Genomic Characterization of Sixteen Yersinia enterocolitica-Infecting Podoviruses of Pig Origin



Abstract
:
1. Introduction
2. Materials and Methods
2.1. Bacterial Strains, Phages, Media and Growth Conditions
2.2. Electron Microscopy
2.3. Isolation of Phage DNA
2.4. Genome Sequencing, Assembly and Bioinformatics
2.5. Determination of the Genome Ends
2.6. Efficiency of Plating
2.7. Phage Inhibition Assay
2.8. One-Step Growth Curve Experiment
2.9. Thermal, pH and Solvent Stability Tests
2.10. Nucleotide Sequence Accession Numbers
3. Results and Discussion
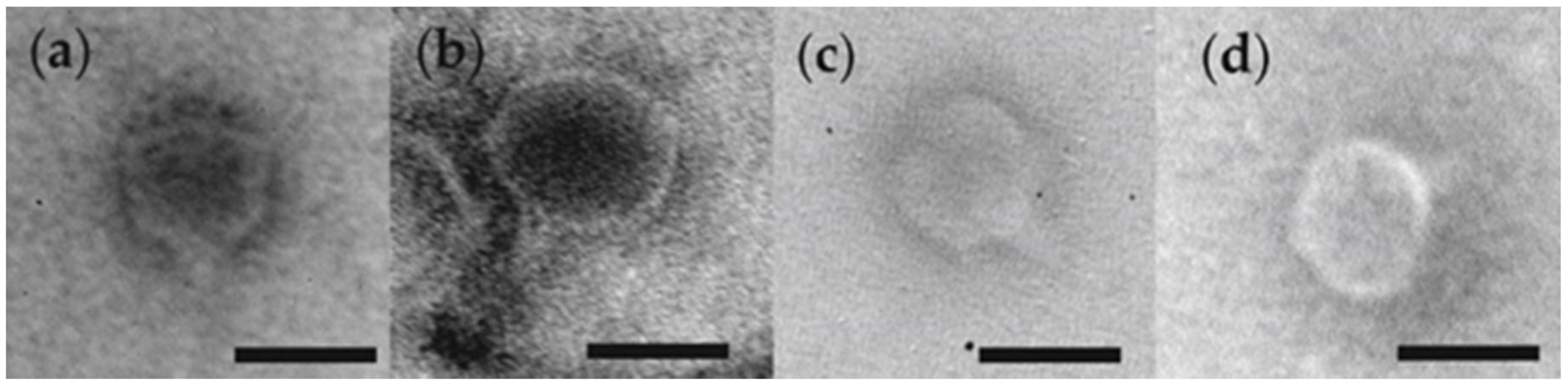
3.1. Phage Morphology
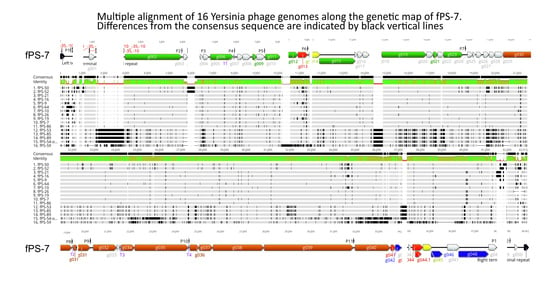
3.2. Analysis of the fPS-phage Genomes
3.2.1. The Phylogenetic Position of the fPS-phages
3.2.2. The Genome Organization of the fPS-phages
The Early Genes
The Middle Genes
The Late Genes
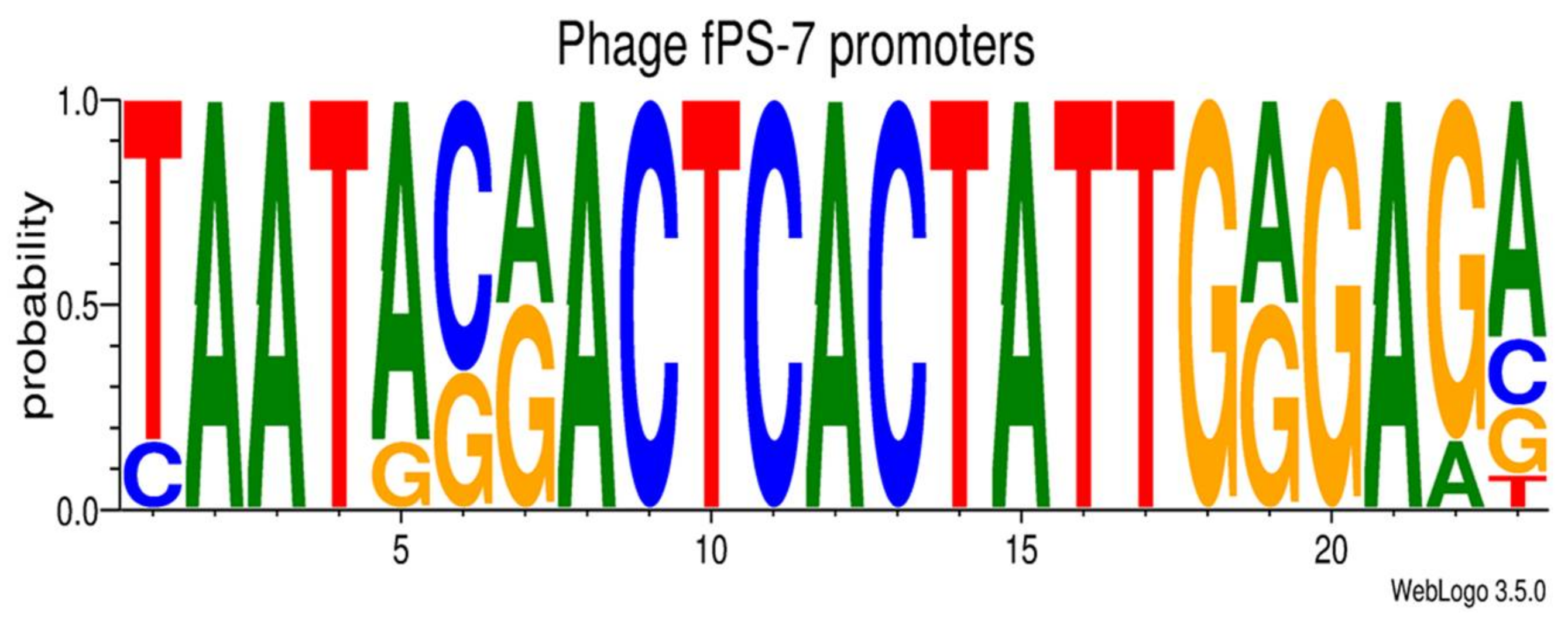
3.2.3. Transcriptional Sequences
3.2.4. The Terminal Repeats (TR)
3.2.5. Microevolution of the fPS-phages
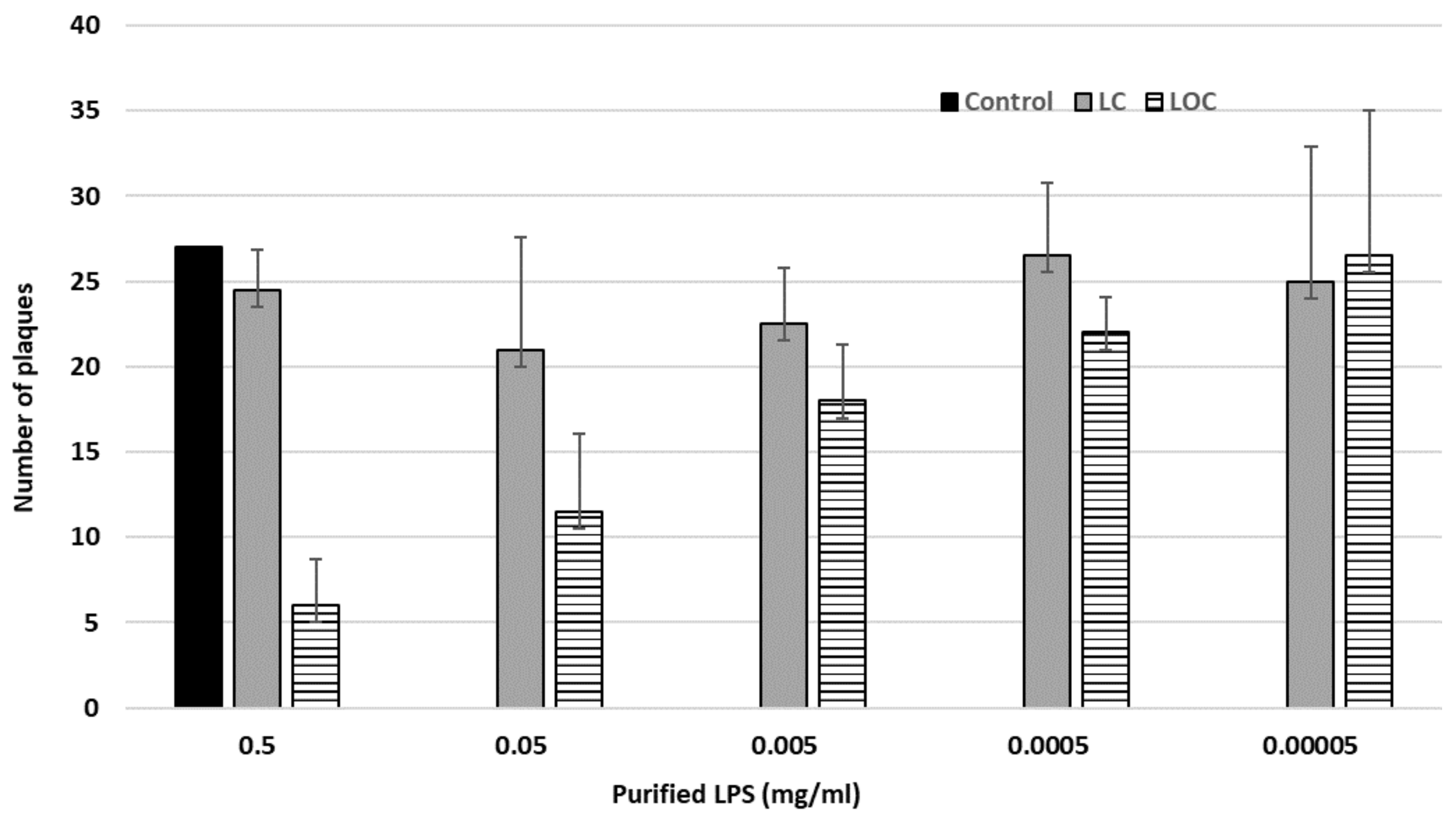
3.3. Characterization of the Phage Receptors
The Phage fPS-7 Uses the O-ag as a Receptor
3.4. One-Step Growth Curves
3.5. Thermal, pH and Solvent Stability of the Phages
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bottone, E.J. Yersinia enterocolitica: Overview and epidemiologic correlates. Microbes Infect. 1999, 1, 323–333. [Google Scholar] [CrossRef]
- Eurosurveillance Editorial Team. The European Union Summary Report on Trends and Sources of Zoonoses, Zoonotic Agents and Food-Borne Outbreaks in 2010; Eurosurveillance: Stockholm, Sweden, 2012. [Google Scholar]
- Fredriksson-Ahomaa, M.; Stolle, A.; Korkeala, H. Molecular epidemiology of Yersinia enterocolitica infections. FEMS Immunol. Med. Microbiol. 2006, 47, 315–329. [Google Scholar] [CrossRef] [PubMed]
- Pham, J.N.; Bell, S.M.; Hardy, M.J.; Martin, L.; Guiyoule, A.; Carniel, E. Susceptibility to β-lactam agents of Yersinia enterocolitica biotype 4, serotype O3 isolated in various parts of the world. J. Med. Microbiol. 1995, 43, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Bhaduri, S.; Wesley, I.; Richards, H.; Draughon, A.; Wallace, M. Clonality and antibiotic susceptibility of Yersinia enterocolitica isolated from U.S. market weight hogs. Foodborne Pathog. Dis. 2009, 6, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Capilla, S.; Goni, P.; Rubio, M.C.; Castillo, J.; Millan, L.; Cerda, P.; Sahagun, J.; Pitart, C.; Beltran, A.; Gomez-Lus, R. Epidemiological study of resistance to nalidixic acid and other antibiotics in clinical Yersinia enterocolitica O:3 isolates. J. Clin. Microbiol. 2003, 41, 4876–4878. [Google Scholar] [CrossRef] [PubMed]
- Falcao, J.P.; Falcao, D.P.; Pitondo-Silva, A.; Malaspina, A.C.; Brocchi, M. Molecular typing and virulence markers of Yersinia enterocolitica strains from human, animal and food origins isolated between 1968 and 2000 in Brazil. J. Med. Microbiol. 2006, 55, 1539–1548. [Google Scholar] [CrossRef] [PubMed]
- Comeau, A.M.; Hatfull, G.F.; Krisch, H.M.; Lindell, D.; Mann, N.H.; Prangishvili, D. Exploring the prokaryotic virosphere. Res. Microbiol. 2008, 159, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Sulakvelidze, A.; Alavidze, Z.; Morris, J.G., Jr. Bacteriophage therapy. Antimicrob. Agents Chemother. 2001, 45, 649–659. [Google Scholar] [CrossRef] [PubMed]
- Golkar, Z.; Bagasra, O.; Pace, D.G. Bacteriophage therapy: A potential solution for the antibiotic resistance crisis. J. Infect. Dev. Ctries 2014, 8, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Abedon, S.T. Information Phage Therapy Research Should Report. Pharmaceuticals 2017, 10, 43. [Google Scholar] [CrossRef] [PubMed]
- Pajunen, M.; Kiljunen, S.; Skurnik, M. Bacteriophage phiYeO3-12, specific for Yersinia enterocolitica serotype O:3, is related to coliphages T3 and T7. J. Bacteriol. 2000, 182, 5114–5120. [Google Scholar] [CrossRef] [PubMed]
- Pajunen, M.I.; Kiljunen, S.J.; Soderholm, M.E.; Skurnik, M. Complete genomic sequence of the lytic bacteriophage phiYeO3-12 of Yersinia enterocolitica serotype O:3. J. Bacteriol. 2001, 183, 1928–1937. [Google Scholar] [CrossRef] [PubMed]
- Kiljunen, S.; Vilen, H.; Pajunen, M.; Savilahti, H.; Skurnik, M. Nonessential genes of phage phiYeO3-12 include genes involved in adaptation to growth on Yersinia enterocolitica serotype O:3. J. Bacteriol. 2005, 187, 1405–1414. [Google Scholar] [CrossRef] [PubMed]
- Leon-Velarde, C.G.; Kropinski, A.M.; Chen, S.; Abbasifar, A.; Griffiths, M.W.; Odumeru, J.A. Complete genome sequence of bacteriophage vB_YenP_AP5 which infects Yersinia enterocolitica of serotype O:3. Virol. J. 2014, 11, 188. [Google Scholar] [CrossRef] [PubMed]
- Kiljunen, S.; Hakala, K.; Pinta, E.; Huttunen, S.; Pluta, P.; Gador, A.; Lonnberg, H.; Skurnik, M. Yersiniophage phiR1-37 is a tailed bacteriophage having a 270 kb DNA genome with thymidine replaced by deoxyuridine. Microbiology 2005, 151, 4093–4102. [Google Scholar] [CrossRef] [PubMed]
- Schwudke, D.; Ergin, A.; Michael, K.; Volkmar, S.; Appel, B.; Knabner, D.; Konietzny, A.; Strauch, E. Broad-host-range Yersinia phage PY100: Genome sequence, proteome analysis of virions, and DNA packaging strategy. J. Bacteriol. 2008, 190, 332–342. [Google Scholar] [CrossRef] [PubMed]
- Leon-Velarde, C.G.; Happonen, L.; Pajunen, M.; Leskinen, K.; Kropinski, A.M.; Mattinen, L.; Rajtor, M.; Zur, J.; Smith, D.; Chen, S.; et al. Yersinia enterocolitica-Specific Infection by Bacteriophages TG1 and varphiR1-RT Is Dependent on Temperature-Regulated Expression of the Phage Host Receptor OmpF. Appl. Environ. Microbiol. 2016, 82, 5340–5353. [Google Scholar] [CrossRef] [PubMed]
- Salem, M.; Virtanen, S.; Korkeala, H.; Skurnik, M. Isolation and characterization of Yersinia-specific bacteriophages from pig stools in Finland. J. Appl. Microbiol. 2015, 118, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Skurnik, M. Lack of correlation between the presence of plasmids and fimbriae in Yersinia enterocolitica and Yersinia pseudotuberculosis. J. Appl. Bacteriol. 1984, 56, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Al-Hendy, A.; Toivanen, P.; Skurnik, M. Lipopolysaccharide O side chain of Yersinia enterocolitica O:3 is an essential virulence factor in an orally infected murine model. Infect. Immun. 1992, 60, 870–875. [Google Scholar] [PubMed]
- Noszczynska, M.; Kasperkiewicz, K.; Duda, K.A.; Podhorodecka, J.; Rabsztyn, K.; Gwizdala, K.; Swierzko, A.S.; Radziejewska-Lebrecht, J.; Holst, O.; Skurnik, M. Serological characterization of the enterobacterial common antigen substitution of the lipopolysaccharide of Yersinia enterocolitica O:3. Microbiology 2015, 161, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Biedzka-Sarek, M.; Venho, R.; Skurnik, M. Role of YadA, Ail, and Lipopolysaccharide in Serum Resistance of Yersinia enterocolitica Serotype O:3. Infect. Immun. 2005, 73, 2232–2244. [Google Scholar] [CrossRef] [PubMed]
- Rabsztyn, K.; Kasperkiewicz, K.; Duda, K.A.; Li, C.M.; Lukasik, M.; Radziejewska-Lebrecht, J.; Skurnik, M. Characterization of anti-ECA antibodies in rabbit antiserum against rough Yersinia enterocolitica O:3. Biochemistry 2011, 76, 832–839. [Google Scholar] [CrossRef] [PubMed]
- FIMM Sequencing Unit. Available online: https://www.fimm.fi/en/services/technology-centre/sequencing (accessed on 15 October 2015).
- Coil, D.; Jospin, G.; Darling, A.E. A5-miseq: An updated pipeline to assemble microbial genomes from Illumina MiSeq data. Bioinformatics 2015, 31, 587–589. [Google Scholar] [CrossRef] [PubMed]
- CSC-IT Center for Science. Available online: https://www.csc.fi/ (accessed on 20 September 2017).
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Chipster. Available online: https://chipster.csc.fi/ (accessed on 25 September 2017).
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid annotations using subsystems technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef] [PubMed]
- Carver, T.; Harris, S.R.; Berriman, M.; Parkhill, J.; McQuillan, J.A. Artemis: An integrated platform for visualization and analysis of high-throughput sequence-based experimental data. Bioinformatics 2012, 28, 464–469. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Alva, V.; Nam, S.Z.; Soding, J.; Lupas, A.N. The MPI bioinformatics Toolkit as an integrative platform for advanced protein sequence and structure analysis. Nucleic Acids Res. 2016, 44, W410–W415. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Lavigne, R.; Sun, W.D.; Volckaert, G. PHIRE, a deterministic approach to reveal regulatory elements in bacteriophage genomes. Bioinformatics 2004, 20, 629–635. [Google Scholar] [CrossRef] [PubMed]
- Crooks, G.E.; Hon, G.; Chandonia, J.M.; Brenner, S.E. WebLogo: A sequence logo generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef] [PubMed]
- Solovyev, V.; Salamov, A. Automatic annotation of microbial genomes and metagenomic sequences. In Metagenomics and Its Applications in Agriculture, Biomedicine and Environmental Studies; Li, R.W., Ed.; Nova Science Publishers: Hauppauge, NY, USA, 2011; pp. 61–78. [Google Scholar]
- Dereeper, A.; Guignon, V.; Blanc, G.; Audic, S.; Buffet, S.; Chevenet, F.; Dufayard, J.F.; Guindon, S.; Lefort, V.; Lescot, M.; et al. Phylogeny.fr: Robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 2008, 36, W465–W469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meier-Kolthoff, J.P.; Goeker, M. VICTOR: Genome-based Phylogeny and Classification of Prokaryotic Viruses. Bioinformatics 2017, 33, 3396–3404. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.P.; Gong, T.; Jost, G.; Liu, W.H.; Ye, D.Z.; Luo, Z.H. Isolation and characterization of five lytic bacteriophages infecting a Vibrio strain closely related to Vibrio owensii. FEMS Microbiol. Lett. 2013, 348, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Birge, E.A. Bacterial and Bacteriophage Genetics: An introduction, 2nd ed.; Springer: New York, NY, USA, 1988. [Google Scholar]
- Kropinski, A.M. Practical advice on the one-step growth curve. In Bacteriophages: Methods and Protocols; Clokie, M.R.J., Kropinski, A.M., Lavigne, R., Eds.; Humana Press: New York, NY, USA, 2018; Volume 3, pp. 41–47. [Google Scholar]
- Park, M.; Lee, J.H.; Shin, H.; Kim, M.; Choi, J.; Kang, D.H.; Heu, S.; Ryu, S. Characterization and comparative genomic analysis of a novel bacteriophage, SFP10, simultaneously inhibiting both Salmonella enterica and Escherichia coli O157:H7. Appl. Environ. Microbiol. 2012, 78, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Verma, V.; Harjai, K.; Chhibber, S. Characterization of a T7-like lytic bacteriophage of Klebsiella pneumoniae B5055: A potential therapeutic agent. Curr. Microbiol. 2009, 59, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Jurczak-Kurek, A.; Gasior, T.; Nejman-Falenczyk, B.; Bloch, S.; Dydecka, A.; Topka, G.; Necel, A.; Jakubowska-Deredas, M.; Narajczyk, M.; Richert, M.; et al. Biodiversity of bacteriophages: Morphological and biological properties of a large group of phages isolated from urban sewage. Sci. Rep. 2016, 6, 34338. [Google Scholar] [CrossRef] [PubMed]
- Maniloff, J.; Ackermann, H.W. Taxonomy of bacterial viruses: Establishment of tailed virus genera and the order Caudovirales. Arch. Virol. 1998, 143, 2051–2063. [Google Scholar] [CrossRef] [PubMed]
- Brenner, D.J.; Ursing, J.; Bercovier, H.; Steigerwalt, A.G.; Fanning, G.R.; Alonso, J.M.; Mollaret, H.H. Deoxyribonucleic acid relatedness in Yersinia enterocolitica and Yersinia enterocolitica-like organisms. Curr. Microbiol. 1980, 4, 195–200. [Google Scholar] [CrossRef]
- Sharp, P.M.; Rogers, M.S.; McConnell, D.J. Selection pressures on codon usage in the complete genome of bacteriophage T7. J. Mol. Evol. 1984, 21, 150–160. [Google Scholar] [CrossRef] [PubMed]
- Molineux, I.J. The T7 Group. In The Bacteriophages, 2nd ed.; Calendar, R.L., Ed.; Oxford University: Oxford, UK, 2006; pp. 277–301. [Google Scholar]
- Figueras, M.J.; Beaz-Hidalgo, R.; Hossain, M.J.; Liles, M.R. Taxonomic affiliation of new genomes should be verified using average nucleotide identity and multilocus phylogenetic analysis. Genome Announc. 2014, 2. [Google Scholar] [CrossRef] [PubMed]
- Garcia, L.R.; Molineux, I.J. Translocation and specific cleavage of bacteriophage T7 DNA in vivo by EcoKI. Proc. Natl. Acad. Sci. USA 1999, 96, 12430–12435. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.; Pulido-Cid, M.; Chagoyen, M.; Arranz, R.; Gonzalez-Garcia, V.A.; Garcia-Doval, C.; Caston, J.R.; Valpuesta, J.M.; van Raaij, M.J.; Martin-Benito, J.; et al. Structural characterization of the bacteriophage T7 tail machinery. J. Biol. Chem. 2013, 288, 26290–26299. [Google Scholar] [CrossRef] [PubMed]
- Dunn, J.J.; Studier, F.W. Complete nucleotide sequence of bacteriophage T7 DNA and the locations of T7 genetic elements. J. Mol. Biol. 1983, 166, 477–535. [Google Scholar] [CrossRef]
- Casjens, S.R.; Gilcrease, E.B. Determining DNA packaging strategy by analysis of the termini of the chromosomes in tailed-bacteriophage virions. Methods Mol. Biol. 2009, 502, 91–111. [Google Scholar] [PubMed]
- Snyder, L.; Peters, J.E.; Henkin, T.M.; Champness, W. Molecular Genetics of Bacteria, 4th ed.; ASM Press: Washington, DC, USA, 2013; pp. 265–321. [Google Scholar]
- Fujisawa, H.; Morita, M. Phage DNA packaging. Genes Cells 1997, 2, 537–545. [Google Scholar] [CrossRef] [PubMed]
- Skurnik, M.; Venho, R.; Toivanen, P.; Al-Hendy, A. A novel locus of Yersinia enterocolitica serotype O:3 involved in lipopolysaccharide outer core biosynthesis. Mol. Microbiol. 1995, 17, 575–594. [Google Scholar] [CrossRef] [PubMed]
- al-Hendy, A.; Toivanen, P.; Skurnik, M. Expression cloning of Yersinia enterocolitica O:3 rfb gene cluster in Escherichia coli K12. Microb. Pathog. 1991, 10, 47–59. [Google Scholar] [CrossRef]
- Kiljunen, S.; Datta, N.; Dentovskaya, S.V.; Anisimov, A.P.; Knirel, Y.A.; Bengoechea, J.A.; Holst, O.; Skurnik, M. Identification of the lipopolysaccharide core of Yersinia pestis and Yersinia pseudotuberculosis as the receptor for bacteriophage phiA1122. J. Bacteriol. 2011, 193, 4963–4972. [Google Scholar] [CrossRef] [PubMed]
- Skurnik, M. Yersinia surface structures and bacteriophages. Adv. Exp. Med. Biol. 2012, 954, 293–301. [Google Scholar] [PubMed]
- Ceyssens, P.J.; Lavigne, R.; Mattheus, W.; Chibeu, A.; Hertveldt, K.; Mast, J.; Robben, J.; Volckaert, G. Genomic analysis of Pseudomonas aeruginosa phages LKD16 and LKA1: Establishment of the phiKMV subgroup within the T7 supergroup. J. Bacteriol. 2006, 188, 6924–6931. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bacterial Strain | Comments | Reference/Source |
|---|---|---|
| 6471/76 (YeO3) | Serotype O:3, wild type. Human stool isolate | [20] |
| 6471/76-c (YeO3-c) | Virulence plasmid-cured derivative of 6471/76 | [20] |
| YeO3-R1 | Spontaneous rough derivative of YeO3-c | [21] |
| YeO3-R1-M164 | waaF::Cat-Mu. derivative of YeO3-R1. ClmR | [22] |
| YeO3-R1-M196 | galU::Cat-Mu derivative of YeO3-R1. ClmR | [22] |
| YeO3-R1-M205 | hldE::Cat-Mu derivative of YeO3-R1. ClmR | [22] |
| YeO3-c-OC | ∆(wzx-wbcQ), derivative of 6471/76-c, a virulence plasmid cured derivative of 6471/76 | [23] |
| YeO3-c-OCR | Spontaneous rough derivative of YeO3-c-OC | [23] |
| YeO3-c-OCR-ECA | Δ(wzx-wbcQ) Δ(wzzE–wzyE). OPS-, outer core- and ECA-negative derivative of 6471/76-c, KmR | [24] |
| Phage | Farm | Genome Size (bp) | GC Content (%) | ORFs (n) | Terminal Repeat (bp) | Promoters (n) | Terminators (n) | Accession Number |
|---|---|---|---|---|---|---|---|---|
| fPS-7 | 3 | 38,966 | 45.6 | 51 | 200 | 12 | 4 | LT961840 |
| fPS-9 | 3 | 39,034 | 45.6 | 52 | 200 | 13 | 5 | LT960606 |
| fPS-10 | 3 | 39,179 | 45.5 | 51 | 202 | 12 | 4 | LT962907 |
| fPS-16 | 3 | 39,227 | 45.5 | 51 | 200 | 12 | 4 | LT962906 |
| fPS-19 | 3 | 38,938 | 45.6 | 51 | 200 | 12 | 4 | LT961838 |
| fPS-21 | 3 | 39,180 | 45.5 | 51 | 202 | 12 | 4 | LT961844 |
| fPS-26 | 5 | 38,792 | 45.7 | 51 | 205 | 12 | 4 | LT961836 |
| fPS-50 | 7 | 39,764 | 45.5 | 50 | 224 | 12 | 4 | LT961843 |
| fPS-52 | 7 | 39,888 | 45.4 | 50 | 224 | 12 | 4 | LT961837 |
| fPS-53 | 7 | 40,451 | 45.4 | 50 | 196 | 11 | 3 | LT962379 |
| fPS-54-ocr | 7 | 40,074 | 45.5 | 49 | 200 | 11 | 4 | LT962475 |
| fPS-59 | 21 | 38,391 | 45.7 | 47 | 190 | 11 | 4 | LT961845 |
| fPS-64 | 5 | 39,326 | 45.5 | 50 | 204 | 12 | 4 | LT961846 |
| fPS-85 | 26 | 40,429 | 45.4 | 50 | 196 | 11 | 3 | LT962380 |
| fPS-86 | 28 | 39,024 | 45.6 | 51 | 215 | 12 | 4 | LT961842 |
| fPS-89 | 25 | 40,405 | 45.4 | 50 | 195 | 11 | 3 | LT961841 |
| Primer | Primer Sequence (5′-3′) |
|---|---|
| fPS-7-F | CCATAGGCCCTCTCAGTCAT |
| fPS-7-R | CAACCTCGTGATGTCTTACCG |
| flic-F3 | TCAACCATCACCAACCTGAA |
| flic-R3 | TCTTTTGCGCTGTTGATACG |
| flic-F4 | GGATGAGCCTGCCGATAATA |
| Box | Description of Differences | Consequence |
|---|---|---|
| 1 | 10 bp repeats | Differences in the length of left terminal repeat (TR) (also valid for right TR) |
| 2 | 12 bp repeats & poly-C tracks | Differences in the length of left TR (also valid for right TR) |
| 3 | Between 11 and 34 repeats of different variations (Table S19) | Different distances between the left TR and phage promoter P1 |
| 4 | 3–5 repeats of 28 bp | Variation in Group Ib phages on promoter P1 left flanking regions |
| 5 | 4–5 repeats of 23 bp | Variation in Group Ib phages on promoter P1 right flanking regions |
| 6 | 1380 bp insertion | Gene g002 of Group II that is absent from Group I and III phages |
| 7 | 274 bp insertion in Group Ia, and 274 + 342 (=616 bp) insertion in Group Ib genomes | Two variants of g003 in Group Ia and Ib genomes. The gene is absent from Groups II and III |
| 8 | 303 bp insertion | g012 of fPS-59, absent in Group I and II |
| 9 | 94 bp insertion | May encode a 30 amino acid long polypeptide in fPS-59 |
| 10 | Poly-G7 to G13 stretch | Part of ribosomal binding site (TAAGG) |
| 11 | 422 bp insertion | Extra gene in Group II and III phages |
| 12 | 136 bp fragment replacing a 127 bp fragment | The g022 in Group II phages has different 5′-end resulting in different N-terminal sequence of 15 amino acids. |
| 13 | 140 bp region | A pseudogene in Groups Ib, II and III corresponding to Group Ia gene g027 |
| 14 | 6 or 21 bp deletions and short duplications within a 40 bp GC-rich stretch | In frame deletions and substitutions in Group Ia gene g028 homologs in corresponding Groups Ib, II and III genes |
| 15 & 16 | 386 bp deletion | The Group I gene g029 is missing from both fPS-54-ocr and fPS-59 |
| 17 | Poly-T7 to T9 stretch | Downstream of Rho-independent terminator |
| 18 | Variable region | 3′-thirds of the genes encoding tail fiber protein in fPS-54-ocr and fPS-59 are highly divergent from the others receptor binding domains |
| 19 | 207 bp deletion | Group I g043 is truncated and fPS-59 lacks the gene |
| 20 | 23 bp duplication | Alters the 3′-end frame of the Group II phage gene thereby altering the last eight codons |
| 21 | 1–5 copies of an 80 bp repeat | Noncoding region downstream of phage promoter P12 |
| 22 | Five different repeat sequences of 10–22 bp in size | Variability in length of the right TR flanking region (see also Table S20) |
| Phage | Strains and LPS Compositions | |||
|---|---|---|---|---|
| YeO3 | YeO3-R1 | YeO3-c-OC | YeO3-c-OCR | |
| LA-IC-OC-Oag | LA-IC-OC | LA-IC-Oag | LA-IC | |
| fPS-7 | 1 | 0 | 0.2 × 10−2 | 0 |
| fPS-9 | 1 | 1.2 × 10−3 | 0.8 × 10−3 | 0 |
| fPS-10 | 1 | 3.3 × 10−5 | 5 × 10−3 | 0 |
| fPS-16 | 1 | 0 | 0.5 × 10−2 | 0 |
| fPS-19 | 1 | 2 × 10−4 | 9.5 × 10−5 | 0 |
| fPS-21 | 1 | 0 | 1.2 × 10−4 | 0 |
| fPS-26 | 1 | 0 | 0.7 × 10−3 | 0 |
| fPS-50 | 1 | 0.3 × 10−4 | 0.8 × 10−3 | 0 |
| fPS-52 | 1 | 0 | 0 | 0 |
| fPS-53 | 0.5 | 1 | 0 | 0 |
| fPS-54-ocr | 0 | 0 | 0 | 1 |
| fPS-59 | 1 | 0 | 1.7 × 10−1 | 0 |
| fPS-64 | 1 | 0.6 × 10−6 | 1.3 × 10−3 | 0 |
| fPS-85 | 1 | 0.5 | 0 | 0 |
| fPS-86 | 1 | 0.1 × 10−4 | 0.5 × 10−2 | 0 |
| fPS-89 | 1 | 0.6 × 10−2 | 0 | 0 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salem, M.; Skurnik, M. Genomic Characterization of Sixteen Yersinia enterocolitica-Infecting Podoviruses of Pig Origin. Viruses 2018, 10, 174. https://doi.org/10.3390/v10040174
Salem M, Skurnik M. Genomic Characterization of Sixteen Yersinia enterocolitica-Infecting Podoviruses of Pig Origin. Viruses. 2018; 10(4):174. https://doi.org/10.3390/v10040174
Chicago/Turabian StyleSalem, Mabruka, and Mikael Skurnik. 2018. "Genomic Characterization of Sixteen Yersinia enterocolitica-Infecting Podoviruses of Pig Origin" Viruses 10, no. 4: 174. https://doi.org/10.3390/v10040174
APA StyleSalem, M., & Skurnik, M. (2018). Genomic Characterization of Sixteen Yersinia enterocolitica-Infecting Podoviruses of Pig Origin. Viruses, 10(4), 174. https://doi.org/10.3390/v10040174