Transposition Behavior Revealed by High-Resolution Description of Pseudomonas Aeruginosa Saltovirus Integration Sites

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Transposable Phages Origin
2.2. DNA Preparation
2.3. Enrichment of the Right End of Phage Ab30 to Produce a Library of Ab30 Transposition Sites
2.4. Sequencing and Sequence Data Management
2.5. Statistical Analysis of the Distribution of Transposition Events
3. Results
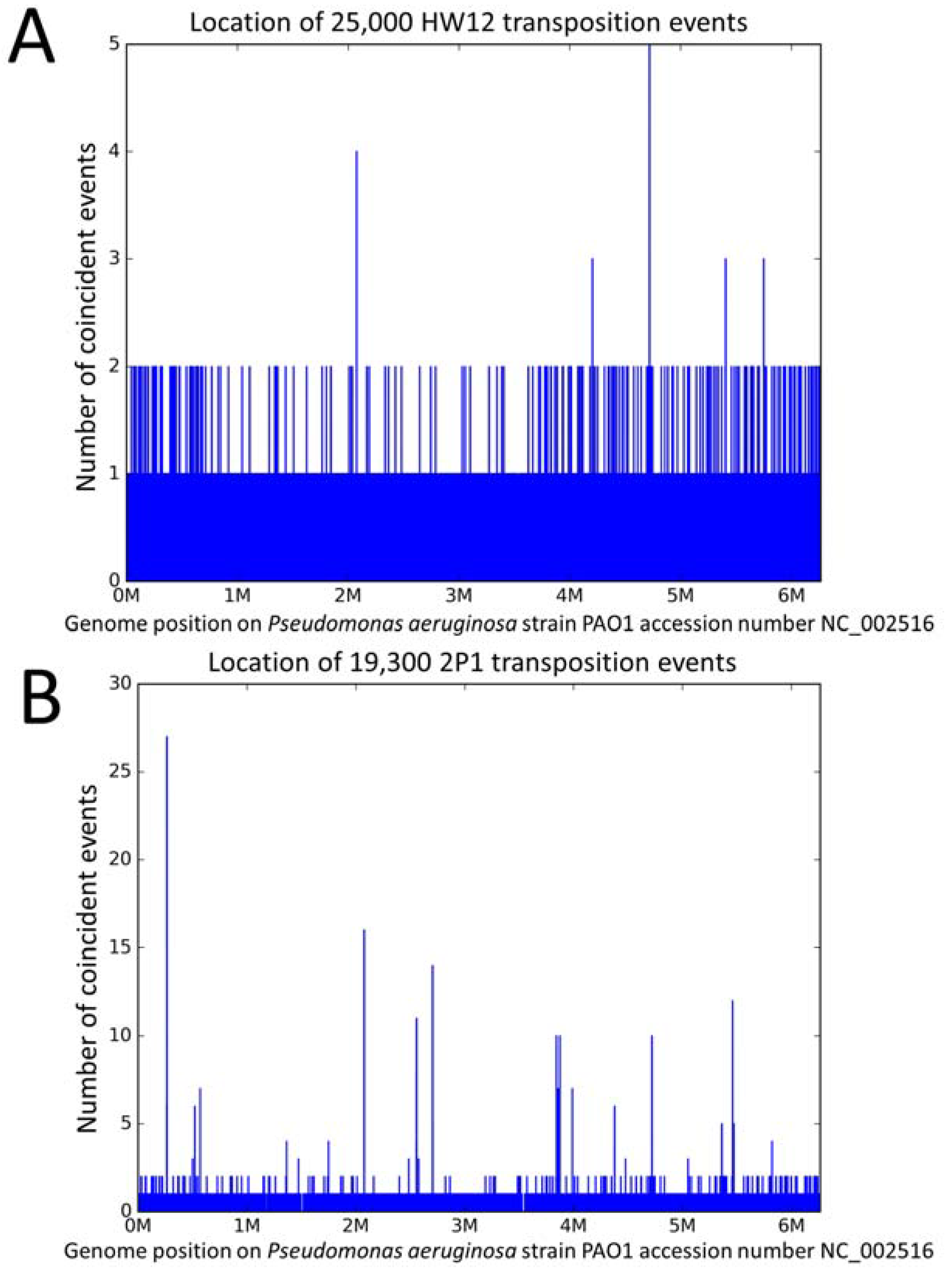
3.1. The Different Transposition Site Distributions Observed in the Three Phages
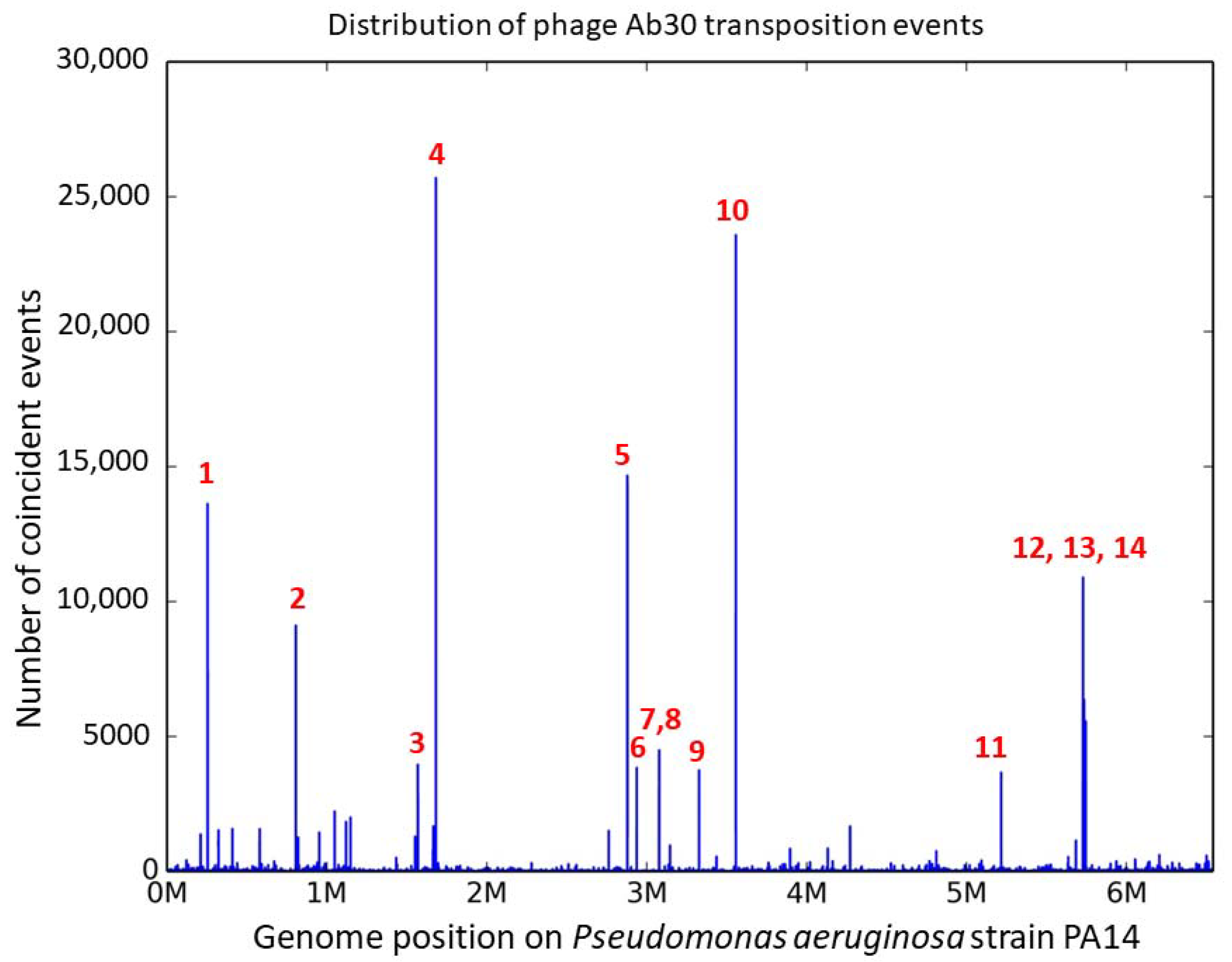
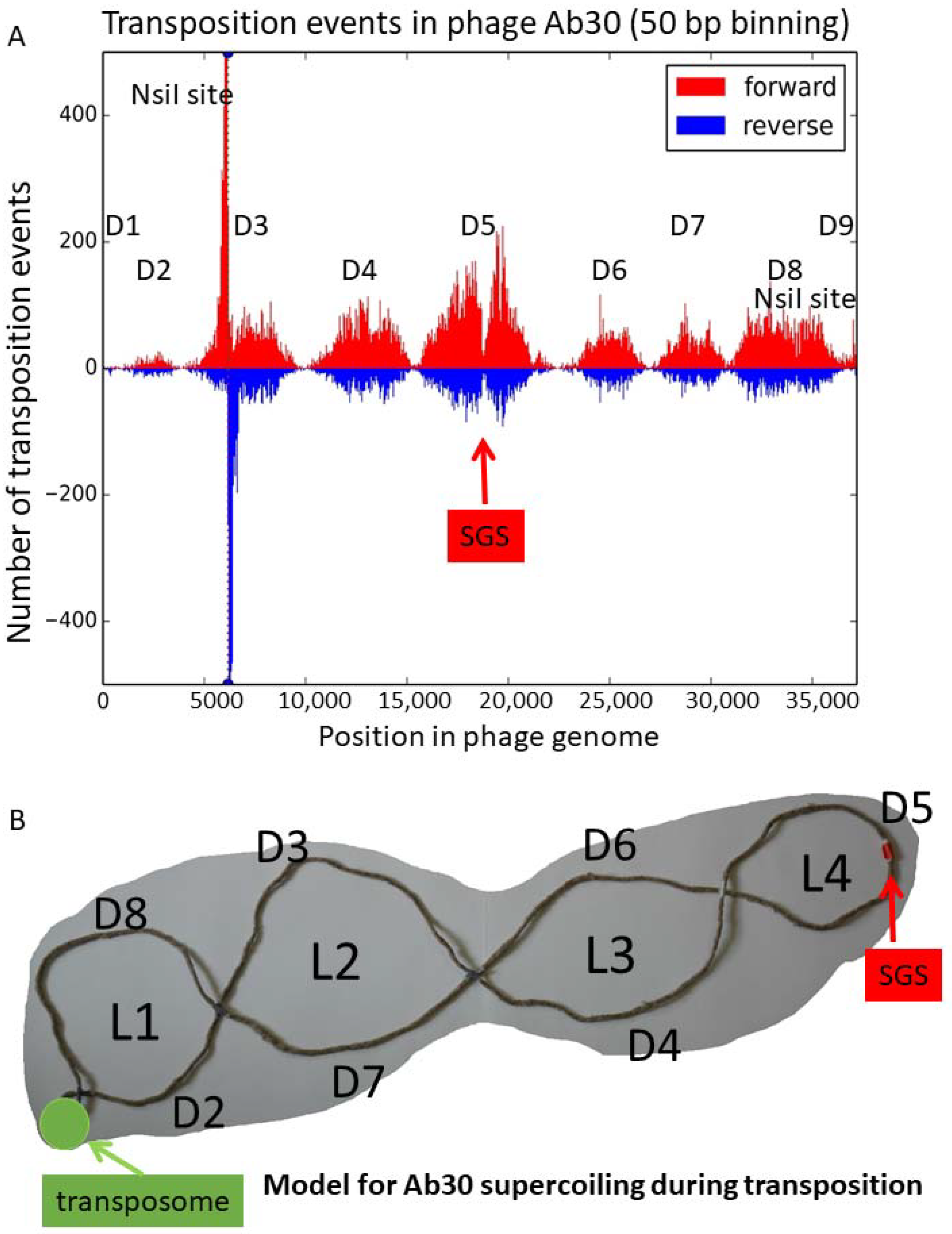
3.2. Distribution of the Ab30 Transposition Sites
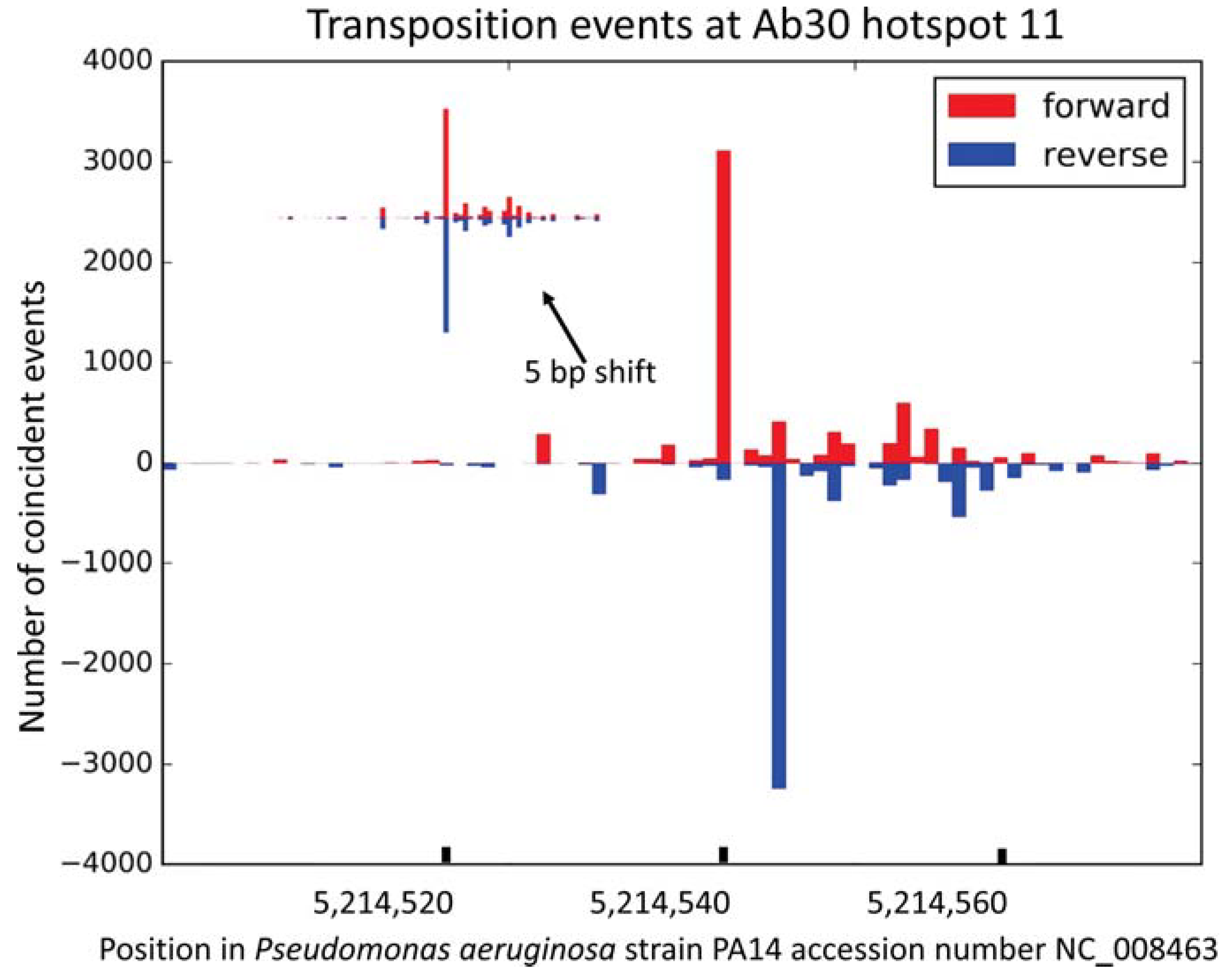
3.3. Presence of a Consensus Sequence Adjacent to the Hotspots
3.4. Coldspots for the Transposition Events Corresponding to Low GC Content Regions
3.5. Insertion of Ab30 into the Already Inserted Copies of Ab30
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Hulo, C.; Masson, P.; Le Mercier, P.; Toussaint, A. A structured annotation frame for the transposable phages: A new proposed family “saltoviridae” within the caudovirales. Virology 2015, 477, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Symonds, N.; Toussaint, A.; Van de Putte, A.P.; Howe, M.M. Phage Mu; Cold Spring Harbor Laboratory: Cold Spring Harbor, NY, USA, 1987. [Google Scholar]
- George, M.; Bukhari, A.I. Heterogeneous host DNA attached to the left end of mature bacteriophage mu DNA. Nature 1981, 292, 175–176. [Google Scholar] [CrossRef] [PubMed]
- Summer, E.J.; Gonzalez, C.F.; Carlisle, T.; Mebane, L.M.; Cass, A.M.; Savva, C.G.; LiPuma, J.; Young, R. Burkholderia cenocepacia phage bcepmu and a family of mu-like phages encoding potential pathogenesis factors. J. Mol. Biol. 2004, 340, 49–65. [Google Scholar] [CrossRef] [PubMed]
- Mizuuchi, K. Transpositional recombination: Mechanistic insights from studies of mu and other elements. Annu. Rev. Biochem. 1992, 61, 1011–1051. [Google Scholar] [CrossRef] [PubMed]
- Harshey, R.M. Transposable phage mu. Microbiol. Spectr. 2014, 2, 1100602544. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, N.; Dramicanin, M.; Mizuuchi, M.; Adam, J.; Wang, Y.; Han, Y.W.; Yang, W.; Steven, A.C.; Mizuuchi, K.; Ramon-Maiques, S. Mub is an aaa+ atpase that forms helical filaments to control target selection for DNA transposition. Proc. Natl. Acad. Sci. USA 2013, 110, E2441–E2450. [Google Scholar] [CrossRef] [PubMed]
- Harshey, R.M. The mu story: How a maverick phage moved the field forward. Mob. DNA 2012, 3, 21. [Google Scholar] [CrossRef] [PubMed]
- Choi, W.; Saha, R.P.; Jang, S.; Harshey, R.M. Controlling DNA degradation from a distance: A new role for the mu transposition enhancer. Mol. Microbiol. 2014, 94, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Greene, E.C.; Mizuuchi, K. Target immunity during mu DNA transposition. Transpososome assembly and DNA looping enhance mua-mediated disassembly of the mub target complex. Mol. Cell 2002, 10, 1367–1378. [Google Scholar] [CrossRef]
- Greene, E.C.; Mizuuchi, K. Visualizing the assembly and disassembly mechanisms of the mub transposition targeting complex. J. Biol. Chem. 2004, 279, 16736–16743. [Google Scholar] [CrossRef] [PubMed]
- Toussaint, A. Transposable mu-like phages in firmicutes: New instances of divergence generating retroelements. Res. Microbiol. 2013, 164, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Ge, J.; Lou, Z.; Cui, H.; Shang, L.; Harshey, R.M. Analysis of phage mu DNA transposition by whole-genome escherichia coli tiling arrays reveals a complex relationship to distribution of target selection protein b, transcription and chromosome architectural elements. J. Biosci. 2011, 36, 587–601. [Google Scholar] [CrossRef] [PubMed]
- Fogg, P.C.; Hynes, A.P.; Digby, E.; Lang, A.S.; Beatty, J.T. Characterization of a newly discovered mu-like bacteriophage, rcapmu, in rhodobacter capsulatus strain sb1003. Virology 2011, 421, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Manna, D.; Breier, A.M.; Higgins, N.P. Microarray analysis of transposition targets in escherichia coli: The impact of transcription. Proc. Natl. Acad. Sci. USA 2004, 101, 9780–9785. [Google Scholar] [CrossRef] [PubMed]
- Manna, D.; Deng, S.; Breier, A.M.; Higgins, N.P. Bacteriophage mu targets the trinucleotide sequence cgg. J. Bacteriol. 2005, 187, 3586–3588. [Google Scholar] [CrossRef] [PubMed]
- Haapa-Paananen, S.; Rita, H.; Savilahti, H. DNA transposition of bacteriophage mu. A quantitative analysis of target site selection in vitro. J. Biol. Chem. 2002, 277, 2843–2851. [Google Scholar] [CrossRef] [PubMed]
- Adzuma, K.; Mizuuchi, K. Target immunity of mu transposition reflects a differential distribution of mu b protein. Cell 1988, 53, 257–266. [Google Scholar] [CrossRef]
- Han, Y.W.; Mizuuchi, K. Phage mu transposition immunity: Protein pattern formation along DNA by a diffusion-ratchet mechanism. Mol. Cell 2010, 39, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Ge, J.; Lou, Z.; Harshey, R.M. Immunity of replicating mu to self-integration: A novel mechanism employing mub protein. Mob. DNA 2010, 1, 8. [Google Scholar] [CrossRef] [PubMed]
- Dramicanin, M.; Ramon-Maiques, S. Mub gives a new twist to target DNA selection. Mob. Genet. Elements 2013, 3, e27515. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.W.; Chu, L.; Guttman, D.S. Complete sequence and evolutionary genomic analysis of the Pseudomonas aeruginosa transposable bacteriophage d3112. J. Bacteriol. 2004, 186, 400–410. [Google Scholar] [CrossRef] [PubMed]
- Cazares, A.; Mendoza-Hernandez, G.; Guarneros, G. Core and accessory genome architecture in a group of Pseudomonas aeruginosa mu-like phages. BMC Genom. 2014, 15, 1146. [Google Scholar] [CrossRef] [PubMed]
- Braid, M.D.; Silhavy, J.L.; Kitts, C.L.; Cano, R.J.; Howe, M.M. Complete genomic sequence of bacteriophage b3, a mu-like phage of Pseudomonas aeruginosa. J. Bacteriol. 2004, 186, 6560–6574. [Google Scholar] [CrossRef] [PubMed]
- Akhverdian, V.Z.; Khrenova, E.A.; Reulets, M.A.; Gerasimova, T.V.; Krylov, V.N. Characteristics of phages-transposons of Pseudomonas aeruginosa belonging to 2 groups distinguished by DNA-DNA homology. Genetika 1985, 21, 735–747. [Google Scholar] [PubMed]
- Toussaint, A.; Rice, P.A. Transposable phages, DNA reorganization and transfer. Curr. Opin. Microbiol. 2017, 38, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Chung, I.Y.; Bae, H.W.; Jang, H.J.; Kim, B.O.; Cho, Y.H. Superinfection exclusion reveals heteroimmunity between pseudomonas aeruginosa temperate phages. J. Microbiol. 2014, 52, 515–520. [Google Scholar] [CrossRef] [PubMed]
- Toussaint, A.; Van Gijsegem, F. Extension of the transposable bacterial virus family: Two genomic organisations among phages and prophages with a tn552-related transposase. Res. Microbiol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Krylov, V.N.; Bogush, V.G.; Ianenko, A.S.; Kirsanov, N.B. Pseudomonas aeruginosa bacteriophages with DNA structure similar to the DNA structure of mu1 phage. Ii. Evidence for similarity between d3112, b3, and b39 bacteriophages: Analysis of DNA splits by restriction endonucleases, isolation of d3112 and b3 recombinant phages. Genetika 1980, 16, 975–984. [Google Scholar] [PubMed]
- Darzins, A.; Casadaban, M.J. Mini-d3112 bacteriophage transposable elements for genetic analysis of Pseudomonas aeruginosa. J. Bacteriol. 1989, 171, 3909–3916. [Google Scholar] [CrossRef] [PubMed]
- Essoh, C.; Latino, L.; Midoux, C.; Blouin, Y.; Loukou, G.; Nguetta, S.P.; Lathro, S.; Cablanmian, A.; Kouassi, A.K.; Vergnaud, G.; et al. Investigation of a large collection of Pseudomonas aeruginosa bacteriophages collected from a single environmental source in abidjan, cote d’ivoire. PLoS ONE 2015, 10, e0130548. [Google Scholar] [CrossRef] [PubMed]
- Essoh, C.; Blouin, Y.; Loukou, G.; Cablanmian, A.; Lathro, S.; Kutter, E.; Thien, H.V.; Vergnaud, G.; Pourcel, C. The susceptibility of Pseudomonas aeruginosa strains from cystic fibrosis patients to bacteriophages. PLoS ONE 2013, 8, e60575. [Google Scholar] [CrossRef] [PubMed]
- Drake, J.W. Too many mutants with multiple mutations. Crit. Rev. Biochem. Mol. Biol. 2007, 42, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Garneau, J.R.; Depardieu, F.; Fortier, L.C.; Bikard, D.; Monot, M. Phageterm: A tool for fast and accurate determination of phage termini and packaging mechanism using next-generation sequencing data. Sci. Rep. 2017, 7, 8292. [Google Scholar] [CrossRef] [PubMed]
- Vu-Thien, H.; Corbineau, G.; Hormigos, K.; Fauroux, B.; Corvol, H.; Clement, A.; Vergnaud, G.; Pourcel, C. Multiple-locus variable-number tandem-repeat analysis for longitudinal survey of sources of pseudomonas aeruginosa infection in cystic fibrosis patients. J. Clin. Microbiol. 2007, 45, 3175–3183. [Google Scholar] [CrossRef] [PubMed]
- Sobral, D.; Mariani-Kurkdjian, P.; Bingen, E.; Vu-Thien, H.; Hormigos, K.; Lebeau, B.; Loisy-Hamon, F.; Munck, A.; Vergnaud, G.; Pourcel, C. A new highly discriminatory multiplex capillary-based mlva assay as a tool for the epidemiological survey of Pseudomonas aeruginosa in cystic fibrosis patients. Eur. J. Clin. Microbiol. Infect. Dis. 2012, 31, 2247–2256. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. Mafft multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Gilson, E.; Saurin, W.; Perrin, D.; Bachellier, S.; Hofnung, M. The bime family of bacterial highly repetitive sequences. Res. Microbiol. 1991, 142, 217–222. [Google Scholar] [CrossRef]
- Raymond, C.K.; Sims, E.H.; Kas, A.; Spencer, D.H.; Kutyavin, T.V.; Ivey, R.G.; Zhou, Y.; Kaul, R.; Clendenning, J.B.; Olson, M.V. Genetic variation at the o-antigen biosynthetic locus in Pseudomonas aeruginosa. J. Bacteriol. 2002, 184, 3614–3622. [Google Scholar] [CrossRef] [PubMed]
- Pato, M.L. Central location of the mu strong gyrase binding site is obligatory for optimal rates of replicative transposition. Proc. Natl. Acad. Sci. USA 1994, 91, 7056–7060. [Google Scholar] [CrossRef] [PubMed]
- Oram, M.; Travers, A.A.; Howells, A.J.; Maxwell, A.; Pato, M.L. Dissection of the bacteriophage mu strong gyrase site (sgs): Significance of the sgs right arm in mu biology and DNA gyrase mechanism. J. Bacteriol. 2006, 188, 619–632. [Google Scholar] [CrossRef] [PubMed]
- Saha, R.P.; Lou, Z.; Meng, L.; Harshey, R.M. Transposable prophage mu is organized as a stable chromosomal domain of E. coli. PLoS Genet. 2013, 9, e1003902. [Google Scholar] [CrossRef] [PubMed]
- Mizuuchi, M.; Mizuuchi, K. Conformational isomerization in phage mu transpososome assembly: Effects of the transpositional enhancer and of mub. EMBO J. 2001, 20, 6927–6935. [Google Scholar] [CrossRef] [PubMed]
- Higgins, C.F.; Ames, G.F.; Barnes, W.M.; Clement, J.M.; Hofnung, M. A novel intercistronic regulatory element of prokaryotic operons. Nature 1982, 298, 760–762. [Google Scholar] [CrossRef] [PubMed]
- Cramton, S.E.; Schnell, N.F.; Götz, F.; Brückner, R. Identification of a new repetitive element in Staphylococcus aureus. Infect. Immun. 2000, 68, 2344–2348. [Google Scholar] [CrossRef] [PubMed]
- Bertels, F.; Rainey, P.B. Within-genome evolution of repins: A new family of miniature mobile DNA in bacteria. PLoS Genet. 2011, 7, e1002132. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.; Ohta, S.; Ohtsubo, E. A novel is element, is621, of the is110/is492 family transposes to a specific site in repetitive extragenic palindromic sequences in Escherichia coli. J. Bacteriol. 2003, 185, 4891–4900. [Google Scholar] [CrossRef] [PubMed]
- Tobes, R.; Pareja, E. Bacterial repetitive extragenic palindromic sequences are DNA targets for insertion sequence elements. BMC Genom. 2006, 7, 62. [Google Scholar] [CrossRef] [PubMed]
- Bachellier, S.; Saurin, W.; Perrin, D.; Hofnung, M.; Gilson, E. Structural and functional diversity among bacterial interspersed mosaic elements (bimes). Mol. Microbiol. 1994, 12, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Espeli, O.; Boccard, F. In vivo cleavage of Escherichia coli bime-2 repeats by DNA gyrase: Genetic characterization of the target and identification of the cut site. Mol. Microbiol. 1997, 26, 767–777. [Google Scholar] [CrossRef] [PubMed]
- Vrljicak, P.; Tao, S.; Varshney, G.K.; Quach, H.N.; Joshi, A.; LaFave, M.C.; Burgess, S.M.; Sampath, K. Genome-wide analysis of transposon and retroviral insertions reveals preferential integrations in regions of DNA flexibility. G3 Genes Genomes Genet. 2016, 6, 805–817. [Google Scholar] [CrossRef] [PubMed]
- Ton-Hoang, B.; Siguier, P.; Quentin, Y.; Onillon, S.; Marty, B.; Fichant, G.; Chandler, M. Structuring the bacterial genome: Y1-transposases associated with rep-bime sequences. Nucleic Acids Res. 2012, 40, 3596–3609. [Google Scholar] [CrossRef] [PubMed]
- Dame, R.T.; Wyman, C.; Goosen, N. Structural basis for preferential binding of h-ns to curved DNA. Biochimie 2001, 83, 231–234. [Google Scholar] [CrossRef]
- Whitfield, C.R.; Wardle, S.J.; Haniford, D.B. The global bacterial regulator h-ns promotes transpososome formation and transposition in the tn5 system. Nucleic Acids Res. 2009, 37, 309–321. [Google Scholar] [CrossRef] [PubMed]
- Manna, D.; Porwollik, S.; McClelland, M.; Tan, R.; Higgins, N.P. Microarray analysis of mu transposition in Salmonella enterica, serovar typhimurium: Transposon exclusion by high-density DNA binding proteins. Mol. Microbiol. 2007, 66, 315–328. [Google Scholar] [CrossRef] [PubMed]
- Castang, S.; McManus, H.R.; Turner, K.H.; Dove, S.L. H-ns family members function coordinately in an opportunistic pathogen. Proc. Natl. Acad. Sci. USA 2008, 105, 18947–18952. [Google Scholar] [CrossRef] [PubMed]
- Lam, J.S.; Taylor, V.L.; Islam, S.T.; Hao, Y.; Kocincova, D. Genetic and functional diversity of Pseudomonas aeruginosa lipopolysaccharide. Front. Microbiol. 2011, 2, 118. [Google Scholar] [CrossRef] [PubMed]
- Pato, M.L.; Karlok, M.; Wall, C.; Higgins, N.P. Characterization of mu prophage lacking the central strong gyrase binding site: Localization of the block in replication. J. Bacteriol. 1995, 177, 5937–5942. [Google Scholar] [CrossRef] [PubMed]
- Leung, P.C.; Teplow, D.B.; Harshey, R.M. Interaction of distinct domains in mu transposase with mu DNA ends and an internal transpositional enhancer. Nature 1989, 338, 656–658. [Google Scholar] [CrossRef] [PubMed]
- Kimura, S.; Hubbard, T.P.; Davis, B.M.; Waldor, M.K. The nucleoid binding protein h-ns biases genome-wide transposon insertion landscapes. mBio 2016, 7, e01351-16. [Google Scholar] [CrossRef] [PubMed]
- Mesarich, C.H.; Rees-George, J.; Gardner, P.P.; Ghomi, F.A.; Gerth, M.L.; Andersen, M.T.; Rikkerink, E.H.; Fineran, P.C.; Templeton, M.D. Transposon insertion libraries for the characterization of mutants from the kiwifruit pathogen Pseudomonas syringae pv. Actinidiae. PLoS ONE 2017, 12, e0172790. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Features | vB_PaeS_PAO1_Ab30 | vB_PaeS_PcyII-10_2P1 | vB_PaeS_PAO1_HW12 |
|---|---|---|---|
| common name | Ab30 | 2P1 | HW12 |
| source | sewage water | sewage water | clinical isolate |
| geographic origin | Abidjan, Côte d’Ivoire | Orsay, France | Moscow, Russia |
| year of isolation | 2012 | 2014 | 1983 * |
| genome size | 37,205 bp | 37,087 bp | 37,496 bp |
| accession number | LN610590 | LN907801 | LT999987 |
| host strain for phage growth | PA14 | PcyII-10 | PAO1 |
| sequencing reads accessions | ERR1596849 | ERR1596851 | ERR2402365 |
| Average phage sequencing coverage | 1200× | 10,000× | 12,000× |
| publication | [31] | this report | [25] |
| # | Position PA14 NC_008463 * | Position PAO1 NC_002516 * | CDS Locus Tag ** | Distance NsiI $ | Ratio Forward $ | Distance from Consensus Motif (bp) £ | Mismatches £ | VNTR § |
|---|---|---|---|---|---|---|---|---|
| 1 | 254,808; 254,824 | 265,008; 265,024 | PA14_RS01160 | 28,587/24,475 | 41% | 56 | 2 | ms211 |
| 2 | 806,635; 806,652 | 4,720,167; 4,720,184 | PA14_RS30080 | 2414/16,824 | 58% | 58 | 0 | |
| 3 | 1,569,481; 1,569,485 | 3,988,644; 3,988,648 | PA14_RS07340 | 68,541/26,332 | 47% | 57 | + | |
| 4 | 1,682,632; 1,682,636 | 3,858,411; 3,858,415 | between PA14_RS07780 and PA14_RS07785 | 299/18,473 | 35% | 57 | 2 | |
| 5 | 2,879,877; 2,879,884 | 2,752,911; 2,752,918 | PA14_RS13425 | 336/4118 | 35% | 57 | 3 | |
| 6 | 2,938,509; 2,938,513 | 2,705,279; 2,705,283 | PA14_RS13610 | 4732/32,396 | 48% | 56 | 0 | ms214 |
| 7 | 3,079,036; 3,079,040 | 2,558,697; 2,558,701 | between PA14_RS14100 and PA14_RS14105 | 1443/34,807 | 50% | 56 | 0 | |
| 8 | 3,079,172 | 2,556,658 | PA14_RS14105 | 1579/34,675 | 47% | 60 | 0 | |
| 9 | 3,325,225; 3,325,229 | 3,874,426; 3,874,430 | between PA14_RS15200 and PA14_RS15205 | 6907/23,919 | 61% | 57 | 0 | |
| 10 | 3,555,249; 3,555,253 | 2,077,078; 2,077,082 | PA14_RS16160 | 495/7668 | 47% | 60 | 0 | |
| 11 | 5,214,540; 5,214,544 | 5,050,834; 5,050,838 | between PA14_RS23875 and PA14_RS23880 | 8737/1571 | 54% | 57 | + | |
| 12 | 5,725,984; 5,725,999 | 5,455,320; 5,455,335 | between PA14_RS26245 and PA14_RS26250 | 44,700/98 | 80% | 54 | 1 | ms223 |
| 13 | 5,732,031; 5,732,035 | 5,461,367; 5,461,371 | PA14_RS26270 | 5927/9363 | 51% | 59 | 1 | |
| 14 | 5,743,027; 5,743,031 | 5,471,620; 5,471,624 | between PA14_RS26335 and PA14_RS26340 | 1622/2499 | 58% | 58 | 1 | ms224 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vergnaud, G.; Midoux, C.; Blouin, Y.; Bourkaltseva, M.; Krylov, V.; Pourcel, C. Transposition Behavior Revealed by High-Resolution Description of Pseudomonas Aeruginosa Saltovirus Integration Sites. Viruses 2018, 10, 245. https://doi.org/10.3390/v10050245
Vergnaud G, Midoux C, Blouin Y, Bourkaltseva M, Krylov V, Pourcel C. Transposition Behavior Revealed by High-Resolution Description of Pseudomonas Aeruginosa Saltovirus Integration Sites. Viruses. 2018; 10(5):245. https://doi.org/10.3390/v10050245
Chicago/Turabian StyleVergnaud, Gilles, Cédric Midoux, Yann Blouin, Maria Bourkaltseva, Victor Krylov, and Christine Pourcel. 2018. "Transposition Behavior Revealed by High-Resolution Description of Pseudomonas Aeruginosa Saltovirus Integration Sites" Viruses 10, no. 5: 245. https://doi.org/10.3390/v10050245
APA StyleVergnaud, G., Midoux, C., Blouin, Y., Bourkaltseva, M., Krylov, V., & Pourcel, C. (2018). Transposition Behavior Revealed by High-Resolution Description of Pseudomonas Aeruginosa Saltovirus Integration Sites. Viruses, 10(5), 245. https://doi.org/10.3390/v10050245