
Molecular Mechanism of the Piezoelectric Response in the β-Phase PVDF Crystals Interpreted by Periodic Boundary Conditions DFT Calculations



Abstract
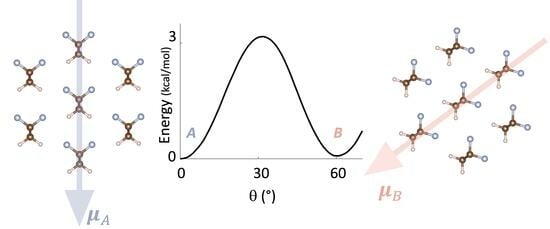
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Characterization
2.2.1. FT-IR Spectroscopy
2.2.2. Raman Spectroscopy
2.3. Computational Details
3. Results and Discussion
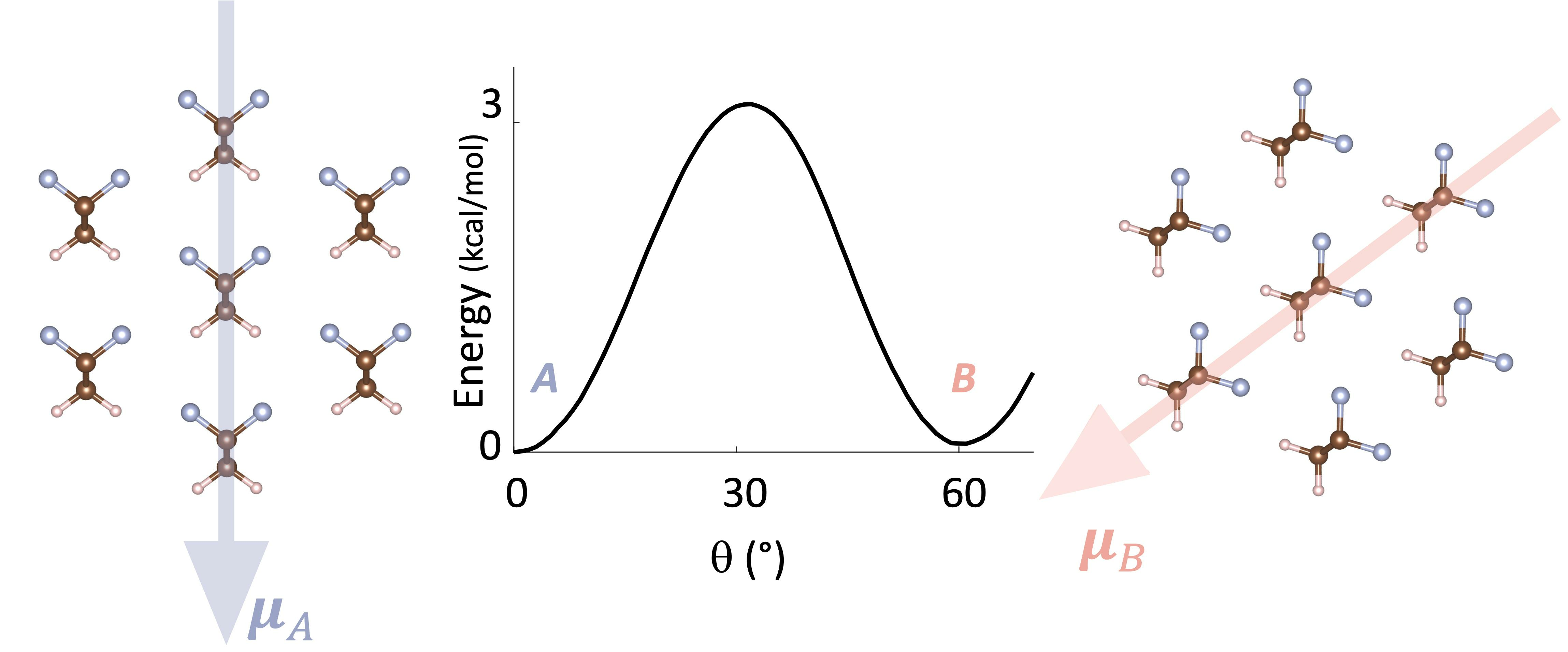
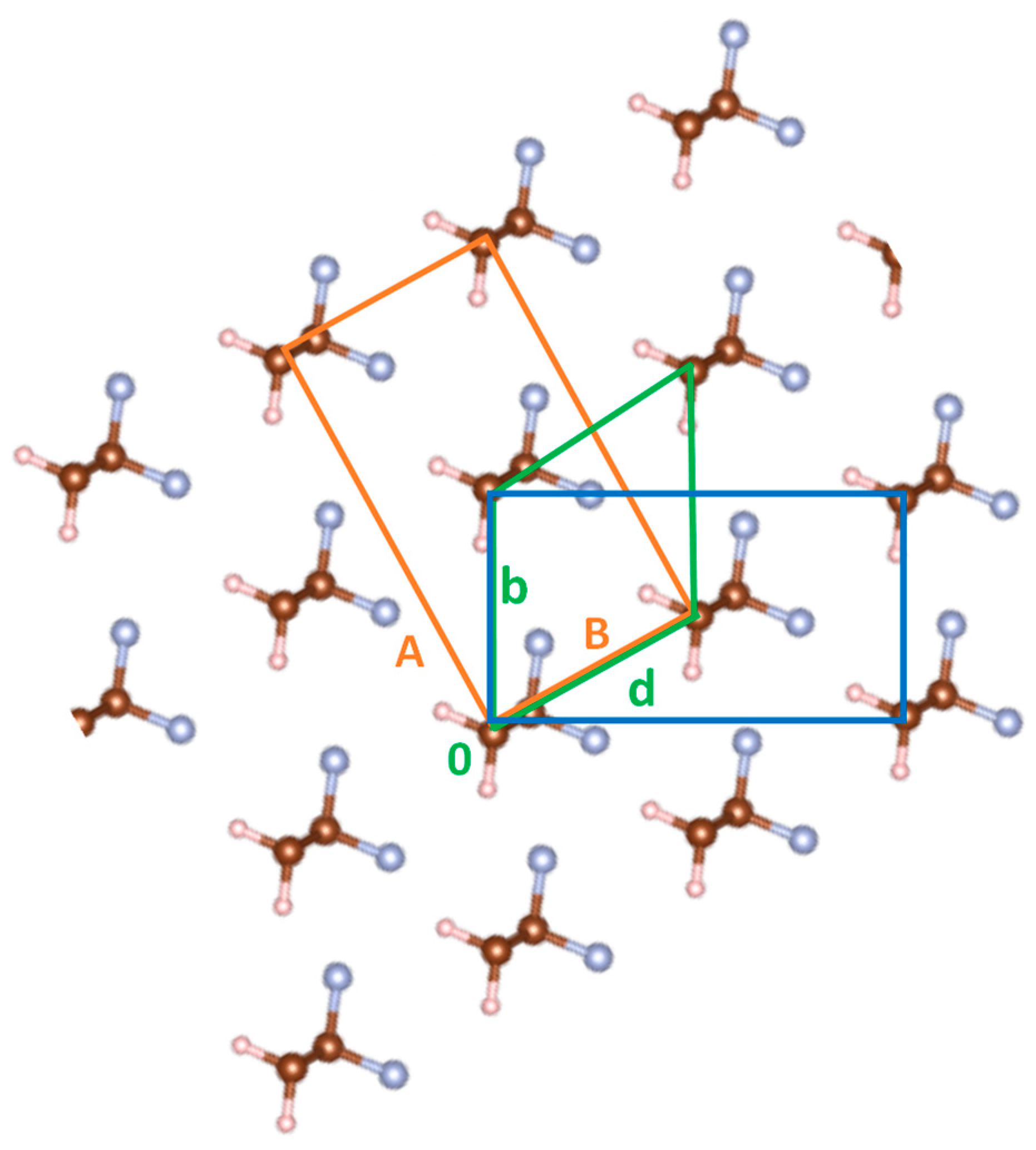
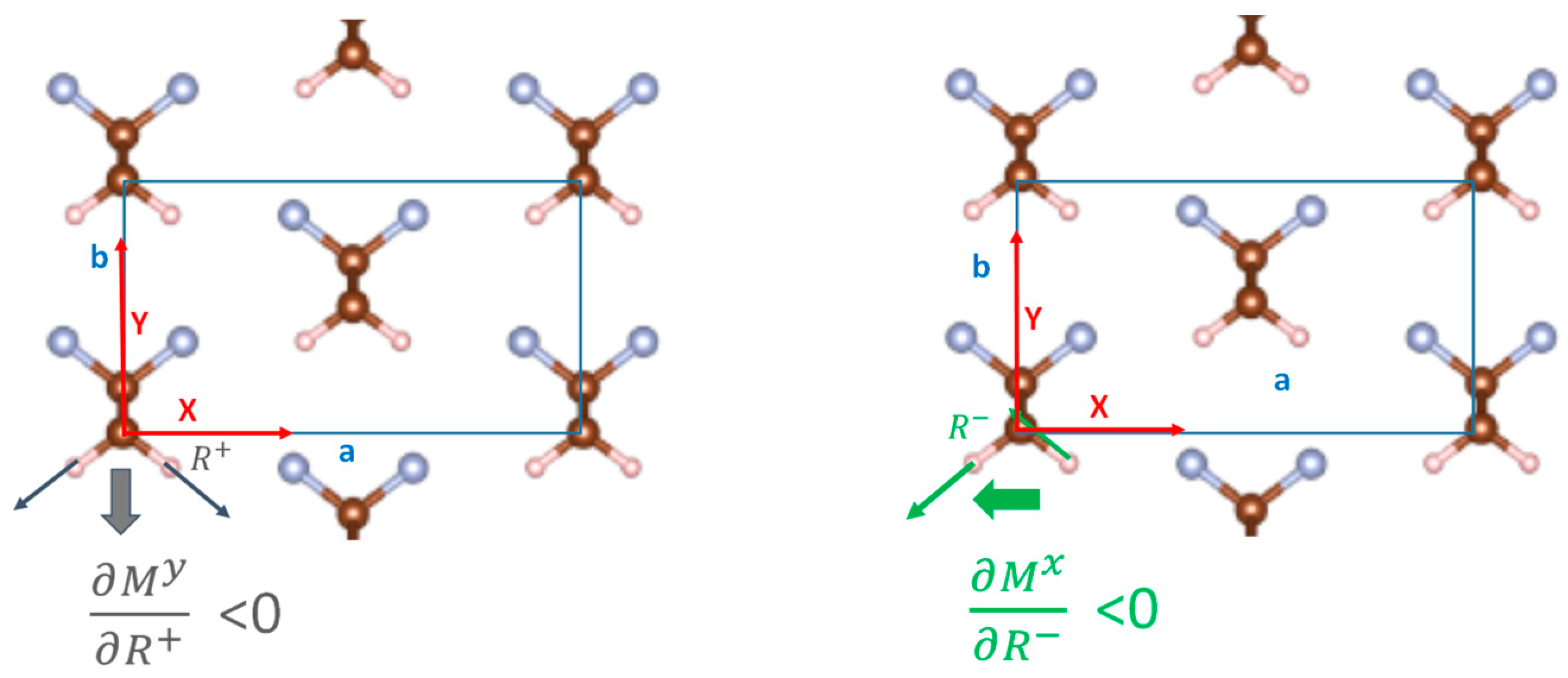
3.1. Energy Landscape for Chain Rotation
3.2. Vibrational Spectra
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kawai, H. The Piezoelectricity of Poly(vinylidene fluoride). Jpn. J. Appl. Phys. 1969, 8, 975−976. [Google Scholar] [CrossRef]
- Saxena, P.; Shukla, P. A comprehensive review on fundamental properties and applications of poly(vinylidene fuoride) (PVDF). Adv. Compos. Hybrid Mater. 2021, 4, 8–26. [Google Scholar] [CrossRef]
- Lovinger, A.J. Ferroelectric Polymers. Science 1983, 220, 1115−1121. [Google Scholar] [CrossRef]
- Furukawa, T.; Date, M.; Fukada, E.; Tajitsu, Y.; Chiba, A. Ferroelectric Behaviour in the Copolymer of Vinylidene Fluoride and Trifluoroethylene. Jpn. J. Appl. Phys. 1980, 19, L109−L112. [Google Scholar] [CrossRef]
- Furukawa, T. Ferroelectric Properties of Vinylidene Fluoride Copolymers. Phase Transit. 1989, 18, 143−211. [Google Scholar] [CrossRef]
- Kochervinskii, V.V. Piezoelectricity in Crystallizing Ferroelectric Polymers: Poly(vinylidene fluoride) and its Copolymers (A Review). Crystallogr. Rep. 2003, 48, 649−675. [Google Scholar] [CrossRef]
- Furukawa, T. Structure and Functional Properties of Ferroelectric Polymers. Adv. Colloid Interface Sci. 1997, 71−72, 183–208. [Google Scholar] [CrossRef]
- Lang, S.B.; Muensit, S. Review of Some Lesser-known Applications of Piezoelectric and Pyroelectric Polymers. Appl. Phys. A Mater. Sci. Process. 2006, 85, 125−134. [Google Scholar] [CrossRef]
- Glass, A.M.; McFee, J.H.; Bergman, J.G., Jr. Pyroelectric Properties of Polyvinylidene Fluoride and Its Use for Infrared Detection. J. Appl. Phys. 1971, 42, 5219–5222. [Google Scholar] [CrossRef]
- Bergman, J.G., Jr.; McFee, J.H.; Crane, G.R. Pyroelectricity and second harmonic generation in polyvinylidene fluoride films. Appl. Phys. Lett. 1971, 18, 203–205. [Google Scholar] [CrossRef]
- MeFee, J.H.; Bergman, J.G., Jr.; Crane, G.R. Pyroelectric and nonlinear optical properties of poled polyvinylidene fluoride films. Ferroelectrics 1972, 3, 305–313. [Google Scholar]
- Ohigashi, H. Electromechanical properties of polarized polyvinylidene fluoride films as studied by the piezoelectric resonance method. J. Appl. Phys. 1976, 47, 949–955. [Google Scholar] [CrossRef]
- Tamura, M.; Hagiwara, S.; Matsumoto, S.; Ono, N. Some aspects of piezoelectricity and pyroelectricity in uniaxially stretched poly(vinylidene fluoride). J. Appl. Phys. 1977, 48, 513–521. [Google Scholar] [CrossRef]
- Furukawa, T.; Date, M.; Fukada, E. Hysteresis phenomena in polyvinylidene fluoride under high electric field. J. Appl. Phys. 1980, 51, 1135–1141. [Google Scholar] [CrossRef]
- Kepler, R.G.; Anderson, R.A. Ferroelectricity in polyvinylidene fluoride. J. Appl. Phys. 1978, 49, 1232–1235. [Google Scholar] [CrossRef]
- Dovesi, R.; Saunders, V.R.; Roetti, C.; Orlando, R.; Zicovich-Wilson, C.M.; Pascale, F.; Civalleri, B.; Doll, K.; Harrison, N.M.; Bush, I.J.; et al. CRYSTAL17 User’s Manual; University of Torino: Torino, Italy, 2017. [Google Scholar]
- Dovesi, R.; Erba, A.; Orlando, R.; Zicovich-Wilson, C.M.; Civalleri, B.; Maschio, L.; Rérat, M.; Casassa, S.; Baima, J.; Salustro, S.; et al. Quantum-mechanical condensed matter simulations with CRYSTAL, Wiley Interdiscip. Rev. Comput. Mol. Sci. 2018, 8, e1360. [Google Scholar] [CrossRef]
- Milani, A.; Castiglioni, C.; Radice, S. Joint Experimental and Computational Investigation of the Structural and Spectroscopic Properties of Poly(vinylidene fluoride) Polymorphs. J. Phys. Chem. B 2015, 119, 4888−4897. [Google Scholar] [CrossRef]
- Itoh, A.; Takahashi, Y.; Furukawa, T.; Yajima, H. Solid-state Calculations of Poly(vinylidene fluoride) Using the Hybrid DFT Method: Spontaneous Polarization of Polymorphs. Polym. J. 2014, 46, 207–211. [Google Scholar] [CrossRef]
- Peintinger, M.F.; Oliveira, D.V.; Bredow, T. Consistent Gaussian Basis Set of Triple-Zeta Valence with Polarization Quality for Solid-State Calculations. J. Comput. Chem. 2013, 34, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Tashiro, K.; Tadokoro, H. Molecular Vibrations of Three Crystal Forms of Poly(vinylidene fluoride). Macromolecules 1975, 8, 159–171. [Google Scholar] [CrossRef]
- Hasegawa, R.; Takahashi, Y.; Chatani, Y.; Tadokoro, H. Crystal Structures of Three Crystalline Forms of Poly(vinylidene fluoride). Polym. J. 1972, 3, 600–610. [Google Scholar] [CrossRef]
- Lando, J.B.; Olf, H.G.; Peterlin, A. Nuclear magnetic resonance and X-ray determination of the structure of poly(vinylidene fluoride). J. Polym. Sci. A-1 Polym. Chem. 1966, 4, 941–951. [Google Scholar] [CrossRef]
- Hasegawa, R.; Kobayashi, M.; Tadokoro, H. Molecular Conformation and Packing of Poly(vinylidene fluoride). Stability of Three Crystalline Forms and the Effect of High Pressure. Polym. J. 1972, 3, 591–599. [Google Scholar] [CrossRef]
- Duan, C.-G.; Mei, W.N.; Hardy, J.R.; Ducharme, S.; Choi, J.; Dowben, P.A. Comparison of the theoretical and experimental band structure of poly(vinylidene fluoride) crystal. Europhys. Lett. 2003, 61, 81–87. [Google Scholar] [CrossRef]
- Byutner, O.G.; Smith, G.D. Quantum Chemistry Based Force Field for Simulations of Poly(vinylidene fluoride). Macromolecules 2000, 33, 4264–4270. [Google Scholar] [CrossRef]
- Hafner, J.; Teuschel, M.; Schneider, M.; Schmid, U. Origin of the strong temperature effect on the piezoelectric response of the ferroelectric (co-)polymer P(VDF70-TrFE30). Polymer 2019, 170, 1–6. [Google Scholar] [CrossRef]
- Hayashi, S.-I.; Imamura, A. A Study of the Polarization Reversal in Poly(vinylidene Fluoride) Using Molecular Orbital Calculations. J. Polym. Sci. B Polym. Phys. 1992, 30, 769–773. [Google Scholar] [CrossRef]
- Tashiro, K.; Kobayashi, M. Structural Phase-Transition in Ferroelectric Fluorine Polymers—X-ray Diffraction and Infrared Raman—Spectroscopic Study. Phase Transit. 1989, 18, 213–246. [Google Scholar] [CrossRef]
- Tashiro, K.; Itoh, Y.; Kobayashi, M.; Tadokoro, H. Polarized Raman spectra and LO-TO splitting of poly (vinylidene fluoride) crystal form I. Macromolecules 1985, 18, 2600–2606. [Google Scholar] [CrossRef]
- Cortili, G.; Zerbi, G. Chain Conformations of Polyvinylidene Fluoride as Derived from its Vibrational Spectrum. Spectrochim. Acta Part A 1967, 23, 285–299. [Google Scholar] [CrossRef]
- Enomoto, S.; Kawai, Y.; Sugita, M. Infrared Spectrum of Poly(vinylidene fluoride). J. Polym. Sci. Part A-2 1968, 6, 861–869. [Google Scholar] [CrossRef]
- Borionetti, G.; Zannoni, G.; Zerbi, G. Lattice Dynamics and Vibrational Spectra of Conformationally Disordered Polymers: Poly(vinylidene fluoride). J. Mol. Struct. 1990, 224, 425–444. [Google Scholar] [CrossRef]
- Benz, M.; Euler, W.B. Determination of the Crystalline Phases of Poly(vinylidene fluoride) Under Different Preparation Conditions Using Differential Scanning Calorimetry and Infrared Spectroscopy. J. Appl. Polym. Sci. 2003, 89, 1093–1100. [Google Scholar] [CrossRef]
- Salimi, A.; Yousefi, A.A. Analysis Method: FTIR Studies of β-phase Crystal Formation in Stretched PVDF Films. Polym. Test. 2003, 22, 699–704. [Google Scholar] [CrossRef]
- Baniasadi, M.; Xu, Z.; Hong, S.; Naraghi, M.; Jolandan, M.M. Thermo-Electromechanical Behavior of Piezoelectric Nanofibers. ACS Appl. Mater. Interfaces 2016, 8, 2540–2551. [Google Scholar] [CrossRef]
- Arrigoni, A.; Brambilla, L.; Bertarelli, C.; Serra, G.; Tommasini, M.; Castiglioni, C. P(VDF-TrFE) nanofibers: Structure of the ferroelectric and paraelectric phases through IR and Raman spectroscopies. RSC Adv. 2020, 10, 37779. [Google Scholar] [CrossRef]
- Nakhmanson, S.M.; Korlacki, R.; Johnston, J.T.; Ducharme, S.; Ge, Z.; Takacs, J.M. Vibrational Properties of Ferroelectric β—Vinylidene fluoride Polymers and Oligomers. Phys. Rev. B Condens. Matter Mater. Phys. 2010, 81, 174120. [Google Scholar] [CrossRef]
- Ramer, N.J.; Raynor, C.M.; Stiso, K.A. Vibrational Frequency and LO−TO Splitting Determination for Planar−Zigzag β-poly- (vinylidene fluoride) Using Density-Functional Theory. Polymer 2006, 47, 424−428. [Google Scholar] [CrossRef]
- Galimberti, D.; Milani, A.; Maschio, L.; Castiglioni, C. Intermolecular modulation of IR intensities in the solid state. The role of weak interactions in polyethylene crystal: A computational DFT study. J. Chem. Phys. 2016, 145, 144901. [Google Scholar] [CrossRef] [PubMed]
- Pimentel, G.C.; McClellan, A.L. The Hydrogen Bond; W.H. Freeman: San Francisco, CA, USA, 1960. [Google Scholar]
- Quarti, C.; Milani, A.; Civalleri, B.; Orlando, R.; Castiglioni, C. Ab Initio Calculation of the Crystalline Structure and IR Spectrum of Polymers: Nylon 6 Polymorphs. J. Phys. Chem. B 2012, 116, 8299−8311. [Google Scholar] [CrossRef]
- Galimberti, D.; Milani, A.; Castiglioni, C. Infrared intensities and charge mobility in hydrogen bonded complexes. J. Chem. Phys. 2013, 139, 074304. [Google Scholar] [CrossRef] [PubMed]
- Sverdlov, L.M.; Kovner, M.A.; Krainov, E.P. Vibrational Intensities of Polyatomic Molecules; Wiley: New York, NY, USA, 1972. [Google Scholar]
- Gribov, L.A. Intensity Theory for Infrared Spectra of Polyatomic Molecules; Consultant’s Bureau: New York, NY, USA, 1964. [Google Scholar]
- Gussoni, M.; Abbate, S. Infrared absorption intensities: Transferability of electro-optical parameters. J. Chem. Phys. 1976, 65, 3439–3445. [Google Scholar] [CrossRef]
- Gussoni, M.; Castiglioni, C.; Zerbi, G. Vibrational Intensities: Interpretation and Use for Diagnostic Purposes. In Handbook of Vibrational Spectroscopy; Chalmers, J.M., Griffiths, P.R., Eds.; Wiley: London, UK, 2001; Volume 3, pp. 2040–2078. [Google Scholar]
- Jona, P.; Gussoni, M.; Zerbi, G. Interpretation of Infrared Intensities of Acetylene, Propyne, and 2-Butyne. A Common Set of Electrooptical Parameters. J. Phys. Chem. 1981, 85, 2210–2218. [Google Scholar]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mode # | Wavenumbers (cm−1) DFT | Wavenumbers (cm−1) DFT, after Scaling | Wavenumbers (cm−1) Experimental | |||
|---|---|---|---|---|---|---|
| 1-D | 3-D | 1-D | 3-D | IR | Raman | |
| 1 (B2) | 75 | 74 | 70 from Ref. [21] | - | ||
| 2 (A2) | 260 | 261 | 254 | 258 | - | 262 (*) |
| 3 (A1) | 429 | 444 | 418 | 439 | - | 445 (*) |
| 4 (B1) | 453 | 452 | 442 | 447 | 474 (*) | - |
| 5 (A1) | 514 | 513 | 500 | 508 | 510 (*) | 513 (*) |
| 6 (A1) | 884 | 862 | 861 | 852 | 840 (*) | 841 |
| 7 (B2) | 927 | 904 | 903 | 894 | 880 | 880 |
| 8 (B1) | 1058 | 1081 | 1031 | 1069 | 1072 (*) | 1076 (*) |
| 9 (B2) | 1273 | 1173 | 1241 | 1160 | 1172 | 1172 |
| 10 (A2) | 1185 | 1204 | 1155 | 1190 | - | 1172 (?) |
| 11 (A1) | 1342 | 1299 | 1307 | 1285 | 1275 (*) | 1275 (*) |
| 12 (B1) | 1385 | 1415 | 1349 | 1399 | 1402 | - |
| 13 (A1) | 1457 | 1448 | 1419 | 1432 | 1429 (*) | 1432 |
| 14 (A1) | 3091 | 3108 | 3011 | 3074 | 2976 | 2979 |
| 15 (B2) | 3149 | 3167 | 3067 | 3132 | 3015 | 3018 |
| Mode # | IR Intensity (km mol−1) | Raman Activity (Å4 amu−1) | IR Intensity (km mol−1) | Raman Activity (Å4 amu−1) | Normalized IR Intensity | Normalized Raman Intensity | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1D | 1D | 3D | 3D | 1D | 3D | exp | 1D | 3D | exp | |
| 1 (B2) | 9.79 | 0.01 | 0.17 | - | 0.01 | - | ||||
| 2 (A2) | - | 0.28 | - | 0.70 | - | - | - | 0.14 | 0.13 | 0.13 |
| 3 (A1) | 0.29 | 0.02 | 9.73 | 0.80 | 0.02 | 0.17 | - | 0.01 | 0.09 | 0.03 |
| 4 (B1) | 26.89 | 0.71 | 35.11 | 1.63 | 2.10 | 0.60 | 0.39 | 0.21 | 0.18 | - |
| 5 (A1) | 3.92 | 0.82 | 14.72 | 3.41 | 0.31 | 0.25 | 0.16 | 0.22 | 0.33 | 0.32 |
| 6 (A1) | 12.83 | 6.49 | 58.06 | 17.59 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| 7 (B2) | 20.08 | 1.63 | 72.64 | 4.16 | 1.57 | 1.25 | 2.07 | 0.24 | 0.23 | 0.19 |
| 8 (B1) | 47.33 | 3.08 | 32.67 | 4.45 | 3.69 | 0.56 | 0.53 | 0.40 | 0.20 | 0.12 |
| 9 (B2) | 0.00 | 7.27 | 303.43 | 4.86 | 10.58 | 5.23 | 4.85 | 0.15 | 0.20 | 0.20 |
| 10 (A2) | 135.70 | 1.42 | 0.00 | 13.28 | - | - | - | 0.83 | 0.54 | |
| 11 (A1) | 98.17 | 11.42 | 218.60 | 10.75 | 7.65 | 3.76 | 1.11 | 1.16 | 0.41 | 0.21 |
| 12 (B1) | 133.54 | 0.24 | 133.91 | 0.21 | 10.41 | 2.31 | 2.14 | 0.02 | 0.01 | - |
| 13 (A1) | 1.82 | 7.49 | 6.40 | 20.09 | 0.14 | 0.11 | 0.10 | 0.70 | 0.68 | 0.54 |
| 14 (A1) | 1.45 | 48.30 | 6.71 | 222.01 | 0.11 | 0.12 | 0.13 | 2.13 | 3.50 | 1.33 |
| 15 (B2) | 2.38 | 25.19 | 8.04 | 85.53 | 0.19 | 0.14 | 0.08 | 1.09 | 1.32 | 1.23 |
| tot | 484.39 | 114.35 | 900.03 | 389.47 | ||||||
| (A) Isotropic cell expansion in the (a, b) plane → isolated chain = 1D crystal | ||||||||||||
| f | 1 | 1.02 | 1.06 | 1.1 | 1.2 | 1.3 | 1.4 | 1.5 | 1.6 | 1.8 | 2 | 1D |
| Irrep | IR intensity (km mol−1) | |||||||||||
| B2 | 404 | 386 | 357 | 331 | 286 | 254 | 235 | 222 | 212 | 199 | 191 | 159 |
| B1 | 202 | 201 | 200 | 199 | 199 | 200 | 201 | 202 | 203 | 203 | 204 | 207 |
| A1 | 304 | 292 | 271 | 253 | 219 | 196 | 181 | 170 | 161 | 150 | 143 | 118 |
| A2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Tot | 910 | 879 | 827 | 784 | 704 | 650 | 616 | 593 | 576 | 553 | 539 | 484 |
| Irrep | Raman activity (Å4 amu−1) | |||||||||||
| B2 | 95 | 95 | 92 | 90 | 78 | 65 | 57 | 51 | 47 | 42 | 39 | 28 |
| B1 | 6 | 6 | 6 | 6 | 6 | 5 | 5 | 5 | 5 | 5 | 4 | 4 |
| A1 | 274 | 264 | 239 | 222 | 180 | 148 | 130 | 117 | 110 | 100 | 94 | 74 |
| A2 | 14 | 14 | 13 | 12 | 11 | 11 | 10 | 10 | 9 | 9 | 9 | 8 |
| Tot | 389 | 380 | 350 | 330 | 275 | 229 | 202 | 183 | 171 | 156 | 147 | 114 |
| (B) Cell expansion along the a-axis → vertical slab | ||||||||||||
| f | 1 | 1.02 | 1.06 | 1.1 | 1.2 | 1.3 | 1.4 | 1.5 | 1.6 | 1.8 | 2 | 1D |
| Irrep | IR intensity (km mol−1) | |||||||||||
| B2 | 404 | 411 | 357 | 326 | 54 | 45 | 231 | 213 | 200 | 182 | 168 | 159 |
| B1 | 202 | 175 | 200 | 200 | 199 | 198 | 198 | 198 | 198 | 198 | 198 | 208 |
| A1 | 304 | 301 | 296 | 294 | 289 | 286 | 285 | 285 | 284 | 284 | 284 | 118 |
| A2 | 0 | 0 | 0 | 0 | 232 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Tot | 910 | 887 | 853 | 820 | 774 | 530 | 714 | 696 | 683 | 664 | 650 | 485 |
| Irrep | Raman activity (Å4 amu−1) | |||||||||||
| B2 | 95 | 94 | 87 | 81 | 76 | 66 | 55 | 50 | 47 | 43 | 40 | 28 |
| B1 | 6 | 5 | 6 | 7 | 7 | 7 | 7 | 7 | 7 | 7 | 7 | 4 |
| A1 | 274 | 266 | 246 | 232 | 203 | 182 | 170 | 161 | 156 | 149 | 145 | 75 |
| A2 | 14 | 13 | 12 | 12 | 5 | 9 | 8 | 7 | 7 | 6 | 6 | 8 |
| Tot | 389 | 378 | 352 | 331 | 290 | 264 | 240 | 225 | 216 | 205 | 197 | 114 |
| Mode | Dipole Derivative (Debye/Å amu1/2) | f = 1 | 1.01 | 1.06 | 1.1 | 1.2 | 1.3 | 1.4 | 1.5 | 1.6 | 1.7 | 1.8 | 2 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 14 (A1) | −0.40 | −0.37 | −0.24 | −0.17 | −0.08 | −0.03 | 0.01 | 0.04 | 0.06 | 0.08 | 0.09 | 0.11 | |
| 15 (B2) | −0.44 | −0.40 | −0.24 | −0.16 | −0.03 | 0.03 | 0.07 | 0.10 | 0.12 | 0.14 | 0.15 | 0.17 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Serra, G.; Arrigoni, A.; Del Zoppo, M.; Castiglioni, C.; Tommasini, M. Molecular Mechanism of the Piezoelectric Response in the β-Phase PVDF Crystals Interpreted by Periodic Boundary Conditions DFT Calculations. Materials 2023, 16, 6004. https://doi.org/10.3390/ma16176004
Serra G, Arrigoni A, Del Zoppo M, Castiglioni C, Tommasini M. Molecular Mechanism of the Piezoelectric Response in the β-Phase PVDF Crystals Interpreted by Periodic Boundary Conditions DFT Calculations. Materials. 2023; 16(17):6004. https://doi.org/10.3390/ma16176004
Chicago/Turabian StyleSerra, Gianluca, Alessia Arrigoni, Mirella Del Zoppo, Chiara Castiglioni, and Matteo Tommasini. 2023. "Molecular Mechanism of the Piezoelectric Response in the β-Phase PVDF Crystals Interpreted by Periodic Boundary Conditions DFT Calculations" Materials 16, no. 17: 6004. https://doi.org/10.3390/ma16176004
APA StyleSerra, G., Arrigoni, A., Del Zoppo, M., Castiglioni, C., & Tommasini, M. (2023). Molecular Mechanism of the Piezoelectric Response in the β-Phase PVDF Crystals Interpreted by Periodic Boundary Conditions DFT Calculations. Materials, 16(17), 6004. https://doi.org/10.3390/ma16176004