3.1. Mechanical Properties
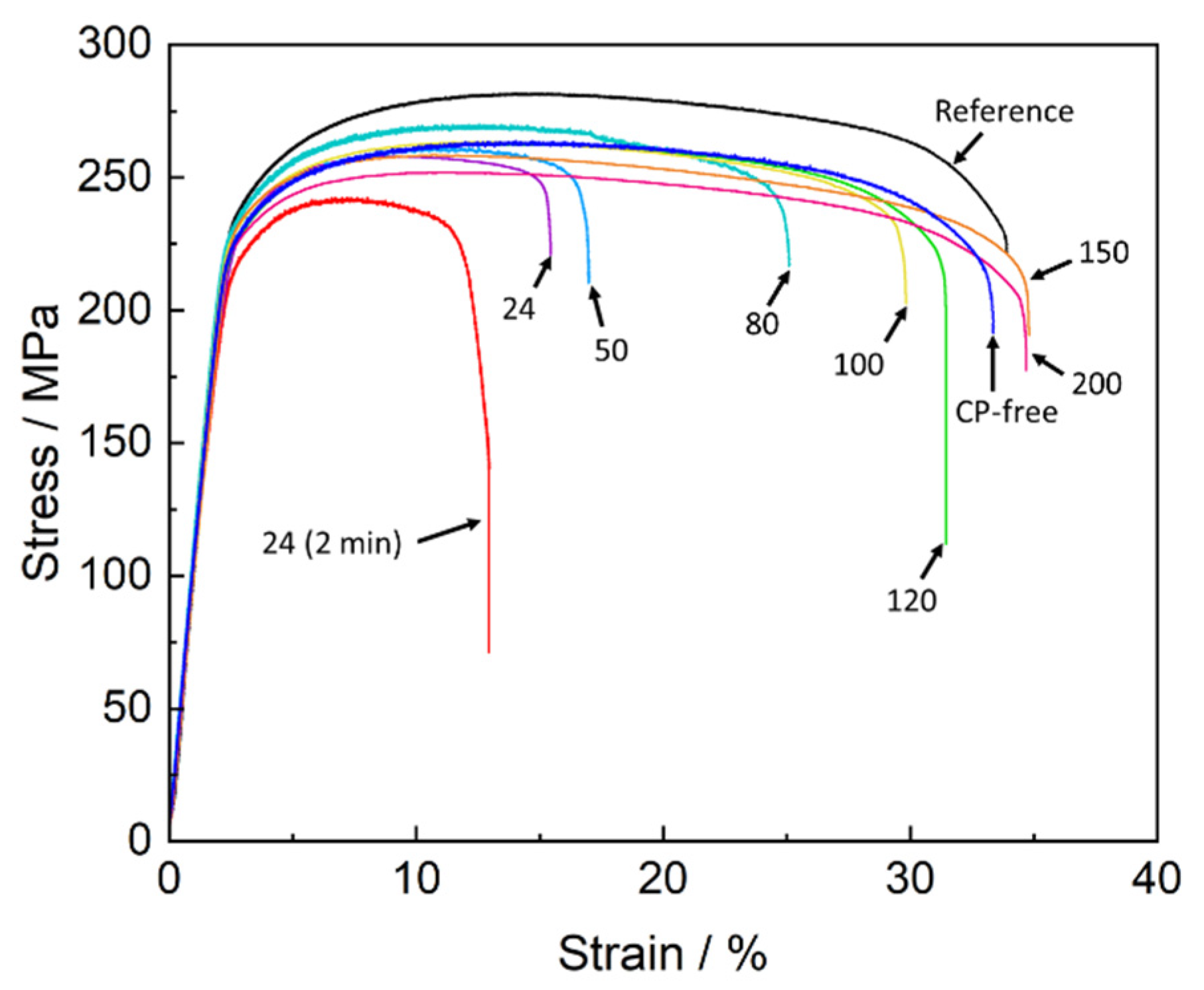
The mechanical testing showed that the specimens which were SSRT tested in air within a few minutes after pre-exposure to the corrosion solution demonstrated a substantial loss in strength and ductility compared to the reference specimens (
Figure 1 and
Figure 2). This is typical behaviour for ZK60 embrittled by PESCC. In harmony with our previous reports [
6,
20,
23], the removal of corrosion products resulted in complete recovery of ductility and partial recovery of strength in all specimens tested. The apparent irreversible loss of strength, which remains after the removal of corrosion products, is not related to embrittlement but rather is due to the reduction in the specimens’ cross-section caused by irreversible corrosion damage [
20]. It was found that similarly to the removal of corrosion products, post-exposure air storage inhibits PESCC. The efficiency of this recovery process, however, depends on the time and temperature of the storage of specimens in air. As can be seen in
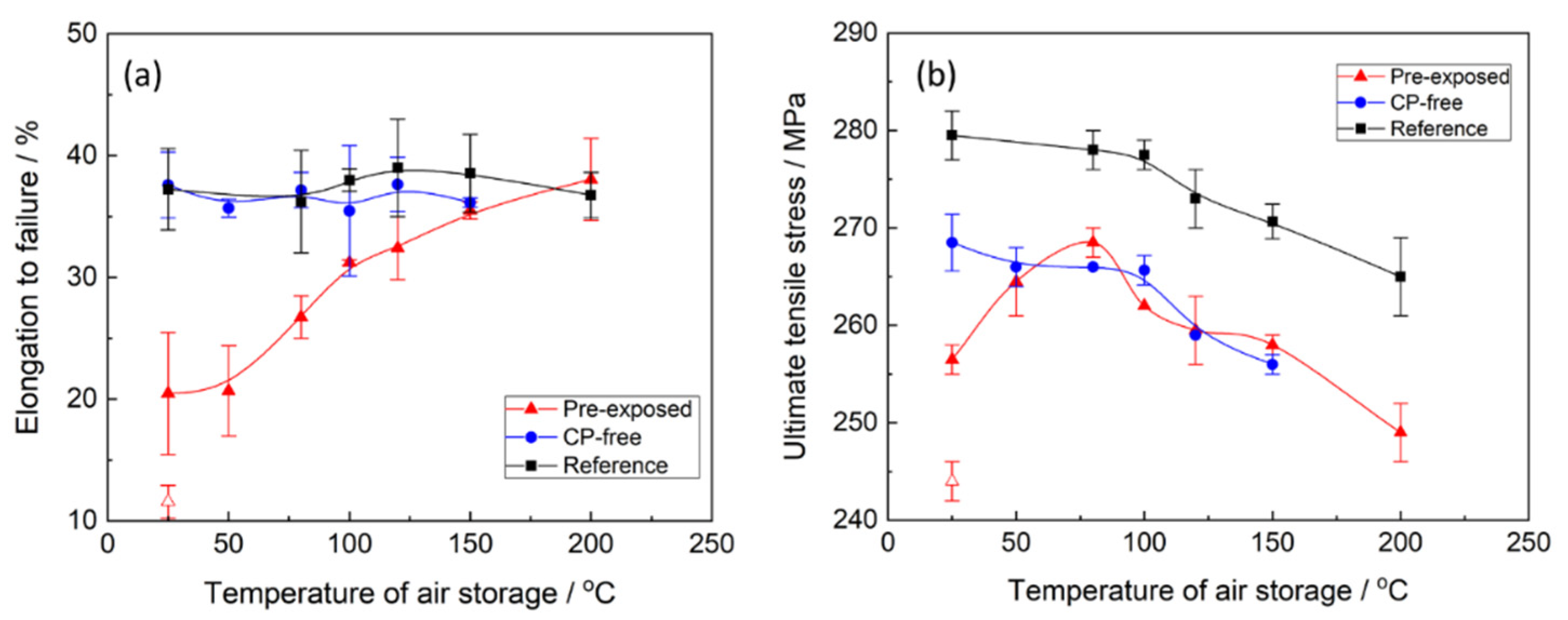
Figure 1 and
Figure 2, both ultimate tensile strength (UTS) and elongation to failure (EF) of the pre-exposed specimens after air storage at 24 °C for 24 h are significantly higher than those after 2 min of air storage at the same temperature. Nevertheless, even after 24 h of storage at room temperature, the pre-exposed specimens show roughly 50% loss of EF and approximately 25 MPa smaller UTS in comparison with their reference counterparts. It was found that the recovery of mechanical properties can be substantially enhanced by storage of the specimens in air at elevated temperatures.
Figure 2a demonstrates that the increased temperature of air storage results in the increase in EF. The improvement in the EF value is non-monotonic and is most pronounced in the temperature range from 50 to 100 °C. After storage in air at 100 °C, the ductility loss of the pre-exposed specimens does not exceed 20%. However, at a chosen post-exposure storage time, the complete recovery of ductility can be achieved only at 150–200 °C. After such treatment, the measured EF values were the same as those of the reference specimens and the specimens with the removed corrosion product layer (referred to hereafter as CP-free specimens).
The effect of storage temperature on the strength of the pre-exposed specimens is more complex than on ductility. It can be seen from
Figure 2b that UTS of the specimens monotonically grows when the storage temperature increases from 25 to 80 °C, while the further temperature increase up to 200 °C results in the non-monotonic decrease in UTS with a short plateau around 120–150 °C. To clarify whether the observed complex behaviour of strength and ductility is associated with temperature-induced microstructural changes or if it is attributed specifically to PESCC, the effect of annealing temperature on mechanical properties of the reference and CP-free specimens was investigated. One can see (
Figure 2b) that the UTS of these specimens is almost unaffected by low-temperature annealing below 100 °C, while at higher temperatures, the strength decreases monotonically with temperature. Apparently, some ageing-related microstructural changes affecting the strength occur in the alloy at temperatures above 100 °C. Therefore, these changes are likely to take some part in the observed deterioration of UTS in the pre-exposed specimens subjected to air storage above 100 °C. However, the opposite trend, i.e., the increase in UTS, in the pre-exposed specimens stored in air in the temperature range below 80 °C, cannot be explained by any microstructural changes in the metallic matrix. Furthermore, the decrease in strength in the pre-exposed specimens, which is observed at higher temperatures, cannot be solely attributed to the microstructural transformations either, because it starts in the range between 80 and 100 °C and not between 100 and 120 °C as for the reference and CP-free specimens. Furthermore, the plateau between 120 and 150 °C featuring the dependence of UTS on the temperature of the pre-exposed specimens is absent on such dependencies for the specimens of other kinds. Thus, the other factors responsible for the decrease in UTS, which is attributed specifically to the specimens embrittled by pre-exposure, do exist. It is important to note that the microstructural changes, which likely affect the strength of the alloy at elevated temperatures, do not influence the ductility. It follows from
Figure 2a that the EF of the reference and CP-free specimens scatters randomly around the nearly constant average value within the whole investigated temperature range. Therefore, the increasing EF of the pre-exposed specimens with post-exposure storage temperature should be entirely associated with the elimination of PESCC.
3.2. Fractographic and Side Surface Observations
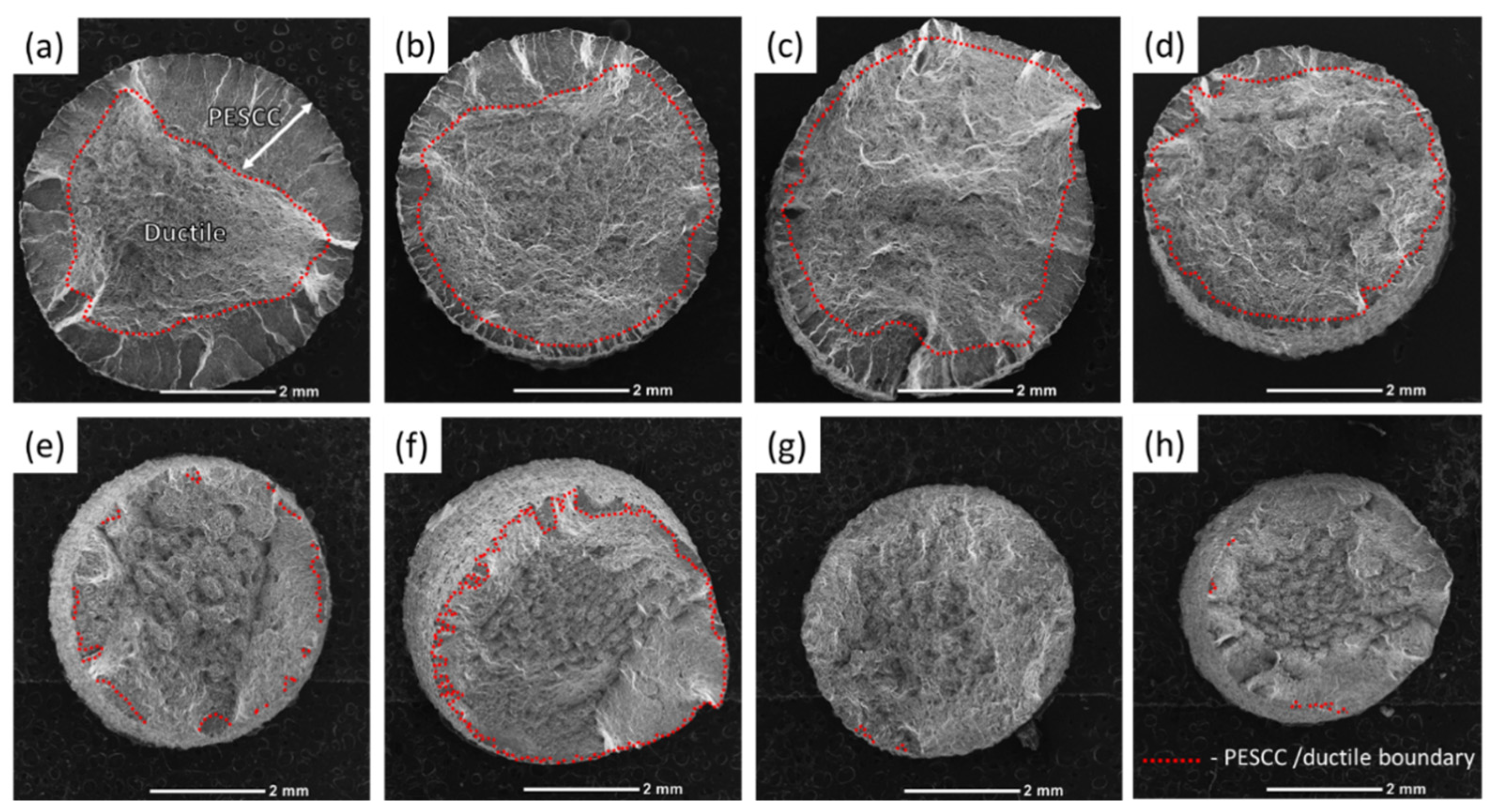
The fracture surface of the specimens suffering from PESCC is characterised by the peripheral brittle-like “PESCC zone” surrounding the dimpled ductile region in the central part of the fracture surface. The similar appearance of the fracture surface was frequently reported to be characteristic of the pre-exposure effects in Mg alloys, including ZK60, used in the present study [
7,
16,
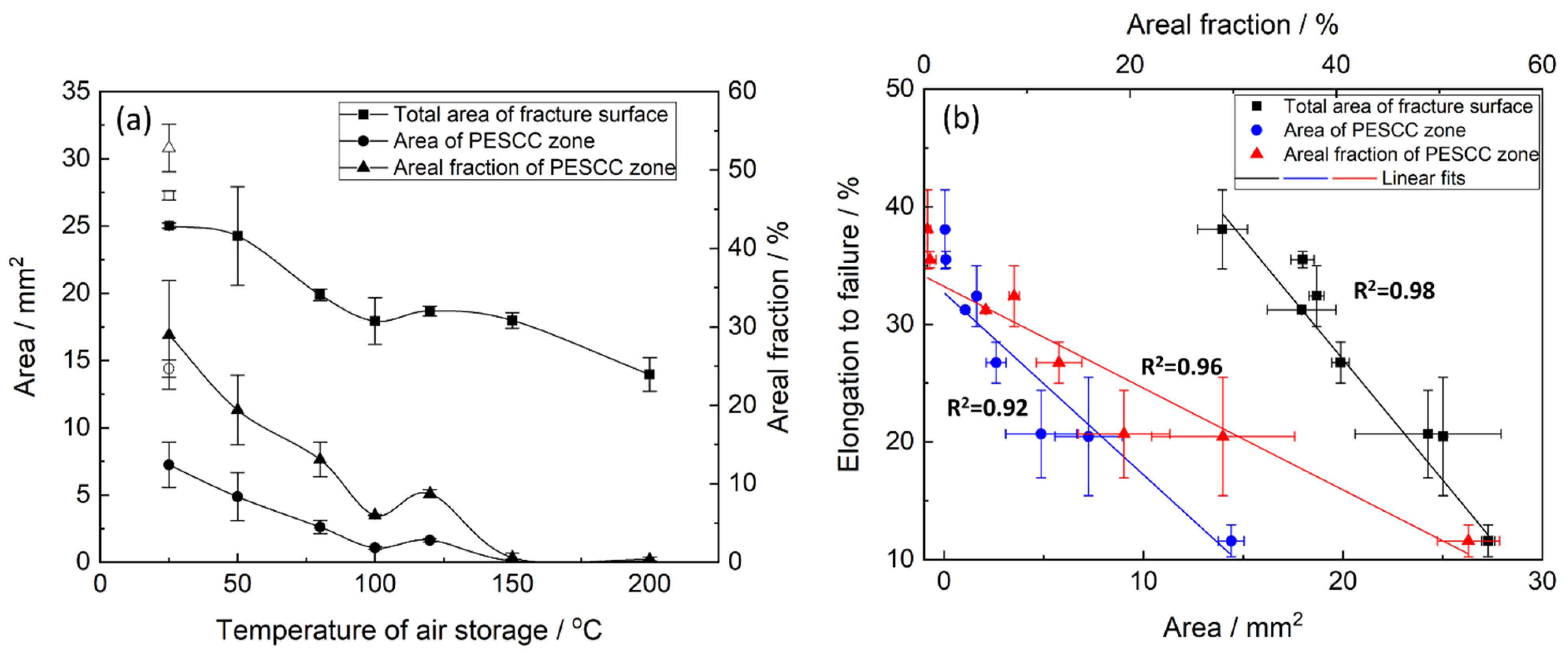
23]. The quantitative fractographic analysis showed that, in general, the increase in the time and temperature of post-exposure storage in air results in the significant reduction in both the total area of the fracture surface and the area of the PESCC zone (as well as its areal fraction with respect to the total area of the fracture surface) (c.f.,
Figure 3 and
Figure 4a). It also follows from
Figure 3f–h that when the temperature of air storage exceeds 80 °C, the PESCC zone becomes discontinuous and is represented by several small isolated “islands” with a brittle-like morphology at the edge of the fracture surface. After post-exposure storage at 150–200 °C, the PESCC zone is almost completely absent. However, some specimens occasionally exhibited a few tiny areas with the brittle topology (e.g.,
Figure 3g,h) even after storage at 150–200 °C. Despite the generally increasing apparent ductility of the fracture surface with the temperature of post-exposure storage, one can notice the anomalous increase in the fraction of the brittle relief on the fracture surface after storage at 120 °C (
Figure 4a). As was shown above, the decrease in strength was supposedly associated with the microstructural changes occurring at the same temperature in the reference specimens.
The fractographic observations corroborate well with the results of mechanical testing showing the increasing ductility of the pre-exposed specimens due to the suppression of PESCC by post-exposure storage in air. The confident linear correlation with Pearson’s R
2 > 0.91 is found between EF and the total area of their fracture surface as well as the area of the PESCC zone and its areal fraction (
Figure 4b), thus confirming the link between the propagation of PESCC and the material’s embrittlement unveiled by SSRT testing in ambient conditions.
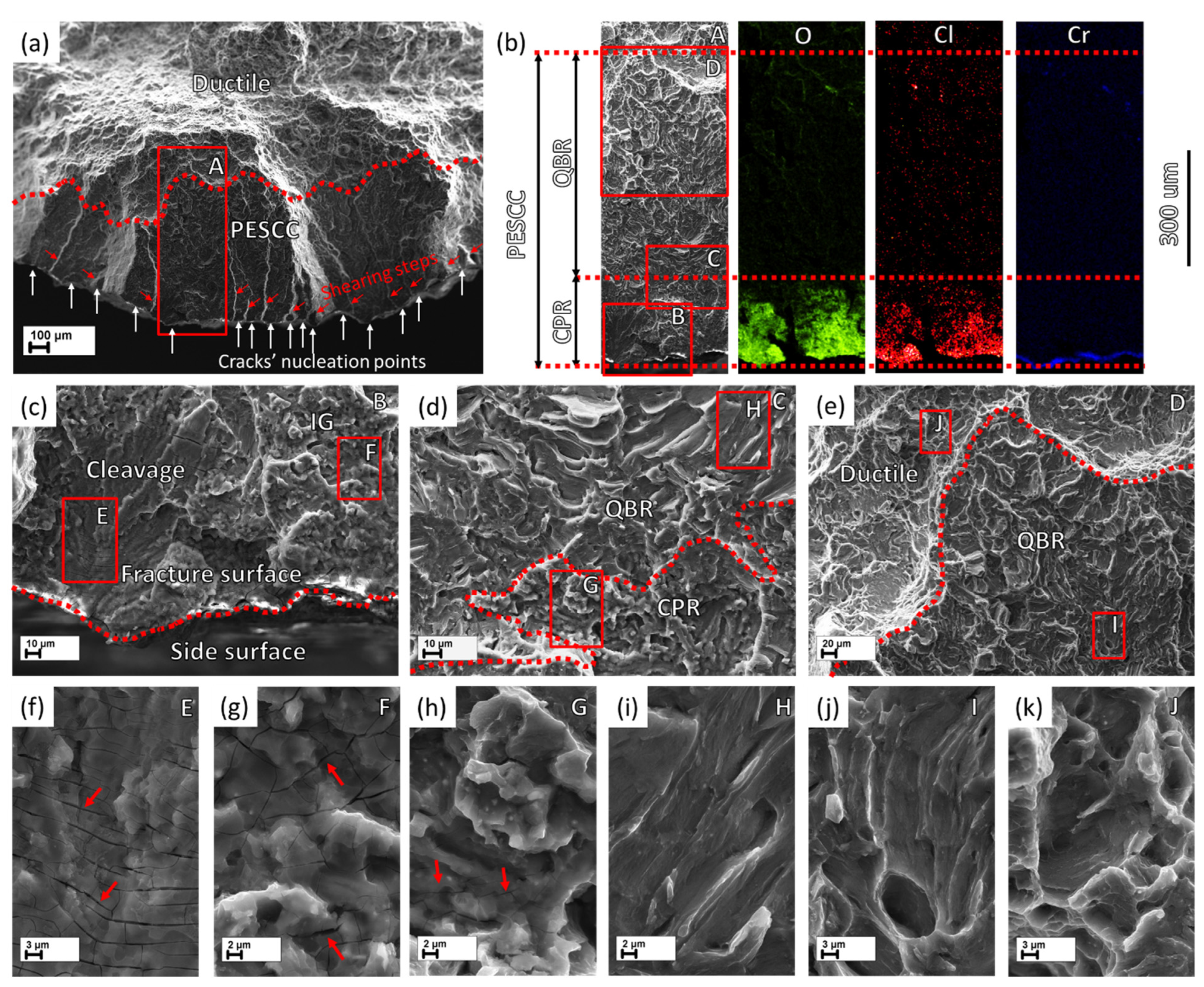
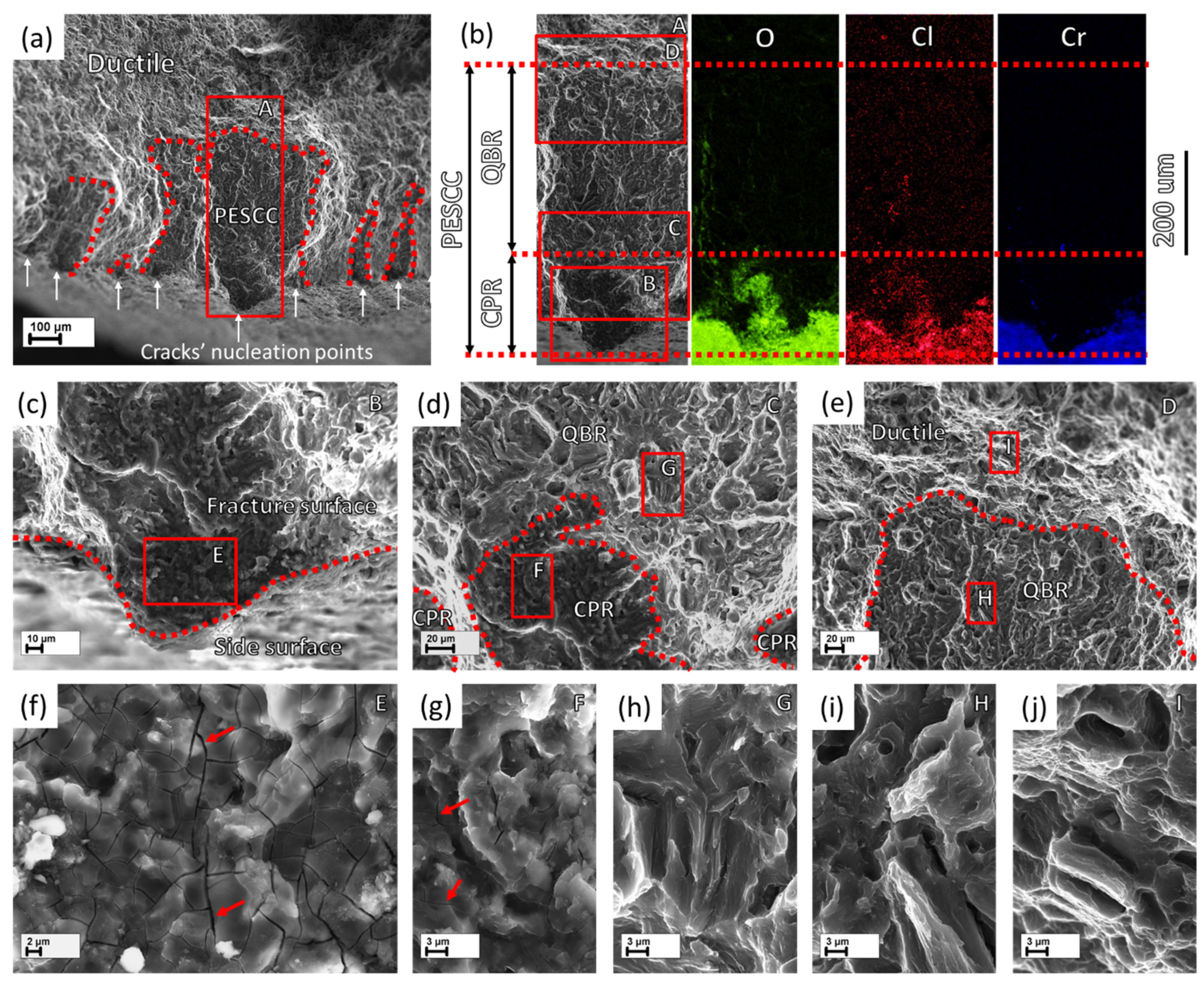
The side surface observations in combination with the fractographic analysis show that the PESCC zone is formed due to the propagation of multiple cracks nucleating at the side surface along the whole gauge part of the specimens (c.f.,
Figure 5 and
Figure 6). These cracks coalesce during their propagation towards the specimen’s centre as well as in the transverse direction. The coalescence of the cracks occurs in a stepwise manner by mutual axial shearing of the two halves of the fracture surface. The location of the cracks’ nucleation sites on the peripheral part of the fracture surface corresponds to relatively flat regions separated from each other by the axial shearing steps. As an example, the shearing steps and the crack initiation sites between them are indicated by the inclined red and vertical white arrows, respectively, in
Figure 7a and
Figure 8a. As is seen in the SEM side surface images in
Figure 5 and
Figure 6, the number and size of the side surface cracks decrease considerably with the temperature of post-exposure storage. This visual observation is testified by the results of the quantitative fractographic analysis provided in
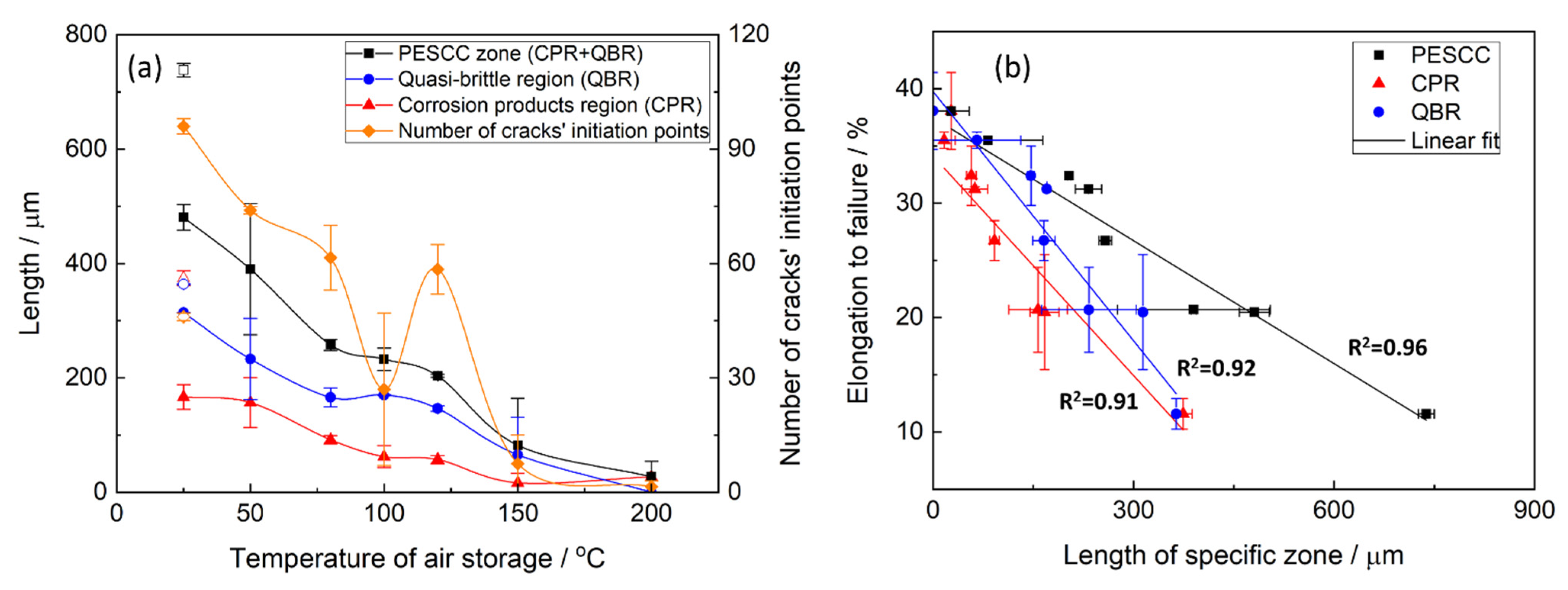
Figure 9a. According to these results, the number of cracks’ nucleation sites on the fracture surface increases considerably along with the storage temperature. Furthermore, as the temperature increases, the side surface cracks become shorter in the transverse direction and acquire a more ductile appearance, as is indicated by their less sharp and more round geometry (c.f.,
Figure 6). It follows from
Figure 5a,b and
Figure 9a that the increase in the time of post-exposure storage in air at 24 °C from 2 min to 24 h results in the remarkable growth in the number of side surface cracks and the cracks’ nucleation sites on the fracture surface. Thus, the number of cracks decreases with temperature and increases with the time of air storage. It is worth noting that, similarly to the area of the PESCC zone, the number of the cracks’ nucleation sites exhibits a sharp increase after air storage at 120 °C (
Figure 9a), whereas just a few cracks can be found in the specimens subjected to holding in air at 150–200 °C.
The PESCC zone in all pre-exposed specimens is composed of two distinct regions referred to as the corrosion product region (CPR) and the quasi-brittle region (QBR), which are seen one after the other from the edge of the specimen to the centre (
Figure 7 and
Figure 8). The corrosion product region starts to develop immediately at the edge of the fracture surface and is covered by the cracked crust of corrosion products (
Figure 7b,c,f–h and
Figure 8b,c,f,g). It has been shown recently that the intergranular cracking assisted by the corrosion solution sealed in the side surface layer of corrosion products can be responsible for the formation of the corrosion product region on the fracture surface [
6]. Thus, the specific “melted” relief, which is observed within this region under the crust of corrosion products (
Figure 7g,h and
Figure 8f,g), is probably composed of fine intergranular facets attacked by corrosion. In addition, the large transgranular cleavage facets exhibiting corrosion damage can also be occasionally distinguished within the corrosion product region on the fracture surface (
Figure 7c,f). The thickness of the corrosion product layer on the fracture surface decreases with the distance from the edge of the fracture surface towards the following quasi-brittle region, as is firmly evidenced by the EDX analysis (
Figure 7b and
Figure 8b). Additionally, the EDX elemental map obtained for the PESCC zone undeniably shows that this layer is strongly enriched with oxygen and chlorine. It should be stressed that the concentration of these elements decreases progressively along the crack path within the corrosion product region; however, it drops down abruptly to the negligible level at the boundary between the corrosion products and quasi-brittle regions. Thereby, the latter region is always completely free of traces of aggressive elements. Alternatively, the CPR–QBR boundary can be reliably distinguished by the morphological features in the SEM images with no aid from the EDX. The characteristic microcracks, such as those indicated by the arrows in
Figure 7f–h and
Figure 8f,g, are found to be indispensable attributes of the crust of corrosion products. As such, they serve as independent markers of corrosion products on the fracture surface. The size and density of these microcracks are gradually reduced with the thickness of the corrosion product layer throughout the respective region (see
Figure 7f–h and
Figure 8f,g). However, the cracks cease to appear in the quasi-brittle region beyond the CPR–QBR boundary (c.f.,
Figure 7i–k and
Figure 8h–j). Moreover, in contrast to the region that has been covered by corrosion products, the morphology of the quasi-brittle region is distinctively characterised by various tear ridges, small dimples, and fluted facets (
Figure 7j,i and
Figure 8h,i). These features indicate the appreciable contribution of plastic deformation to the crack growth mechanism.
Notably, the CPR–QBR boundary recognised by the topological features on SEM images matches precisely with that distinguished by the EDX analysis (
Figure 7b and
Figure 8b). Thus, both methods can be used to characterise and identify two morphologically distinct regions. Apparently, the secondary electron imaging is less laborious in comparison with the EDX mapping. That is why the former was prioritised for further measurements. The length of the corrosion product zone, l
CPR, as well as the length of the quasi-brittle region, l
QBR, and the PESCC zone, l
PESCC, were measured for each crack within the PESCC zone, as is schematically shown in
Figure 7b and
Figure 8b. The average values of these characteristics were calculated for the specimens tested under specific experimental conditions. The effect of air storage temperature and time on the measured fractographic properties is illustrated in
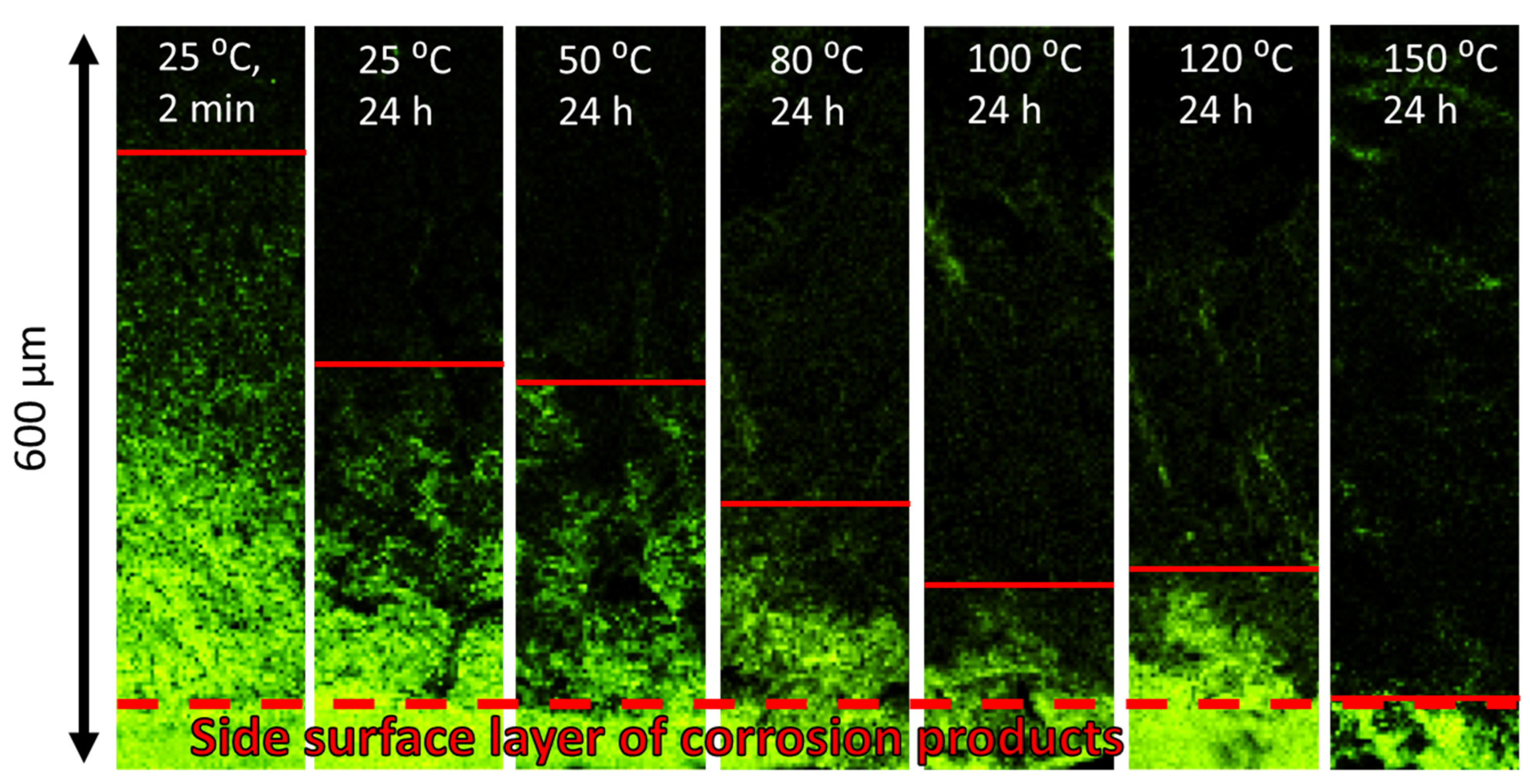
Figure 9a: the lengths of all specific zones, including the corrosion product region, decrease significantly with temperature and time. In particular, the corresponding reduction in l
CPR is also evidently illustrated by the EDX oxygen maps shown in
Figure 10. The obtained results corroborate the conclusions made in the previous study that the corrosion products on the fracture surface are produced during the SSRT testing. Indeed, if the corrosion product region was formed during pre-exposure, the size of this region would not be affected by subsequent air storage. It is worth noting that reduction in l
CPR becomes notable at the temperatures of air storage higher than 50 °C, while the l
QBR considerably decreases after storage at a lower temperature. In the temperature range of 100–120 °C, the plateau followed by the further decay up to near complete vanishing at higher temperatures of air storage is seen on the l
CPR, l
QBR, and l
PESCC curves in
Figure 9a. It is established that the lengths of all three characteristic regions, including the corrosion product region, correlate well with the elongation to failure of the specimens embrittled by pre-exposure to the aggressive environment (
Figure 9b). This finding suggests the crucially active role of the corrosion solution sealed/stored within the side surface layer of corrosion products in the mechanism of PESCC.
As has been mentioned, the corrosion products on the fracture surface can be associated with the corrosion solution, which is likely sealed within the corrosion product layer on the side surface. In conjunction with this suggestion, it is important to note that according to the results of EDX analysis, the corrosion product region on the fracture surface almost does not contain chromium (
Figure 7b and
Figure 8b), which is, however, abundantly present in the side surface layer of corrosion products (
Figure 11a). This feature of the corrosion product region also drastically differs the fracture surface of the tensile tested pre-exposed specimens from that of the specimens which have been SSRT tested right in the corrosive solution containing chromates. The EDX maps obtained from the fracture surface of the specimen SSRT tested in the 4% NaCl + 4% K
2Cr
2O
7 solution in the previous study [
19] are provided in
Figure 11b. It is clear that the fracture surface of this specimen demonstrates high concentrations of Cl, O, and Cr. This finding implies that the corrosion solution responsible for the formation of corrosion products on the fracture surface of the pre-exposed specimens is Cr-depleted and thus, its chemical composition can be different from that of the liquid initially used in the pre-exposure process.
3.3. Hydrogen Desorption Analysis
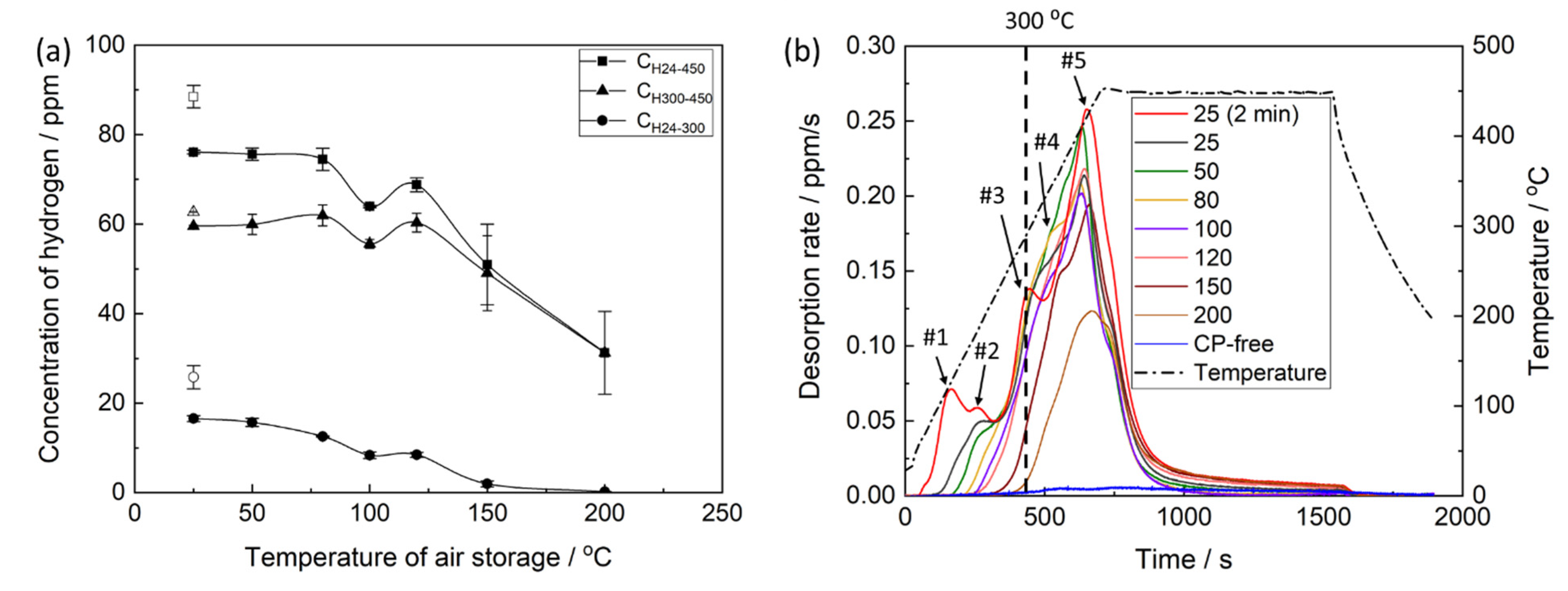
The thermal desorption analysis showed that the concentration of hydrogen extracted from the pre-exposed samples in the temperature range between 24 and 450 °C,
CH24–450, decreases notably with increasing temperature and time of air storage. The dependence of
CH24–450 on the temperature of air storage is non-linear and is characterised by the plateau within the interval between 24 and 80 °C, followed by the remarkable decay at higher temperatures (
Figure 12a). Moreover, the anomalous increase in the hydrogen concentration is observed after air storage at 120 °C. The correlation between
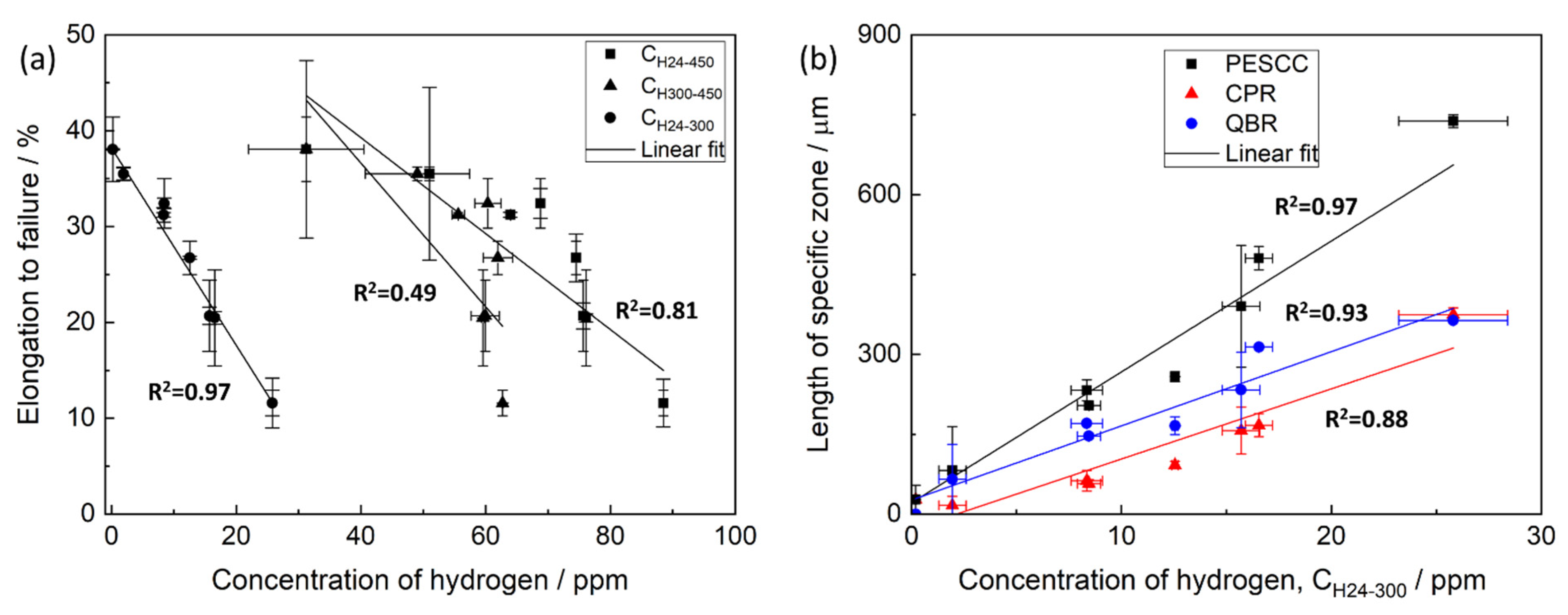
CH24–450 and elongation to failure of the pre-exposed samples subjected to air storage at different temperatures is poor (
Figure 13a), indicating that the relationship between
CH24–450 and the degree of PESCC is unlikely. It is generally accepted that diffusible hydrogen, i.e., chemically free hydrogen possessing high mobility within a metal, is responsible for HE in steels and other alloys [
32]. It has been well established that this hydrogen completely escapes from metals at temperatures below 300 °C [
33]. As was shown in our previous reports, the concentration of diffusible hydrogen in the matrix of the pre-exposed specimens of ZK60 and AZ31 alloys was negligible [
20,
23]. However, chemically free hydrogen can be, probably, accumulated within the layer of corrosion products. The extraction temperature of this hydrogen is also expected to be below 300 °C. The hydrogen extracted from the metals at temperatures above 300 °C is usually considered to be immobile, and hence, its role in hydrogen-assisted cracking is relatively insignificant. Thus, the concentrations of hydrogen extracted from the pre-exposed samples in the temperature intervals of 24–300,
CH24–300, and 300–450 °C,
CH300–450, were additionally assessed using the thermal desorption curves shown in
Figure 12b. It was found that the behaviour of
CH300–450, depending on the temperature of air storage, is similar to that of
CH24–450 exhibiting the plateau at 24–80 °C, followed by the overall decay with the hump at 120 °C. In contrast,
CH24–300 notably decreases in the whole temperature range of air storage, including the 24–80 °C interval, where
CH24–450 and
CH300–450 remain almost unchanged. Furthermore, the concentration of hydrogen extracted below 300 °C during the thermal desorption analysis is negligible in the samples subjected to air storage at 150 and 200 °C. A strong correlation is found between
CH24–300 and EF of the pre-exposed specimens, which were exposed to air at different temperatures and times (
Figure 13a), whereas the correlation between
CH300–450 and EF is even worse than that of
CH24–450. Moreover,
CH24–300 appreciably correlates with the size of specific zones on the fracture surface of the pre-exposed specimens, including the PESCC zone, corrosion product region and quasi-brittle region (
Figure 13b).
The thermal desorption spectra of hydrogen obtained from the specimens, which were subjected to pre-exposure, exhibit a few distinct superimposed peaks, as can be seen in
Figure 12b. It was conclusively established in the previous studies [
20,
23,
24] that all the observed peaks are associated with the corrosion products on the side surface layer because no considerable desorption of hydrogen from the matrix of the pre-exposed specimens occurs after chemical removal of those corrosion products. In support of this observation, the desorption spectrum for the specimen with the removed corrosion product layer is provided in
Figure 12b (the plot is denoted as “CP-free”) for reference. Thus, all peaks observed on the thermal desorption diagram in
Figure 12b should be associated either with chemically bonded hydrogen evolving from the thermally decomposing components of corrosion products or with the chemically free hydrogen. The latter can be either trapped in the corrosion product layer or produced during the corrosion of the Mg matrix interacting with the corrosion solution sealed within that layer. All hydrogen other than that linked to chemical components of corrosion products can be considered to be potentially capable for facilitating crack growth and, thus, inducing embrittlement associated with PESCC. The large peaks #3–5 appearing above 300 °C likely correspond to such components of corrosion products as Mg(OH)
2 and/or MgH
2, which decompose in the temperature range of 280–450 °C [
13,
34,
35]. It can be seen that at least peaks #4 and #5 are still present on the desorption spectra after air storage at 150 and 200 °C, though PESCC is fully eliminated at these temperatures. Thus, hydrogen related to the high-temperature peaks #4 and #5 is not likely responsible for PESCC. This conclusion corroborates the fact that the concentration of hydrogen extracted in the temperature intervals 25–450 and 300–450 °C poorly correlates with the propagation of PESCC (
Figure 13a). The low-temperature peak indicated as #1 in
Figure 12b is attributed to hydrogen, which freely evolves from the CP layer at room temperature. This is evidenced by the absence of this peak on the thermal desorption spectra corresponding to the pre-exposed specimens subjected to air storage at 24 °C for 24 h. Moreover, the bubbles of hydrogen gas emanating from the pre-exposed specimen can be seen by the naked eye when this specimen is submerged in an inert liquid such as CCl
4 [
26]. This hydrogen can be associated with the molecular or atomic hydrogen liberated from the corrosion product layer as well as with hydrogen, which is produced in situ by the corrosion reaction [
26]. Since the pre-exposed specimens do suffer from appreciable embrittlement after 24 h of air storage at 24 °C but do not exhibit peak #1 in the thermal desorption spectra, the hydrogen associated with this peak is not sufficient to be the sole reason for PESCC. Nevertheless, the contribution of this hydrogen to the observed embrittlement is possible because the reduction in peak #1 due to air storage at 24 °C is accompanied by the partial recovery of ductility. The nature of peaks #2 and #3 is still unknown. Presumably, they can be associated with the desorption of atomic or molecular hydrogen stored within the corrosion product layer or with the thermal decomposition of some hydrogen-containing components in this layer. As follows from
Figure 12b, both peaks contribute considerably to the concentration of hydrogen extracted below 300 °C, which is found to be correlated with the extension of PESCC (
Figure 13a), as well as with the size of the specific zones on the fracture surface (
Figure 13b). Thus, hydrogen associated with peaks #1–3 can likely be involved in the PESCC phenomenon. However, the detailed analysis of origin and activation energies of hydrogen in different traps in the surface layer with corrosion products has yet to be carried out, e.g., methodologically similarly to that used in [
36] to clarify the hydrogen distribution in the environmentally embrittled Al-Cu-Mg alloy.
3.5. The Corrosion Solution in the Corrosion Product Layer
The present and other reports [
6,
16,
20,
23] have documented that the peripheral part of the fracture surface of ZK60 specimens, which were SSRT tested in air after pre-exposure, is abundantly covered by corrosion products. Several independent experimental observations convincingly demonstrated that these products are created during the SSRT testing in air, when none of the external aggressive environments interact with the surface of the specimens. To be more specific, it is strongly supported by the following observations: (i) no secondary cracks containing corrosion products inside are found in the pre-exposed specimens before mechanical testing [
6], and (ii) the length and area of corrosion product region on the fracture surface are reduced with the increasing strain rate [
6] and (iii) the temperature of air storage [present study]. If the corrosion product regions were formed during pre-exposure, they would be observed in the cross-section of the specimen right after pre-exposure, while their size would not be affected by the strain rate or conditions of air storage. Assuming that ambient air, in which the SSRT testing is carried out, cannot cause the formation of extensive corrosion products, we can conclude that there must be some corrosive medium present in the side surface layer of corrosion products or at the interface between this layer and the surface of the specimen during SSRT testing. The interaction of this corrosive medium with the crack tip is, therefore, decided to be responsible for the formation of the corrosion product region on the fracture surface. Moreover, the correlation between the length of this region and the degree of PESCC observed in the present study suggests that the corrosion solution contained within the corrosion product layer plays a crucial role in the mechanism of PESCC.
Although the nature of the corrosive medium being stored within the corrosion product layer is questionable and additional comprehensive investigation addressing this issue is required, some plausible explanations can be proposed. For example, it might be supposed that during pre-exposure, some amount of the original corrosion solution is sealed within the discontinuities, such as microcracks and voids, which are abundantly present within the corrosion product layer. Alternatively, the corrosive medium can probably be produced inside this layer after extraction of the specimen from the corrosion solution. It has been reported that the Mg hydroxide can react with CO
2 from ambient air, leading to the formation of magnesite MgCO
3 and water [
37,
38]. Probably, some components of corrosion products, such as MgCl
2, can be dissolved in this water, thus making the solution even more aggressive. In favour of this scenario, one may recall the fact that the corrosion product region is always Cr-free. The original corrosion solution contains K
2Cr
2O
7 producing the passive film on the Mg surface. The presence of this film is evidenced by the high concentration of Cr in the side surface layer of corrosion products in the pre-exposed specimens as well as on the fracture surface of the specimens tested in the corrosion solution. In contrast to MgCl
2, the chromates, which are formed on the Mg surface, are sparingly soluble in water. Thus, the lack of Cr in the newly formed corrosion solution is explainable. The reaction of the saline water with the crack surface would produce MgO, Mg(OH)
2, and MgCl
2, providing the high concentration of oxygen and chlorine on the elemental maps obtained by EDX from the corrosion product region on the fracture surface.
Regardless of its origin, the corrosion solution within the corrosion product layer should contain water to activate and maintain the electrochemical corrosion reaction producing corrosion products on the fracture surface. It can be suggested that air storage after pre-exposure is accompanied by the evaporation of this water from the corrosion product layer. The amount of evaporated water should grow with increasing temperature and time of air storage, causing consumption of the available corrosion solution. This can explain the reduction in corrosion product region on the fracture surface with time and temperature of air storage. As has been discussed above, the size of this region decreases progressively within the temperature range from 50 to 120 °C and almost vanishes after storage in air at 150–200 °C. The complete evaporation of water in this temperature range is well expected. The size of the corrosion product region is also reduced with the increasing time of air storage. However, unlike the effect of temperature, the longer time of air storage results in an increasing number of surface cracks. Probably, during longer post-exposure storage, more local volumes of hydroxide transform into magnesite with the release of water, thus creating a greater number of favourable sites for crack initiation. Nevertheless, the size of the corrosion product region decreases with the time of air storage because the total amount of the corrosion solution available at the cracks’ initiation sites decreases due to the evaporation of water.
Despite the obvious interaction of the corrosive medium with the surface of the propagating cracks during mechanical testing of the pre-exposed specimens, the exact role of this interaction in the mechanism of the crack growth remains unclear. It was shown previously that the corrosion product region is mainly produced by intergranular cracking [
6]. For example, the anodic dissolution along the grain boundaries, which are enriched with noble secondary phase particles, might be responsible for this kind of fracture mode. On the other hand, the anodic dissolution of Mg is always accompanied by the cathodic reaction of hydrogen evolution. The adsorption of this hydrogen at the crack tip or its absorption within a few atomic layers beneath the surface can cause intergranular cracking, as well as transgranular cleavage through the HE mechanism, referred to as adsorption-induced dislocation emission (AIDE) [
21,
39]. Furthermore, the corrosion solution being in contact with the juvenile metal surface at the crack tip can possibly act as the surface active liquid, inducing the embrittlement through Rehbinder’s effect [
40]. Thus, it is not necessary that the anodic dissolution accompanied by the formation of corrosion products on the fracture surface is the rate-controlling factor for the crack propagation.
3.6. Hydrogen in the Corrosion Product Layer
It is found that the corrosion product region is always followed by the quasi-brittle region, which exhibits no signs of any interaction with the corrosion solution. All features indicating the signs of corrosion on the fracture surface, including the elevated concentration of oxygen and chlorine as well as the microcracks attributed to the crust of corrosion products, cease to appear abruptly at the boundary between the corrosion product region and the quasi-brittle region. Thus, there should be a factor other than the direct contact of the crack tip with the corrosion solution, which drives the crack growth producing the quasi-brittle region. This factor should also be associated with the side surface corrosion product layer because the quasi-brittle region is absent on the fracture surfaces of the reference and corrosion product-free specimens. Furthermore, similarly to that responsible for the corrosion product region, this factor should be eliminated by storage in air, since the quasi-brittle region is found to be reduced under increasing time and temperature of air storage. Probably, the only factor which might act this way is hydrogen.
As was mentioned above, the evolution of hydrogen is an inalienable part of the corrosion process of Mg in aqueous solutions. Hydrogen can be produced during pre-exposure of the specimen as well as during the subsequent air storage if the corrosion solution sealed within the corrosion product layer interacts with the bare metal of the specimen. Indeed, it was shown recently that a large portion of hydrogen gas evolved from the pre-exposed specimen during its 24 h storage in CCl
4 [
26]. The volume of this hydrogen summed with that of hydrogen extracted from the same specimen during subsequent gas analysis was twice as high as the volume of hydrogen extracted from the counterpart specimen subjected to the gas analysis right after pre-exposure. This was concluded to be evidence for the generation of hydrogen via the corrosion reaction, which occurs within the surface layer of the pre-exposed specimen stored in the inert environment. The part of hydrogen being produced both during pre-exposure and subsequent air storage can likely be accumulated inside the discontinuities of the side surface corrosion product layer or at the interface between this layer and the bare metal. Presumably, this hydrogen can stay within the corrosion product layer in the molecular and atomic forms. Along with time after extraction of the specimen from the corrosion solution, the weakly bonded part of this hydrogen desorbs from the corrosion product layer even at room temperature. This is witnessed by the low-temperature desorption peak in
Figure 12b as well as by the naked-eye-visible hydrogen bubbles released from the specimen immersed in CCl
4 right after pre-exposure [
26]. The remaining part of hydrogen sitting within the corrosion product layer is bonded more strongly and, therefore, desorbs only at higher temperatures and longer times. At least a part of hydrogen extracted from the specimen below 300 °C during the thermal desorption analysis can be associated with this strongly bonded hydrogen. It is established that the concentration of hydrogen extracted below 300 °C correlates well with both ductility and fractographic features of the pre-exposed specimens; these features include the lengths of the PESCC zone, corrosion product region, and quasi-brittle region. The observed correlation favours the suggestion that hydrogen being stored inside the corrosion product layer and extracted at relatively low temperatures can be involved in the mechanism of PESCC. Apparently, the post-exposure air storage results in the desorption of hydrogen from the corrosion product layer, thus suppressing PESCC. The correlation between the concentration of hydrogen and the size of the corrosion product region indicates that there is also a relationship between the amount of hydrogen and the corrosion solution stored within the corrosion product layer. Probably, both substances are contained together inside the same collectors because the corrosion solution can generate hydrogen.
As has been mentioned, it is unclear whether hydrogen is responsible for the intergranular crack growth producing the corrosion product region on the fracture surface or not. However, the formation of the quasi-brittle region is likely a hydrogen-assisted process. Various morphological signatures of ductile fracture, which are typical of the quasi-brittle region, indicate that plastic deformation should be involved to some extent into the mechanism of hydrogen-assisted cracking observed. Taking into account that adsorbed hydrogen, rather than absorbed one, plays the key role in this process, one can plausibly suggest that AIDE is the most probable mechanism of the crack growth producing the quasi-brittle region. This mechanism implies that the dislocation emission from the crack tip is facilitated due to the adsorption of hydrogen atoms, thus promoting the locally ductile crack growth [
21,
41]. The propagation of such crack produces fine slip markings, dimples, and tear ridges on the fracture surface, which represent the time-extended plastic processes rather than instant brittle failures. It was also suggested that AIDE is responsible for the fluted facets on the fracture surface of pure Mg failing due to SCC [
39]. A similar fluted morphology is also commonly observed within the quasi-brittle region on the fracture surface of the ZK60 and AZ31 alloys SSRT tested in the corrosion solution [
24,
42] or after pre-exposure [
6,
20,
23].


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}












