkó, A.; Solymosi, F. Spectroscopic study on the formation of CO−2 on K-promoted Mo2C/Mo(100) surface. Surf. Sci. 2000, 461, 177–190. [Google Scholar] [CrossRef]
- Porosoff, M.D.; Baldwin, J.W.; Peng, X.; Mpourmpakis, G.; Willauer, H.D. Potassium-promoted molybdenum carbide as a highly active and selective catalyst for CO2 conversion to CO. ChemSusChem 2017, 10, 2408–2415. [Google Scholar] [CrossRef]
- Xu, J.; Gong, X.; Hu, R.; Liu, Z.-W.; Liu, Z.-T. Highly active K-promoted Cu/β-Mo2C catalysts for reverse water gas shift reaction: Effect of potassium. Mol. Catal. 2021, 516, 111954. [Google Scholar] [CrossRef]
- Ye, X.; Ma, J.; Yu, W.; Pan, X.; Yang, C.; Wang, C.; Liu, Q.; Huang, Y. Construction of bifunctional single-atom catalysts on the optimized β-Mo2C surface for highly selective hydrogenation of CO2 into ethanol. J. Energy Chem. 2022, 67, 184–192. [Google Scholar] [CrossRef]
- Kowalik, P.; Próchniak, W.; Borowiecki, T. The effect of alkali metals doping on properties of Cu/ZnO/Al2O3 catalyst for water gas shift. Catal. Today 2011, 176, 144–148. [Google Scholar] [CrossRef]
- Delley, B. From molecules to solids with the DMol3 approach. J. Chem. Phys. 2000, 113, 7756–7764. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Delley, B. An all-electron numerical method for solving the local density functional for polyatomic molecules. J. Chem. Phys. 1990, 92, 508–517. [Google Scholar] [CrossRef]
- Halgren, T.A.; Lipscomb, W.N. The synchronous-transit method for determining reaction pathways and locating molecular transition states. Chem. Phys. Lett. 1977, 49, 225–232. [Google Scholar] [CrossRef]
- Henkelman, G.; Jónsson, H. Improved tangent estimate in the nudged elastic band method for finding minimum energy paths and saddle points. J. Chem. Phys. 2000, 113, 9978–9985. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Guo, K.; Yuan, D.; Cheng, J.; Wang, B. Unraveling Reaction Mechanisms of Mo2C as Cathode Catalyst in a Li-CO2 Battery. J. Am. Chem. Soc. 2020, 142, 6983–6990. [Google Scholar] [CrossRef]
- Hammer, B.; Nørskov, J.K. Electronic factors determining the reactivity of metal surfaces. Surf. Sci. 1995, 343, 211–220. [Google Scholar] [CrossRef]
- Mortensen, J.J.; Hammer, B.; Nørskovi, J.K. A theoretical study of adsorbate–adsorbate interactions on Ru(0001). Surf. Sci. 1998, 414, 315–329. [Google Scholar] [CrossRef]
- Günther, S.; Esch, F.; del Turco, M.; Africh, C.; Comelli, G.; Kiskinova, M. K-stabilized high-oxygen-coverage states on Rh(110): A low-pressure pathway to formation of surface oxide. J. Phys. Chem. B 2005, 109, 11980–11985. [Google Scholar] [CrossRef] [PubMed]
- McGuire, N.K.; O’Keeffe, M. Bond lengths in alkali metal oxides. J. Solid State Chem. 1984, 54, 49–53. [Google Scholar] [CrossRef]
- Tai, J.; Ge, Q.; Davis, R.J.; Neurock, M. Adsorption of CO2 on Model Surfaces of Cesium Oxides Determined from First Principles. J. Phys. Chem. B 2004, 108, 16798–16805. [Google Scholar] [CrossRef]
- Porosoff, M.D.; Yang, X.; Boscoboinik, J.A.; Chen, J.G. Molybdenum carbide as alternative catalysts to precious metals for highly selective reduction of CO2 to CO. Angew. Chem. Int. Ed. 2014, 53, 6705–6709. [Google Scholar] [CrossRef]
- Porosoff, M.D.; Kattel, S.; Li, W.; Liu, P.; Chen, J.G. Identifying trends and descriptors for selective CO2 conversion to CO over transition metal carbides. Chem. Commun. 2015, 51, 6988–6991. [Google Scholar] [CrossRef]
- Lee, J.S.; Kim, S.; Kim, Y.G. Electronic and geometric effects of alkali promoters in CO hydrogenation over K/Mo2C catalysts. Top. Catal. 1995, 2, 127–140. [Google Scholar] [CrossRef]
- Bugyi, L.; Solymosi, F. Effects of Potassium on the Chemisorption of CO on the Mo2C/Mo(100) Surface. J. Phys. Chem. B 2001, 105, 4337–4342. [Google Scholar] [CrossRef]
- Solymosi, F.; Bugyi, L. Effects of potassium on the chemisorption of CO2 and CO on the Mo2C/Mo (100) surface. Catal. Lett. 2000, 66, 227–230. [Google Scholar] [CrossRef]
- Hammer, B. Adsorption, diffusion, and dissociation of NO, N and O on flat and stepped Ru(0001). Surf. Sci. 2000, 459, 323–348. [Google Scholar] [CrossRef]
- Wang, Y.-X.; Wang, G.-C. A systematic theoretical study of the water gas shift reaction on the Pt/ZrO2 interface and Pt(111) face: Key role of a potassium additive. Catal. Sci. Technol. 2020, 10, 876–892. [Google Scholar] [CrossRef]
- Austin, N.; Ye, J.; Mpourmpakis, G. CO2 activation on Cu-based Zr-decorated nanoparticles. Catal. Sci. Technol. 2017, 7, 2245–2251. [Google Scholar] [CrossRef]
- Freund, H.-J.; Roberts, M.W. Surface chemistry of carbon dioxide. Surf. Sci. Rep. 1996, 25, 225–273. [Google Scholar] [CrossRef] [Green Version]
- Wurth, W.; Stöhr, J.; Feulner, P.; Pan, X.; Bauchspiess, K.R.; Baba, Y.; Hudel, E.; Rocker, G.; Menzel, D. Bonding, structure, and magnetism of physisorbed and chemisorbed O2 on Pt(111). Phy. Rev. Lett. 1990, 65, 2426–2429. [Google Scholar] [CrossRef] [PubMed]
- Austin, N.; Butina, B.; Mpourmpakis, G. CO2 activation on bimetallic CuNi nanoparticles. Prog. Nat. Sci. Mater. 2016, 26, 487–492. [Google Scholar] [CrossRef] [Green Version]
- Dixit, M.; Peng, X.; Porosoff, M.D.; Willauer, H.D.; Mpourmpakis, G. Elucidating the role of oxygen coverage in CO2 reduction on Mo2C. Catal. Sci. Technol. 2017, 7, 5521–5529. [Google Scholar] [CrossRef]





| Catalysts | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Mo2C | 12.45 | −50.78 | −44.89 | −72.33 | −68.64 | 0.00 | −44.89 | 0.00 | −72.33 | 0.00 | 0.00 |
| Na-Mo2C | 11.30 | −60.28 | −47.39 | −74.22 | −75.04 | −0.09 | −47.29 | −0.28 | −73.94 | −2.50 | −1.89 |
| K-Mo2C | 9.45 | −60.44 | −51.82 | −73.31 | −73.38 | −0.12 | −51.71 | −0.32 | −73.00 | −6.93 | 0.99 |
| Rb-Mo2C | 10.61 | −61.34 | −47.14 | −75.08 | −76.17 | −0.12 | −47.03 | −0.25 | −74.82 | −2.25 | −2.75 |
| Cs-Mo2C | 10.38 | −60.11 | −45.36 | −76.04 | −75.89 | −5.77 | −45.32 | −0.25 | −75.77 | −0.47 | −3.71 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, R.; Chen, C.; Chu, W.; Sun, W. Unveiling the Origin of Alkali Metal (Na, K, Rb, and Cs) Promotion in CO2 Dissociation over Mo2C Catalysts. Materials 2022, 15, 3775. https://doi.org/10.3390/ma15113775
Liu R, Chen C, Chu W, Sun W. Unveiling the Origin of Alkali Metal (Na, K, Rb, and Cs) Promotion in CO2 Dissociation over Mo2C Catalysts. Materials. 2022; 15(11):3775. https://doi.org/10.3390/ma15113775
Chicago/Turabian StyleLiu, Renmin, Congmei Chen, Wei Chu, and Wenjing Sun. 2022. "Unveiling the Origin of Alkali Metal (Na, K, Rb, and Cs) Promotion in CO2 Dissociation over Mo2C Catalysts" Materials 15, no. 11: 3775. https://doi.org/10.3390/ma15113775
APA StyleLiu, R., Chen, C., Chu, W., & Sun, W. (2022). Unveiling the Origin of Alkali Metal (Na, K, Rb, and Cs) Promotion in CO2 Dissociation over Mo2C Catalysts. Materials, 15(11), 3775. https://doi.org/10.3390/ma15113775