
Synthesis, X-ray Structure, Optical, and Electrochemical Properties of a White-Light-Emitting Molecule


Abstract
:
1. Introduction

2. Experimental Section
2.1. General
2.2. Synthesis
2.2.1. Synthesis of 3-Bromo-7-methoxy-2,3-dihydro-1H-inden-1-one (5)
2.2.2. Synthesis of 7-Methoxy-1H-inden-1-one (4)
2.2.3. Synthesis of 7-Methoxy-1H-inden-1-one (3)
2.2.4. Synthesis of 1-Hydroxy-11H-benzo[b]fluoren-11-one (2)
2.2.5. Synthesis of 4-tert-Butyl-1-hydroxy-11H-benzo[b]fluoren-11-one (1)
2.3. Crystal Structural Determination
2.4. Steady State Spectral Measurements
2.5. Computational Methods
3. Results and Discussion
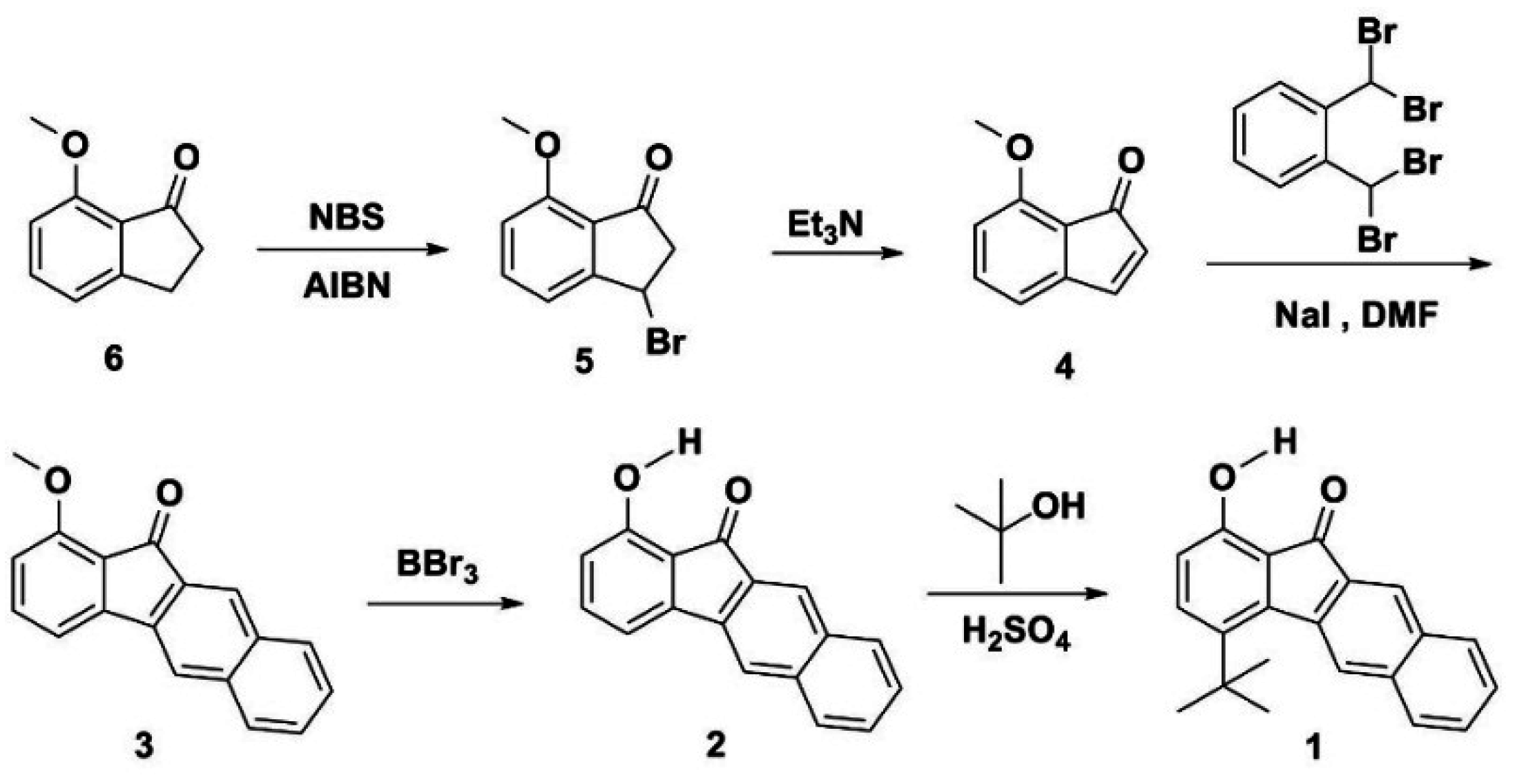
3.1. Synthesis
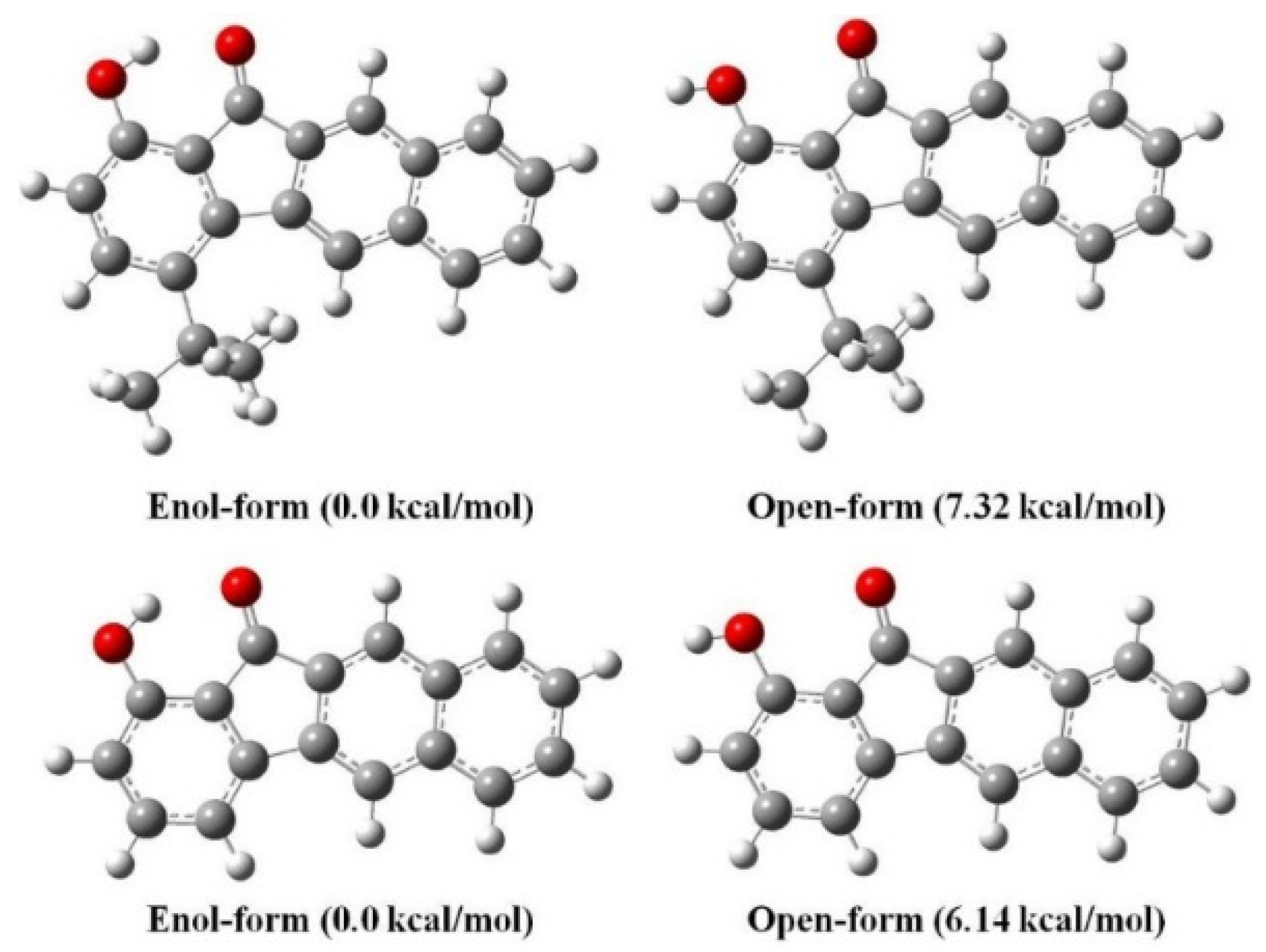
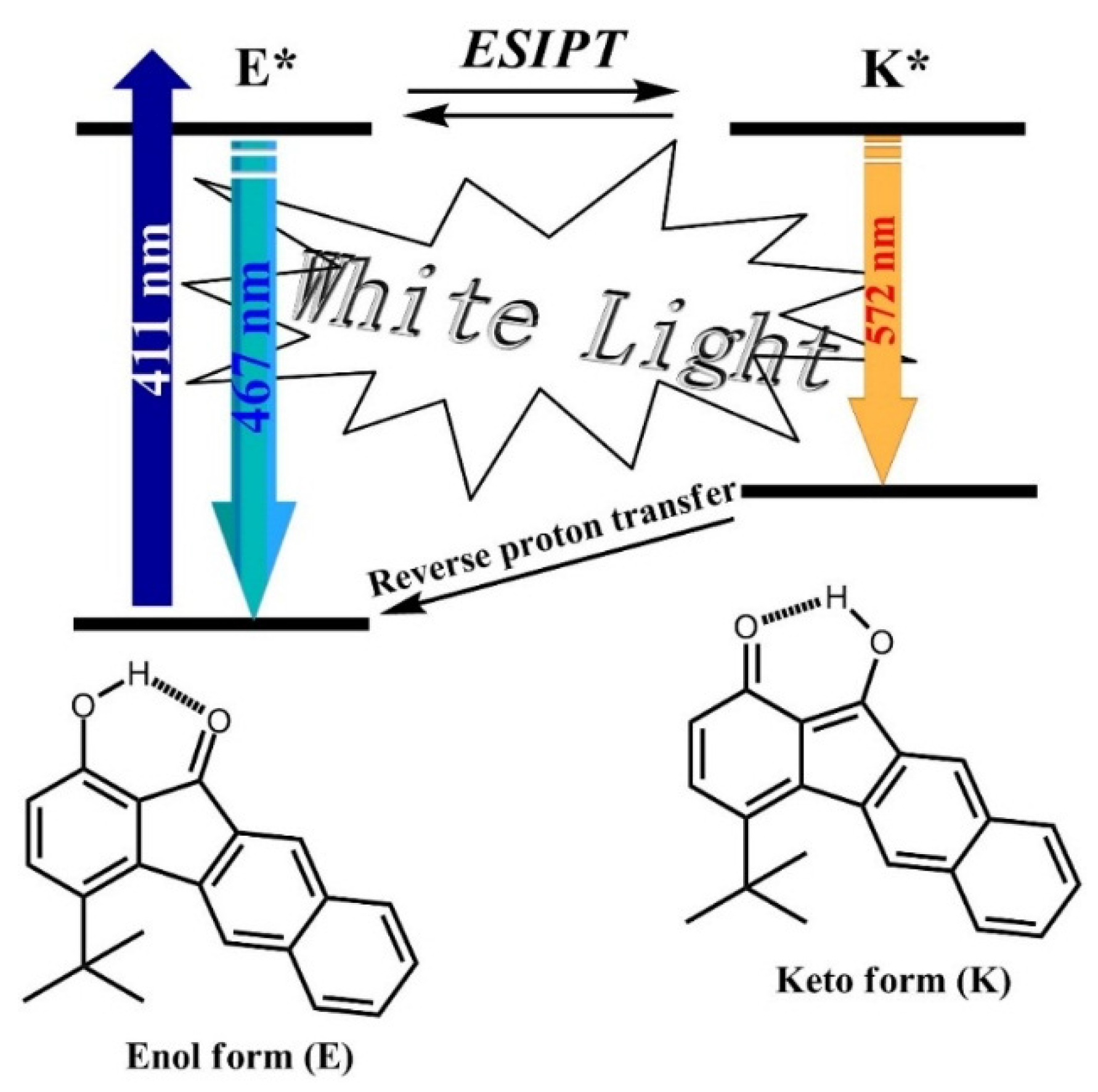
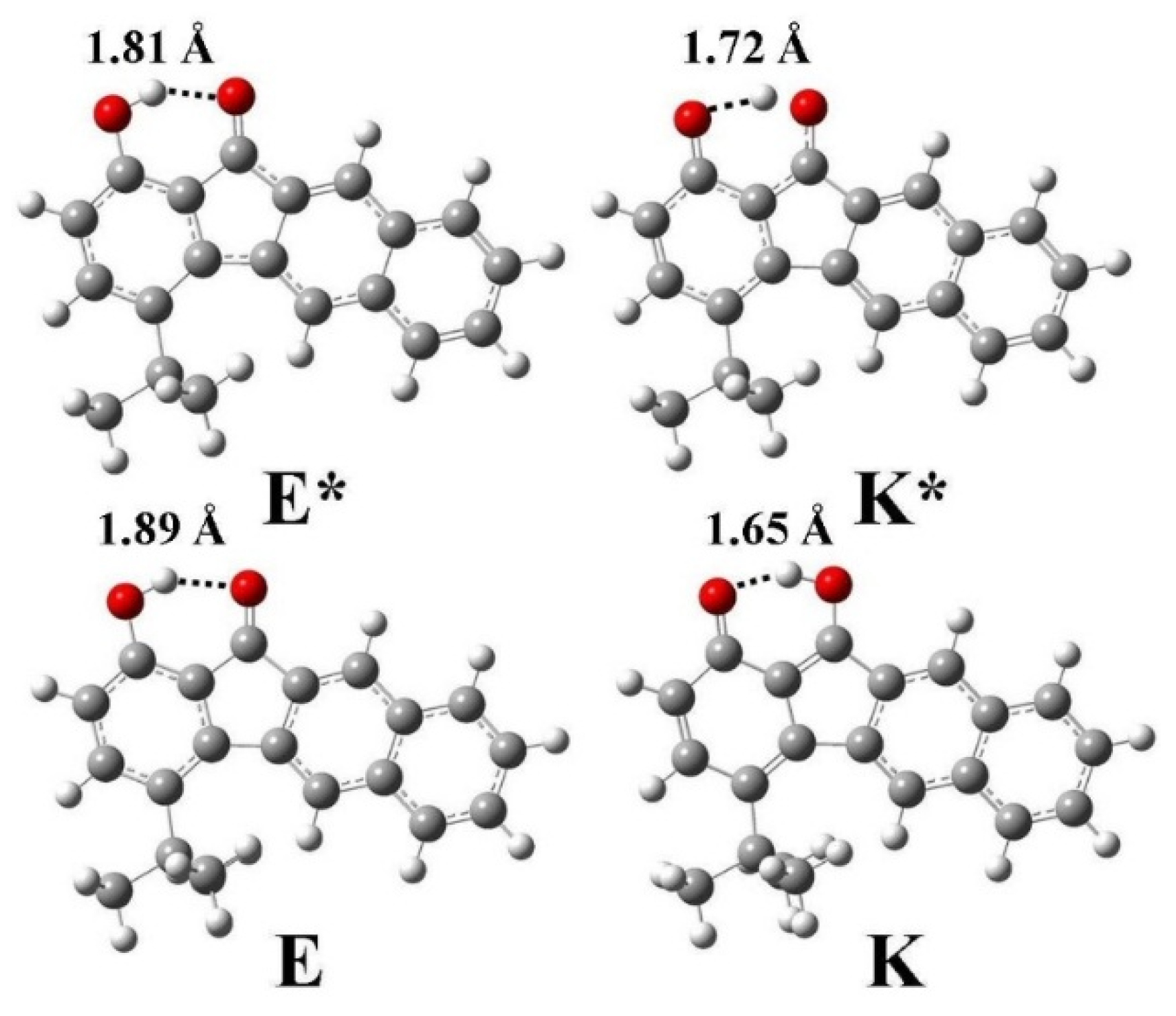
3.2. Hydrogen Bond Studies

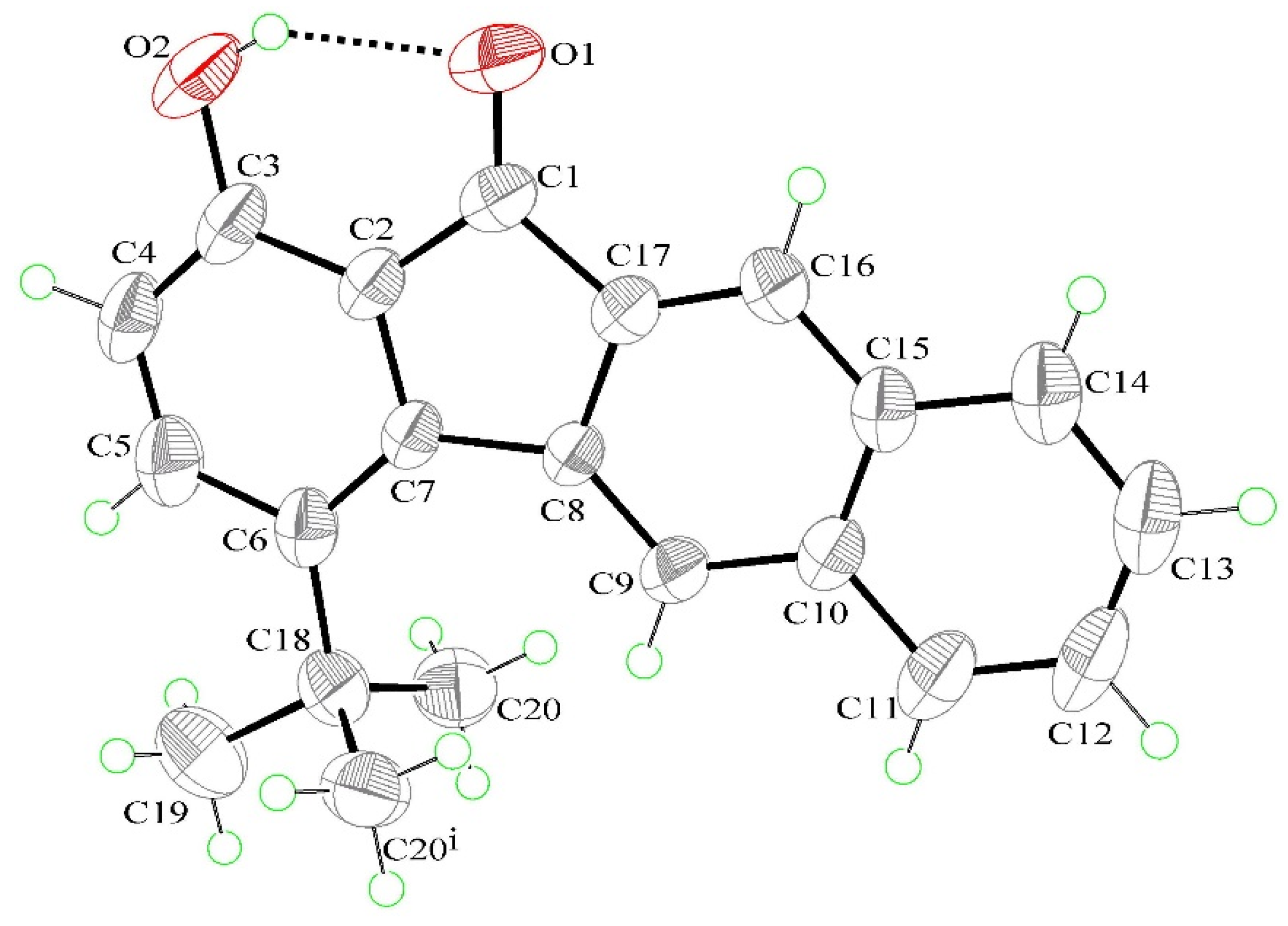
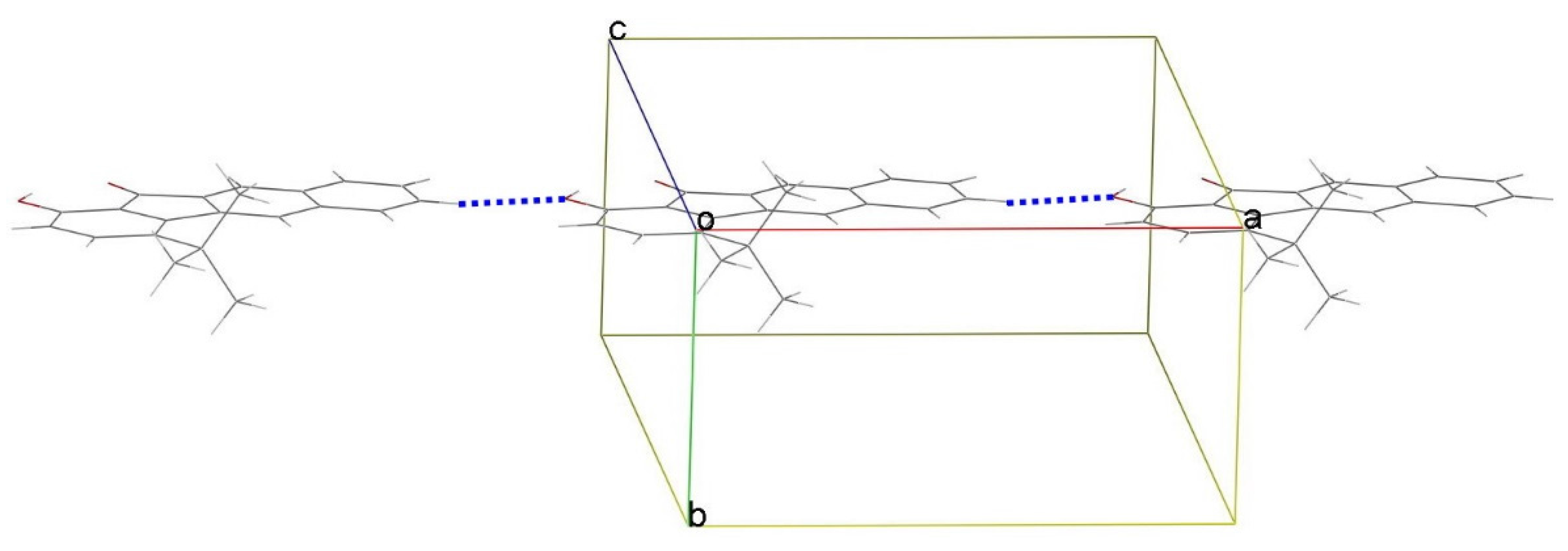
3.3. X-ray Structures

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | 1 | 2 |
|---|---|---|
| Chemical Formula | C21H17O2 | C17H10O2 |
| Formula Weight | 301.35 | 246.25 |
| Crystal System | Orthorhombic | Monoclinic |
| Space Group | Pnma | P21/c |
| a (Å) | 12.6814(6) | 12.474(2) |
| b (Å) | 7.0824(4) | 6.4401(12) |
| c (Å) | 17.4628(9) | 15.601(3) |
| α (°) | 90 | 90 |
| β (°) | 90 | 109.188(3) |
| γ (°) | 90 | 90 |
| Volume (Å3) | 1568.42(14) | 1183.6(4) |
| Z | 4 | 4 |
| Dcalc (g cm−3) | 1.276 | 1.382 |
| μ (mm−1) | 0.081 | 0.090 |
| F000 | 636 | 512 |
| Crystal Size (mm3) | 0.56 × 0.40 × 0.25 | 0.42 × 0.22 × 0.12 |
| θ range (°) | 2.83–29.16 | 1.73–26.00 |
| Index ranges | −9 ≤ h ≤ 17 | −14 ≤ h ≤ 15 |
| −9 ≤ k ≤ 9 | −7 ≤ k ≤ 7 | |
| −13 ≤ l ≤ 23 | −19 ≤ l ≤ 16 | |
| Reflections Collected | 6225 | 6331 |
| Independent Reflections (Rint) | 2013 (0.0961) | 2310 (0.0445) |
| Refinement Method on F2 | Full-matrix least-squares | Full-matrix least-squares |
| GOF on F2 | 1.055 | 1.014 |
| R1 [I > 2σ (I)] | 0.0752 | 0.0571 |
| wR2 [I > 2σ (I)] | 0.2021 | 0.1488 |
| R1 (All Data) | 0.1017 | 0.1047 |
| wR2 (All Data) | 0.2334 | 0.1877 |
| Residual (e Å−3) | 0.345 and −0.259 | 0.235 and −0.183 |

| Compound 1 | X-ray | DFT |
|---|---|---|
| Bond Lengths (Å) | ||
| O(2)-C(3) | 1.352(4) | 1.345 |
| O(1)-C(1) | 1.229(3) | 1.237 |
| C(1)-C(2) | 1.459(4) | 1.458 |
| C(2)-C(3) | 1.391(4) | 1.395 |
| C(7)-C(8) | 1.505(3) | 1.509 |
| C(8)-C(9) | 1.364(3) | 1.376 |
| C(11)-C(12) | 1.367(5) | 1.380 |
| C(18)-C(19) | 1.548(4) | 1.546 |
| Bond Angles (°) | ||
| O(1)-C(1)-C(2) | 126.0(3) | 125.9 |
| O(2)-C(3)-C(4) | 121.1(3) | 121.3 |
| C(1)-C(2)-C(3) | 124.8(3) | 123.5 |
| C(2)-C(7)-C(8) | 106.1(2) | 105.6 |
| C(9)-C(10)-C(11) | 121.1(3) | 121.1 |
| C(13)-C(14)-C(15) | 120.3(3) | 120.7 |
| Torsion Angles (°) | ||
| O(1)-C(1)-C(2)-C(3) | 0.0 | 0.0 |
| O(2)-C(3)-C(4)-C(5) | 0.0 | 0.0 |
| C(8)-C(9)-C(10)-C(11) | 0.0 | 0.0 |
| C(14)-C(15)-C(16)-C(17) | 0.0 | 0.0 |

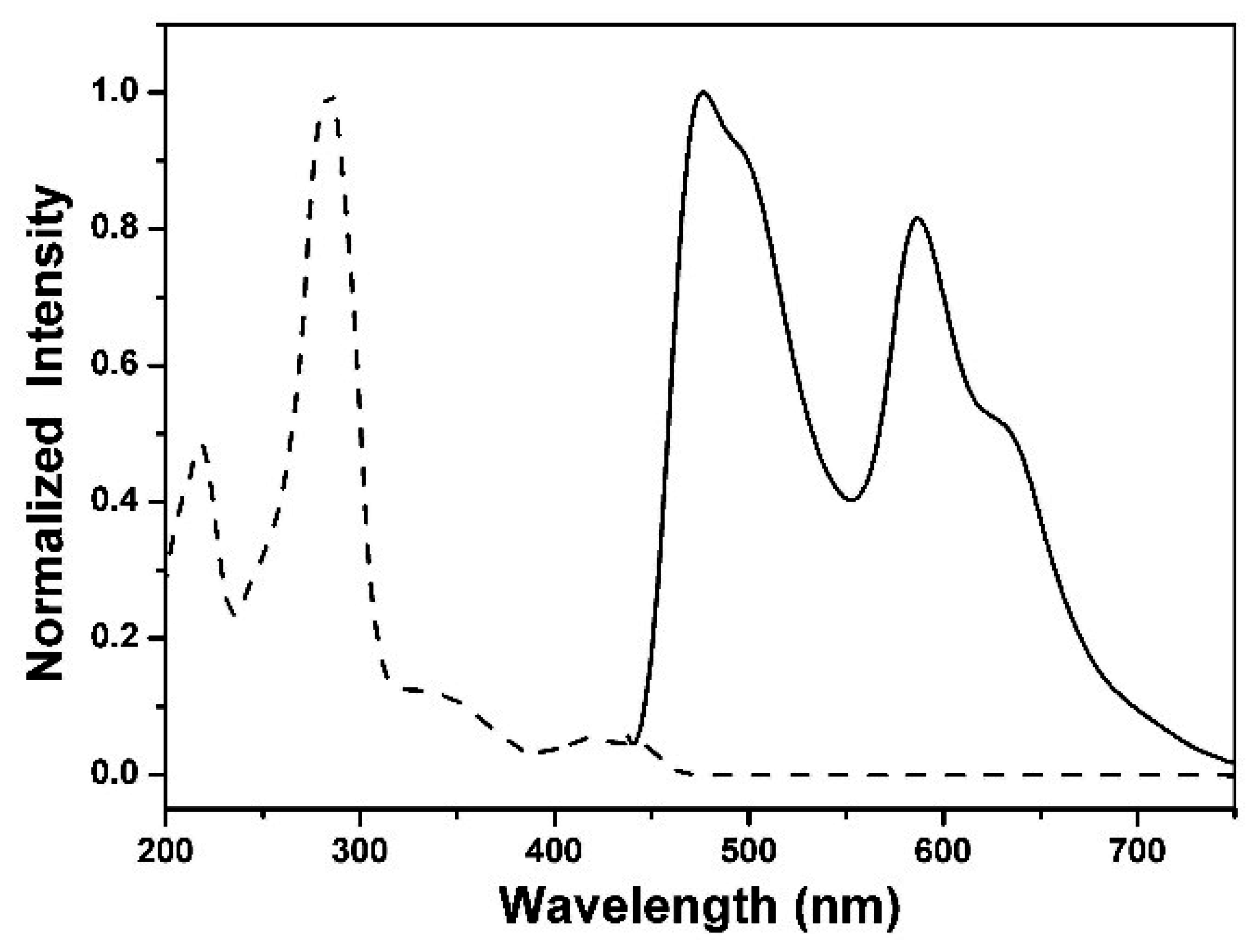
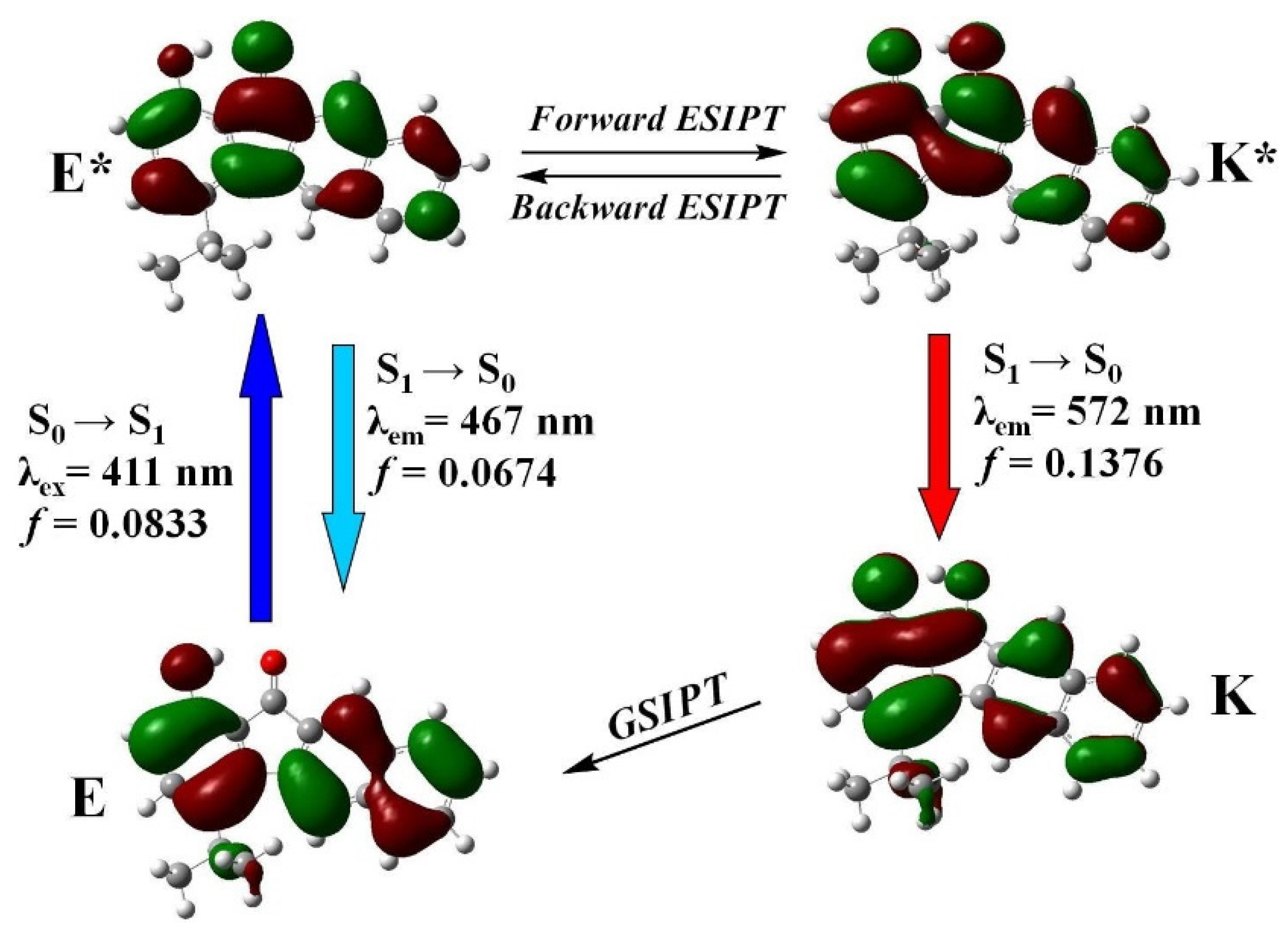
3.4. Photophysical Properties


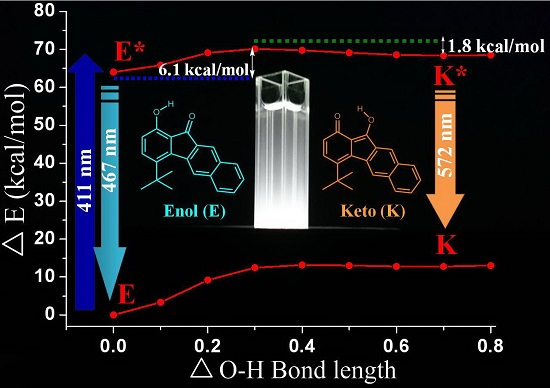
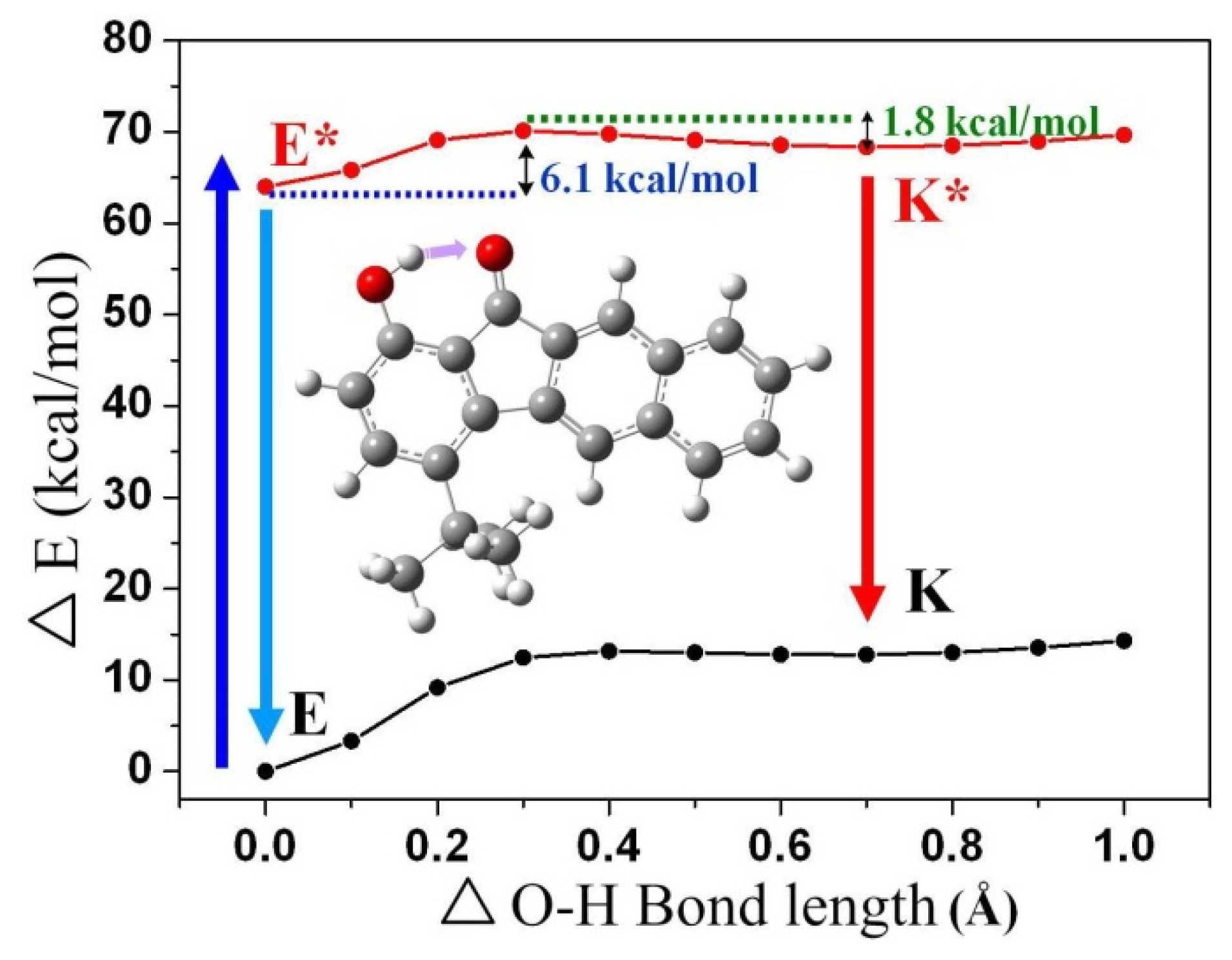
3.5. Quantum Chemistry Computation



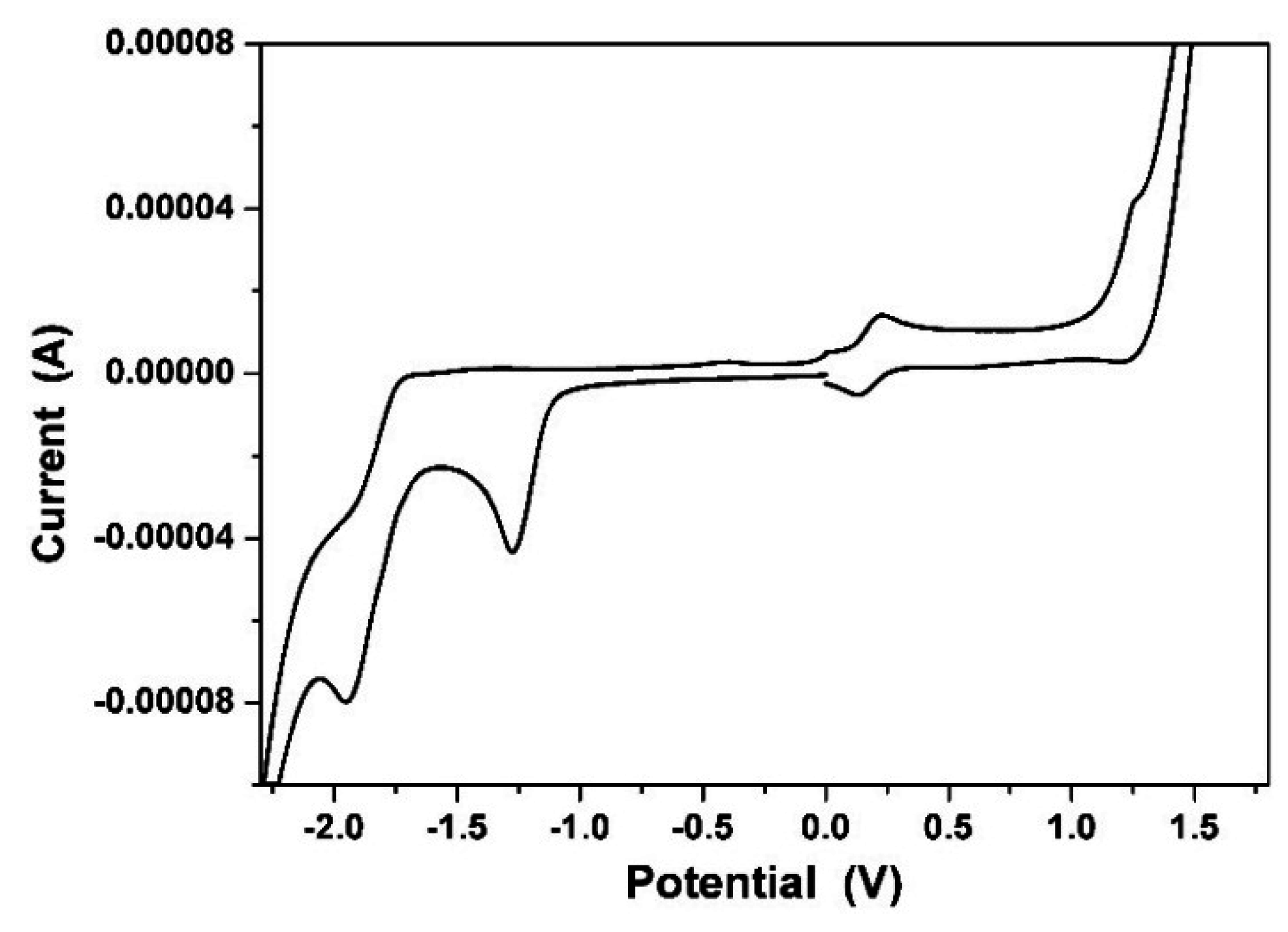
3.6. Electrochemical Properties

| Compound | λabs a | λem a | Φ b | Eg c | E(1) d | E(-1) d | EHOMO/ELUMO e | EHOMO/ELUMO f | Eg g |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 423 | 477/587 | 0.15 | 2.93 | 1.30 | −1.26 | −5.87/−2.94 | −5.91/−2.30 | 3.02 |
| 2 | 427 | 480/589 | 0.04 | 2.90 | 1.39 | −1.28 | −5.96/−3.06 | −5.98/−2.31 | 3.00 |
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Xie, L.; Chen, Y.; Wu, W.; Guo, H.; Zhao, J.; Yu, X. Fluorescent coumarin derivatives with large stokes shift, dual emission and solid state luminescent properties: An experimental and theoretical study. Dyes Pigment. 2012, 92, 1361–1369. [Google Scholar] [CrossRef]
- Yang, P.; Zhao, J.; Wu, W.; Yu, X.; Liu, Y. Accessing the long-lived triplet excited states in bodipy-conjugated 2-(2-hydroxyphenyl) benzothiazole/benzoxazoles and applications as organic triplet photosensitizers for photooxidations. J. Org. Chem. 2012, 77, 6166–6678. [Google Scholar] [CrossRef] [PubMed]
- Satam, M.A.; Raut, R.K.; Sekar, N. Fluorescent azo disperse dyes from 3-(1,3-benzothiazol-2-yl)naphthalen-2-ol and comparison with 2-naphthol analogs. Dyes Pigment. 2013, 96, 92–103. [Google Scholar] [CrossRef]
- Zhao, J.; Ji, S.; Chen, Y.; Guo, H.; Yang, P. Excited state intramolecular proton transfer (ESIPT): From principal photophysics to the development of new chromophores and applications in fluorescent molecular probes and luminescent materials. Phys. Chem. Chem. Phys. 2012, 14, 8803–8817. [Google Scholar] [CrossRef] [PubMed]
- Satam, M.A.; Raut, R.K.; Telore, R.D.; Sekar, N. Fluorescent acid azo dyes from 3-(1,3-benzothiazol-2-yl)naphthalen-2-ol and comparison with 2-naphthol analogs. Dyes Pigment. 2013, 97, 32–42. [Google Scholar] [CrossRef]
- Guo, Z.Q.; Chen, W.Q.; Duan, X.M. Seven-membered ring excited-state intramolecular proton-transfer in 2-benzamido-3-(pyridin-2-yl)acrylic acid. Dyes Pigment. 2011, 92, 619–625. [Google Scholar] [CrossRef]
- Luo, M.H.; Tsai, H.Y.; Lin, H.Y.; Fang, S.K.; Chen, K.Y. Extensive spectral tuning of the proton transfer emission from green to red via a rational derivatization of salicylideneaniline. Chin. Chem. Lett. 2012, 23, 1279–1282. [Google Scholar] [CrossRef]
- Kwon, J.E.; Park, S.; Park, S.Y. Realizing molecular pixel system for full-color fluorescence reproduction: RGB-emitting molecular mixture free from energy transfer crosstalk. J. Am. Chem. Soc. 2013, 135, 11239–11246. [Google Scholar] [CrossRef] [PubMed]
- Fang, T.C.; Tsai, H.Y.; Luo, M.H.; Chang, C.W.; Chen, K.Y. Excited-state charge coupled proton transfer reaction via the dipolar functionality of salicylideneaniline. Chin. Chem. Lett. 2013, 24, 145–148. [Google Scholar] [CrossRef]
- Khan, A.U.; Kasha, M. Mechanism of four-level laser action in solution excimer and excited-state proton-transfer cases. Proc. Natl. Acad. Sci. USA 1983, 80, 1767–1770. [Google Scholar] [CrossRef] [PubMed]
- Lins, G.O.W.; Campo, L.F.; Rodembusch, F.S.; Stefani, V. Novel ESIPT fluorescent benzazolyl-4-quinolones: Synthesis, spectroscopic characterization and photophysical properties. Dyes Pigment. 2010, 84, 114–120. [Google Scholar] [CrossRef]
- Chen, W.H.; Pang, Y. Excited-state intramolecular proton transfer in 2-(2′,6′-dihydroxyphenyl)benzoxazole: Effect of dual hydrogen bonding on the optical properties. Tetrahedron Lett. 2010, 51, 1914–1918. [Google Scholar] [CrossRef]
- Maupin, C.M.; Castillo, N.; Taraphder, S.; Tu, C.; McKenna, R.; Silverman, D.N.; Voth, G.A. Chemical rescue of enzymes: Proton transfer in mutants of human carbonic anhydrase II. J. Am. Chem. Soc. 2011, 133, 6223–6234. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.K.; Seo, J.; Kim, S.; Kwon, I.C.; Ahn, C.H.; Park, S.Y. Concentration and pH-modulated dual fluorescence in self-assembled nanoparticles of phototautomerizable biopolymeric amphiphile. Dyes Pigment. 2011, 90, 284–289. [Google Scholar] [CrossRef]
- Santosa, R.C.; Silva Faleiro, N.V.; Campo, L.F.; Scroferneker, M.L.; Corbellini, V.A.; Rodembusch, F.S.; Stefani, V. Synthesis and photophysical properties of novel succinimidyl benzazole derivatives, evaluated by Candida albicans ATCC 10231 fluorescent staining. Tetrahedron Lett. 2011, 52, 3048–3053. [Google Scholar] [CrossRef]
- Ito, Y.; Amimoto, K.; Kawato, T. Prototropic tautomerism and solid-state photochromism of N-phenyl-2-aminotropones. Dyes Pigment. 2011, 89, 319–323. [Google Scholar] [CrossRef]
- Hong, W.H.; Lin, C.C.; Hsieh, T.S.; Chang, C.C. Preparation of fluoroionophores based on diamine-salicylaldehyde derivatives. Dyes Pigment. 2012, 94, 371–379. [Google Scholar] [CrossRef]
- Huang, Q.; Yang, X.F.; Li, H. A ratiometric fluorescent probe for hydrogen sulfide based on an excited-state intramolecular proton transfer mechanism. Dyes Pigment. 2013, 99, 871–877. [Google Scholar] [CrossRef]
- Lin, W.C.; Fang, S.K.; Hu, J.W.; Tsai, H.Y.; Chen, K.Y. Ratiometric fluorescent/colorimetric cyanide-selective sensor based on excited-state intramolecular charge transfer-excited-state intramolecular proton transfer switching. Anal. Chem. 2014, 86, 4648–4652. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.J.; He, X.P.; Hu, M.; Li, Z.; Shi, X.X.; Chen, G.R. Highly optically selective and electrochemically active chemosensor for copper (II) based on triazole-linked glucosyl anthraquinone. Dyes Pigment. 2011, 88, 391–395. [Google Scholar] [CrossRef]
- Li, T.; Yang, Z.; Li, Y.; Liu, Z.; Qi, G.; Wang, B. A novel fluorescein derivative as a colorimetric chemosensor for detecting copper (II) ion. Dyes Pigment. 2011, 88, 103–108. [Google Scholar] [CrossRef]
- Ashraf, M.; Teshome, A.; Kay, A.J.; Gainsford, G.J.; Bhuiyan, M.D.H.; Asselberghs, I.; Clays, K. Synthesis and optical properties of NLO chromophores containing an indoline donor and azo linker. Dyes Pigment. 2012, 95, 455–464. [Google Scholar] [CrossRef]
- Ikeda, S.; Toganoh, M.; Easwaramoorthi, S. Synthesis and photophysical properties of N-fused tetraphenylporphyrin derivatives: Near-infrared organic Dye of [18]annulenic compounds. J. Org. Chem. 2010, 75, 8637–8649. [Google Scholar] [CrossRef] [PubMed]
- Chuang, W.-T.; Hsieh, C.-C.; Lai, C.-H.; Lai, C.-H.; Shih, C.-W.; Chen, K.-Y.; Hung, W.-Y.; Hsu, Y.-H.; Chou, P.-T. Excited-state intramolecular proton transfer molecules bearing o-hydroxy analogues of green fluorescent protein chromophore. J. Org. Chem. 2011, 76, 8189–8202. [Google Scholar] [CrossRef] [PubMed]
- Kwon, J.E.; Park, S.Y. Advanced organic optoelectronic materials: Harnessing excited-state intramolecular proton transfer (ESIPT) process. Adv. Mater. 2011, 23, 3615–3642. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Seo, J.; Kim, S.H.; Park, S.Y. Tetraphenylimidazole-based excited-state intramolecular proton-transfer molecules for highly efficient blue electroluminescence. Adv. Funct. Mater. 2008, 18, 726–731. [Google Scholar] [CrossRef]
- Kido, J.; Kimura, M.; Nagai, K. Multilayer white light-emitting organic electroluminescent device. Science 1995, 267, 1332–1334. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.-L.; Huang, S.-C. Combustion synthesis and photoluminescence properties of red-emitting CaAlSiN3:Eu2+ phosphor for white-LEDs. Materials 2014, 7, 7828–7842. [Google Scholar] [CrossRef]
- D’Andrade, B.W.; Forrest, S.R. White organic light-emitting devices for solid-state lighting. Adv. Mater. 2004, 16, 1585–1595. [Google Scholar] [CrossRef]
- Hussain, I.; Bano, N.; Hussain, S.; Soomro, Y.; Nur, O.; Willander, M. Study of the distribution of radiative defects and reabsorption of the UV in ZnO nanorods-organic hybrid white light emitting diodes (LEDs). Materials 2011, 4, 1260–1270. [Google Scholar] [CrossRef]
- Sun, Y.; Giebink, N.C.; Knanno, H.; Ma, B.; Thompson, M.E.; Forrest, S.R. Management of singlet and triplet excitons for efficient white organic light-emitting devices. Nature 2006, 440, 908–912. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Meng, G.; Zhou, Y.; Tang, H.; Zhao, J.; Wang, Z. The photoluminescent properties of new cationic iridium(III) complexes using different anions and their applications in white light-emitting diodes. Materials 2015, 8, 6105–6116. [Google Scholar] [CrossRef]
- Kanno, H.; Sun, Y.; Forrest, S.R. White organic light-emitting device based on a compound fluorescent-phosphor-sensitized-fluorescent emission layer. Appl. Phys. Lett. 2006, 89. [Google Scholar] [CrossRef] [Green Version]
- Gong, X.; Wang, S.; Moses, D.; Bazan, G.C.; Heeger, A.J. Multilayer polymer light-emitting diodes: White-light emission with high efficiency. Adv. Mater. 2005, 17, 2053–2058. [Google Scholar] [CrossRef]
- Liu, Y.; Guo, J.; Zhang, H.; Wang, Y. Highly efficient white organic electroluminescence from a double-layer device based on a boron hydroxyphenylpyridine complex. Angew. Chem. Int. Ed. 2002, 41, 182–184. [Google Scholar] [CrossRef]
- Jou, J.-H.; Kumar, S.; Agrawal, A.; Li, T.-H.; Sahoo, S. Approaches for fabricating high efficiency organic light emitting diodes. J. Mater. Chem. C 2015, 3, 2974–3002. [Google Scholar] [CrossRef]
- Yang, X.; Zhou, G.; Wong, W.-Y. Functionalization of phosphorescent emitters and their host materials by main-group elements for phosphorescent organic light-emitting devices. Chem. Soc. Rev. 2015, 44, 8484–8575. [Google Scholar] [CrossRef] [PubMed]
- Mazzeo, M.; Vitale, V.; Della Sala, F.; Anni, M.; Barbarella, G.; Favaretto, L.; Sotgiu, G.; Cingolani, R.; Gigli, G. Bright white organic light-emitting devices from a single active molecular material. Adv. Mater. 2005, 17, 34–39. [Google Scholar] [CrossRef]
- Liu, Y.; Nishiura, M.; Wang, Y.; Hou, Z. π-conjugated aromatic enynes as a single-emitting component for white electroluminescence. J. Am. Chem. Soc. 2006, 128, 5592–5593. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Kwon, J.E.; Kim, S.H.; Seo, J.; Chung, K.; Park, S.Y.; Jang, D.J.; Medina, B.M.; Gierschner, J.; Park, S.Y. A white-light-emitting molecule: Frustrated energy transfer between constituent emitting centers. J. Am. Chem. Soc. 2009, 131, 14043–14049. [Google Scholar] [CrossRef] [PubMed]
- Tang, K.-C.; Chang, M.-J.; Lin, T.-Y.; Pan, H.-A.; Fang, T.-C.; Chen, K.-Y.; Hung, W.-Y.; Hsu, Y.-H.; Chou, P.-T. Fine tuning the energetics of excited-state intramolecular proton transfer (ESIPT): White light generation in a single ESIPT system. J. Am. Chem. Soc. 2011, 133, 17738–17745. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Seo, J.; Jung, H.K.; Kim, J.J.; Park, S.Y. White luminescence from polymer thin films containing excited-state intramolecular proton-transfer dyes. Adv. Mater. 2005, 17, 2077–2082. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXS97, A Program for Automatic Solution of Crystal Structure; University of Göttingen: Göttingen, Germany, 1997. [Google Scholar]
- Sheldrick, G.M. SHELX97, A Program for Crystal Structure Refinement; University of Göttingen: Göttingen, Germany, 1997. [Google Scholar]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy; Springer: New York, NY, USA, 2006. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A.; Vreven, T., Jr.; Kudin, K.N.; Burant, J.C.; et al. Gaussian 03; Gaussian, Inc.: Pittsburgh, PA, USA, 2003. [Google Scholar]
- Koné, M.; Illien, B.; Graton, J.; Laurenc, C. B3LYP and MP2 calculations of the enthalpies of hydrogen-bonded complexes of methanol with neutral bases and anions: Comparison with experimental data. J. Phys. Chem. A 2005, 109, 11907–11913. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, T. Relation between hydroxyl proton chemical shifts and torsional frequencies in some ortho-substituted phenol derivatives. J. Phys. Chem. 1975, 79, 1888–1890. [Google Scholar] [CrossRef]
- Chen, K.-Y.; Cheng, Y.-M.; Lai, C.-H.; Hsu, C.-C.; Ho, M.-L.; Lee, G.-H.; Chou, P.-T. Ortho green fluorescence protein synthetic chromophore; excited-state intramolecular proton transfer via a seven-membered-ring hydrogen-bonding system. J. Am. Chem. Soc. 2007, 129, 4534–4535. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.-Y.; Hsieh, C.-C.; Cheng, Y.-M.; Lai, C.-H.; Chou, P.-T. Extensive spectral tuning of the proton transfer emission from 550 to 675 nm via a rational derivatization of 10-hydroxybenzo[h]quinoline. Chem. Commun. 2006, 42, 4395–4397. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.-Y.; Tsai, H.-Y.; Lin, W.-C.; Chu, H.-H.; Weng, Y.-C.; Chan, C.-C. ESIPT fluorescent dyes with adjustable optical properties: Substituent and conjugation effects. J. Lumin. 2014, 154, 168–177. [Google Scholar] [CrossRef]
- Chen, K.-Y.; Hu, J.-W. Excited-state charge coupled proton transfer reaction in dipole-functionalized salicylideneaniline. J. Lumin. 2015, 159, 171–177. [Google Scholar] [CrossRef]
- Chen, K.-Y.; Tsai, H.-Y. Synthesis, X-ray structure, spectroscopic properties and DFT studies of a novel Schiff base. Int. J. Mol. Sci. 2014, 15, 18706–18724. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Yang, D.; Ren, B.; Wang, D. A TDDFT study on the excited-state intramolecular proton transfer (ESIPT): Excited-state equilibrium induced by electron density swing. J. Fluoresc. 2013, 23, 761–766. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, J.-W.; Wu, Y.-H.; Tsai, H.-Y.; Chen, K.-Y. Synthesis, X-ray Structure, Optical, and Electrochemical Properties of a White-Light-Emitting Molecule. Materials 2016, 9, 48. https://doi.org/10.3390/ma9010048
Hu J-W, Wu Y-H, Tsai H-Y, Chen K-Y. Synthesis, X-ray Structure, Optical, and Electrochemical Properties of a White-Light-Emitting Molecule. Materials. 2016; 9(1):48. https://doi.org/10.3390/ma9010048
Chicago/Turabian StyleHu, Jiun-Wei, Ying-Hsuan Wu, Hsing-Yang Tsai, and Kew-Yu Chen. 2016. "Synthesis, X-ray Structure, Optical, and Electrochemical Properties of a White-Light-Emitting Molecule" Materials 9, no. 1: 48. https://doi.org/10.3390/ma9010048
APA StyleHu, J. -W., Wu, Y. -H., Tsai, H. -Y., & Chen, K. -Y. (2016). Synthesis, X-ray Structure, Optical, and Electrochemical Properties of a White-Light-Emitting Molecule. Materials, 9(1), 48. https://doi.org/10.3390/ma9010048