Potential Therapeutic Agents for Feline Calicivirus Infection

 ,
,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Cells and FCV
2.2. NNIs and NAs
2.3. FCV Pro-Pol Cloning, Expression and Purification
2.4. Cytotoxicity Study
2.5. Antiviral Screening Using Fluroescent RdRp Assays
2.6. Protease FRET Activity Assay and Antiviral Screening
2.7. Inhibition of FCV Plaque Formation in Cell Culture
2.8. FCV Genome Reduction Assay Using Reverse Transcription Quantitative Polymerase Chain Reaction (RT-qPCR)
2.9. Statistical Analysis
3. Results
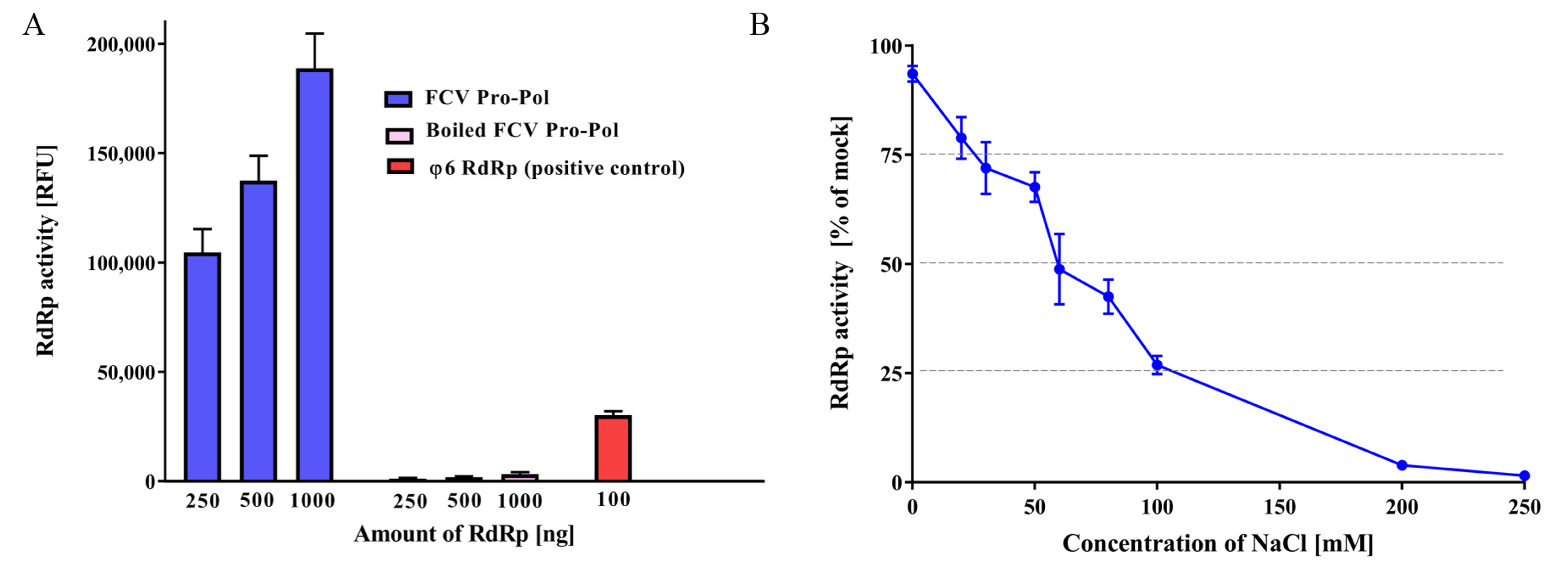
3.1. FCV Pro-Pol Expression
3.2. RdRp In Vitro Assay
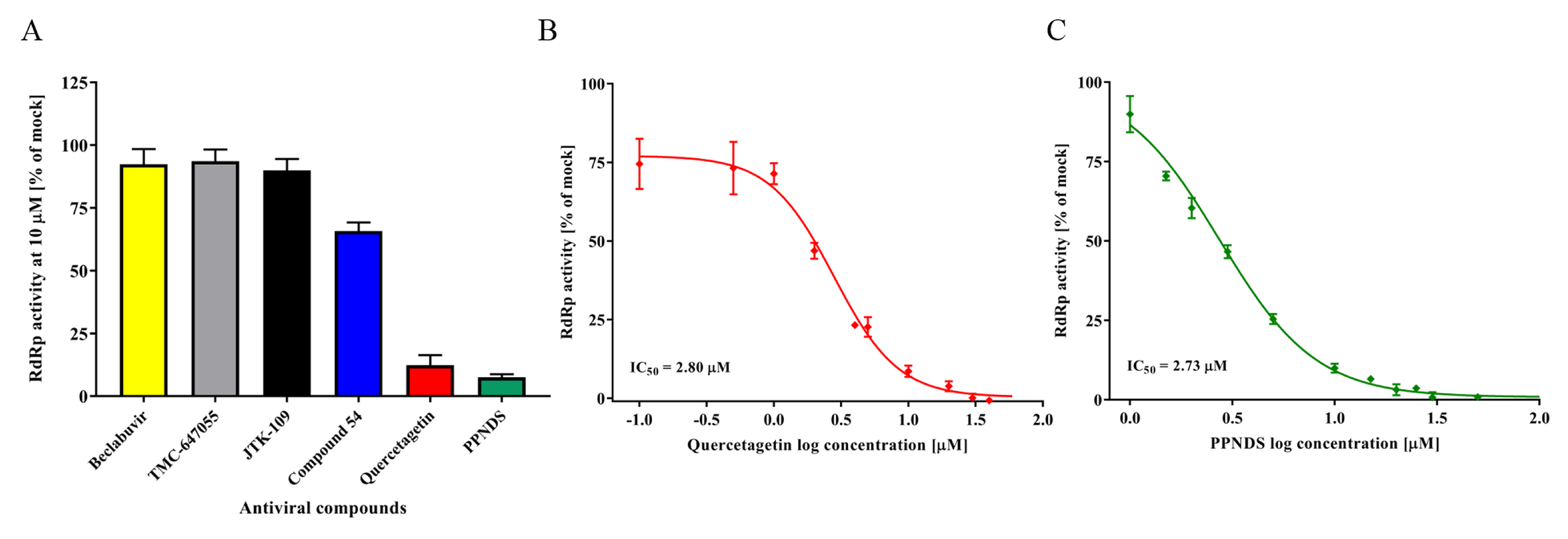
3.3. Inhibition of RdRp Activity Using NNIs
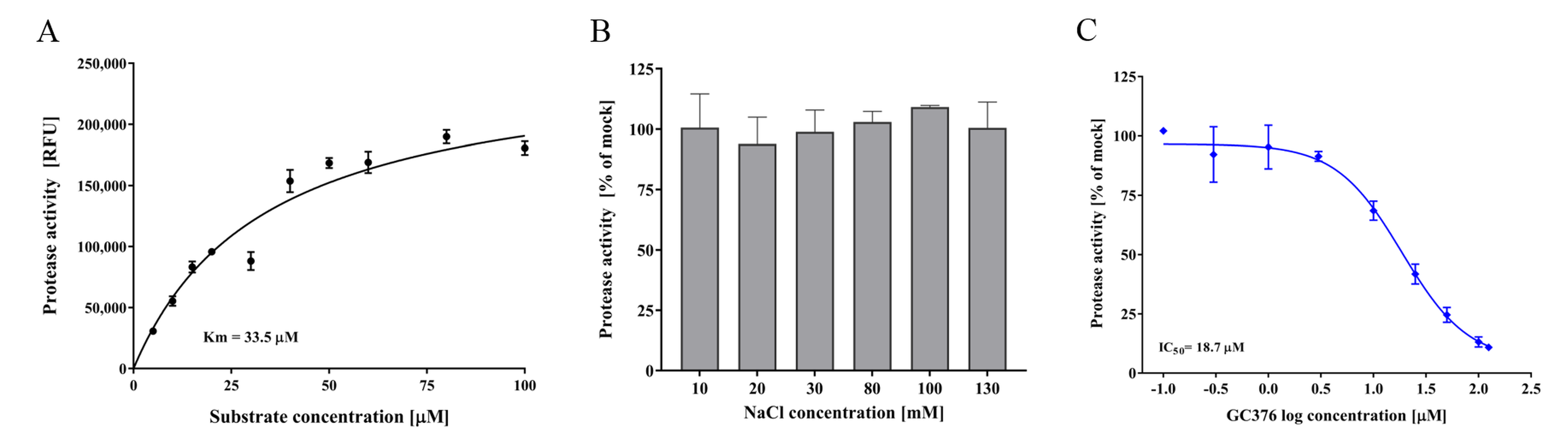
3.4. FCV Protease In Vitro Assay and Test Compounds
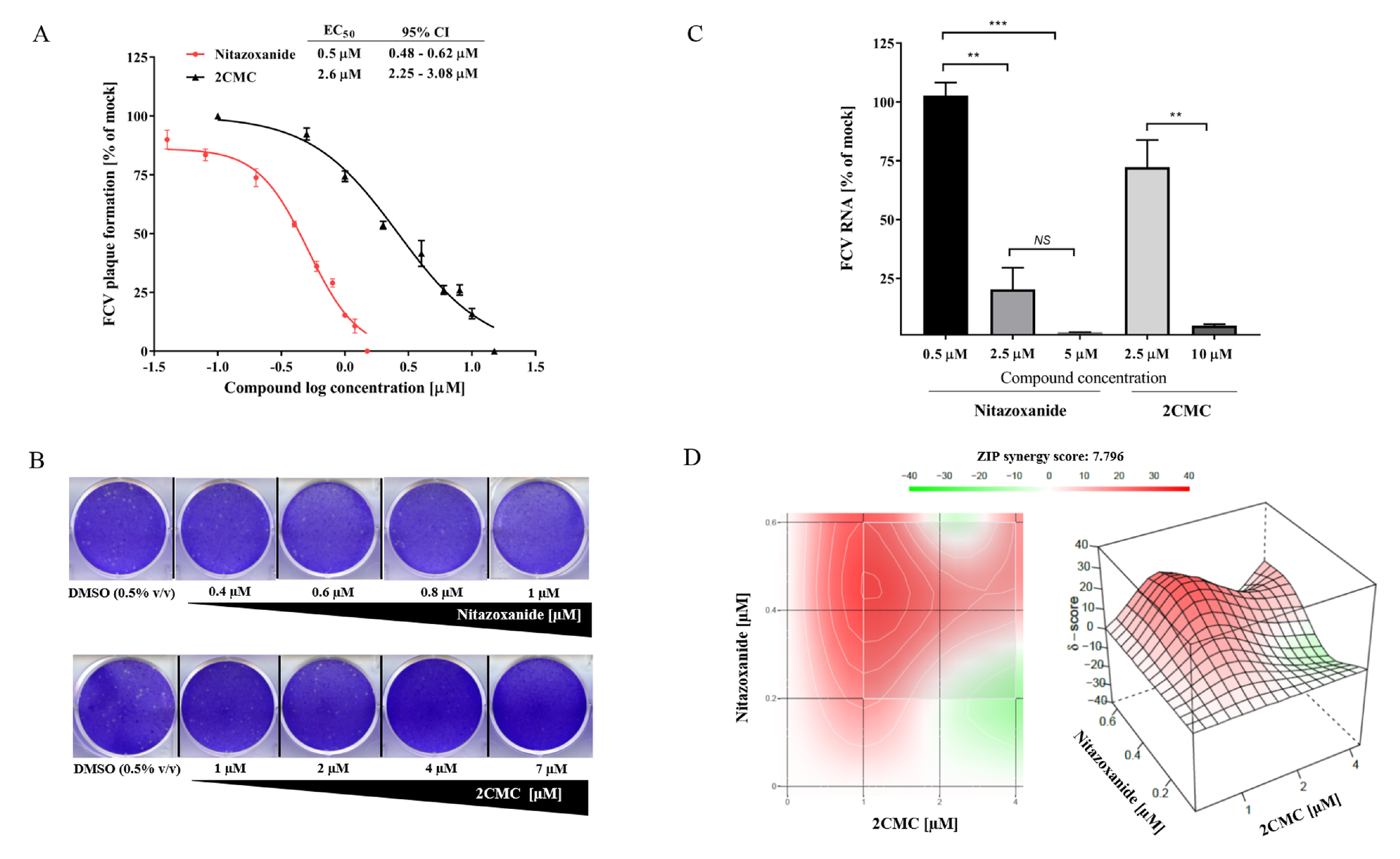
3.5. 2CMC and Nitazoxanide Inhibit FCV Infectivity
3.6. Combinational Treatment with Nitazoxanide and 2CMC Showed Synergistic Antiviral Effects
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cai, Y.; Fukushi, H.; Koyasu, S.; Kuroda, E.; Yamaguchi, T.; Hirai, K. An etiological investigation of domestic cats with conjunctivitis and upper respiratory tract disease in japan. J. Vet. Med. Sci. 2002, 64, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Radford, A.D.; Coyne, K.P.; Dawson, S.; Porter, C.J.; Gaskell, R.M. Feline calicivirus. Vet. Res. 2007, 38, 319–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bannasch, M.J.; Foley, J.E. Epidemiologic evaluation of multiple respiratory pathogens in cats in animal shelters. J. Feline Med. Surg. 2005, 7, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Pesavento, P.A.; Chang, K.O.; Parker, J.S. Molecular virology of feline calicivirus. Vet. Clin. N. Am. Small Anim. Pract. 2008, 38, 775–786. [Google Scholar] [CrossRef] [PubMed]
- Dawson, S.; Bennett, D.; Carter, S.D.; Bennett, M.; Meanger, J.; Turner, P.C.; Carter, M.J.; Milton, I.; Gaskell, R.M. Acute arthritis of cats associated with feline calicivirus infection. Res. Vet. Sci. 1994, 56, 133–143. [Google Scholar] [CrossRef]
- Pedersen, N.C.; Elliott, J.B.; Glasgow, A.; Poland, A.; Keel, K. An isolated epizootic of hemorrhagic-like fever in cats caused by a novel and highly virulent strain of feline calicivirus. Vet. Microbiol. 2000, 73, 281–300. [Google Scholar] [CrossRef]
- Radford, A.D.; Gaskell, R.M. Dealing with a potential case of fcv-associated virulent systemic disease. Vet. Rec. 2011, 168, 585–586. [Google Scholar] [CrossRef] [PubMed]
- Schulz, B.S.; Hartmann, K.; Unterer, S.; Eichhorn, W.; Majzoub, M.; Homeier-Bachmann, T.; Truyen, U.; Ellenberger, C.; Huebner, J. Two outbreaks of virulent systemic feline calicivirus infection in cats in germany. Berl. Munch. Tierarztl. Wochenschr. 2011, 124, 186–193. [Google Scholar] [PubMed]
- Battilani, M.; Vaccari, F.; Carelle, M.S.; Morandi, F.; Benazzi, C.; Kipar, A.; Dondi, F.; Scagliarini, A. Virulent feline calicivirus disease in a shelter in italy: A case description. Res. Vet. Sci. 2013, 95, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Meyer, A.; Kershaw, O.; Klopfleisch, R. Feline calicivirus-associated virulent systemic disease: Not necessarily a local epizootic problem. Vet. Rec. 2011, 168, 589. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, B.S.; Poulet, H.; Pingret, J.L.; Jas, D.; Brunet, S.; Lemeter, C.; Etievant, M.; Boucraut-Baralon, C. A nosocomial outbreak of feline calicivirus associated virulent systemic disease in france. J. Feline Med. Surg. 2009, 11, 633–644. [Google Scholar] [CrossRef] [PubMed]
- Coyne, K.P.; Jones, B.R.; Kipar, A.; Chantrey, J.; Porter, C.J.; Barber, P.J.; Dawson, S.; Gaskell, R.M.; Radford, A.D. Lethal outbreak of disease associated with feline calicivirus infection in cats. Vet. Rec. 2006, 158, 544–550. [Google Scholar] [CrossRef] [PubMed]
- Willi, B.; Spiri, A.M.; Meli, M.L.; Samman, A.; Hoffmann, K.; Sydler, T.; Cattori, V.; Graf, F.; Diserens, K.A.; Padrutt, I.; et al. Molecular characterization and virus neutralization patterns of severe, non-epizootic forms of feline calicivirus infections resembling virulent systemic disease in cats in switzerland and in liechtenstein. Vet. Microbiol. 2016, 182, 202–212. [Google Scholar] [CrossRef] [PubMed]
- Foley, J.; Hurley, K.; Pesavento, P.A.; Poland, A.; Pedersen, N.C. Virulent systemic feline calicivirus infection: Local cytokine modulation and contribution of viral mutants. J. Feline Med. Surg. 2006, 8, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Hurley, K.E.; Pesavento, P.A.; Pedersen, N.C.; Poland, A.M.; Wilson, E.; Foley, J.E. An outbreak of virulent systemic feline calicivirus disease. J. Am. Vet. Med. Assoc. 2004, 224, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Herbert, T.P.; Brierley, I.; Brown, T.D. Identification of a protein linked to the genomic and subgenomic mRNAs of feline calicivirus and its role in translation. J. Gen. Virol. 1997, 78 Pt 5, 1033–1040. [Google Scholar] [CrossRef]
- Carter, M.J.; Milton, I.D.; Meanger, J.; Bennett, M.; Gaskell, R.M.; Turner, P.C. The complete nucleotide sequence of a feline calicivirus. Virology 1992, 190, 443–448. [Google Scholar] [CrossRef]
- Wei, L.; Huhn, J.S.; Mory, A.; Pathak, H.B.; Sosnovtsev, S.V.; Green, K.Y.; Cameron, C.E. Proteinase-polymerase precursor as the active form of feline calicivirus RNA-dependent RNA polymerase. J. Virol. 2001, 75, 1211–1219. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Sanchez-Vizcaino, F.; McGahie, D.; Lesbros, C.; Almeras, T.; Howarth, D.; O’Hara, V.; Dawson, S.; Radford, A.D. European molecular epidemiology and strain diversity of feline calicivirus. Vet. Rec. 2016, 178, 114–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coyne, K.P.; Gaskell, R.M.; Dawson, S.; Porter, C.J.; Radford, A.D. Evolutionary mechanisms of persistence and diversification of a calicivirus within endemically infected natural host populations. J. Virol. 2007, 81, 1961–1971. [Google Scholar] [CrossRef] [PubMed]
- Glenn, M.; Radford, A.D.; Turner, P.C.; Carter, M.; Lowery, D.; DeSilver, D.A.; Meanger, J.; Baulch-Brown, C.; Bennett, M.; Gaskell, R.M. Nucleotide sequence of uk and australian isolates of feline calicivirus (FCV) and phylogenetic analysis of FCVs. Vet. Microbiol. 1999, 67, 175–193. [Google Scholar] [CrossRef]
- Prikhodko, V.G.; Sandoval-Jaime, C.; Abente, E.J.; Bok, K.; Parra, G.I.; Rogozin, I.B.; Ostlund, E.N.; Green, K.Y.; Sosnovtsev, S.V. Genetic characterization of feline calicivirus strains associated with varying disease manifestations during an outbreak season in missouri (1995–1996). Virus Genes 2014, 48, 96–110. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Ohe, K.; Murakami, M.; Fukuyama, M.; Furuhata, K.; Kishikawa, S.; Suzuki, Y.; Kiuchi, A.; Hara, M.; Ishikawa, Y.; et al. Phylogenetic analysis of field isolates of feline calcivirus (FCV) in japan by sequencing part of its capsid gene. Vet. Res. Commun. 2002, 26, 205–219. [Google Scholar] [CrossRef] [PubMed]
- Radford, A.D.; Addie, D.; Belak, S.; Boucraut-Baralon, C.; Egberink, H.; Frymus, T.; Gruffydd-Jones, T.; Hartmann, K.; Hosie, M.J.; Lloret, A.; et al. Feline calicivirus infection. Abcd guidelines on prevention and management. J. Feline Med. Surg. 2009, 11, 556–564. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Chang, K.O. Fexaramine as an entry blocker for feline caliciviruses. Antivir. Res. 2018, 152, 76–83. [Google Scholar] [CrossRef] [PubMed]
- McDonagh, P.; Sheehy, P.A.; Fawcett, A.; Norris, J.M. Antiviral effect of mefloquine on feline calicivirus in vitro. Vet. Microbiol. 2015, 176, 370–377. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.W.; Iversen, P.L.; O’Hanley, P.D.; Skilling, D.E.; Christensen, J.R.; Weaver, S.S.; Longley, K.; Stone, M.A.; Poet, S.E.; Matson, D.O. Virus-specific antiviral treatment for controlling severe and fatal outbreaks of feline calicivirus infection. Am. J. Vet. Res. 2008, 69, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Liu, Y.; Zu, S.; Sun, X.; Liu, C.; Liu, D.; Zhang, X.; Tian, J.; Qu, L. In vitro antiviral effect of germacrone on feline calicivirus. Arch. Virol. 2016, 161, 1559–1567. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Lovell, S.; Tiew, K.C.; Mandadapu, S.R.; Alliston, K.R.; Battaile, K.P.; Groutas, W.C.; Chang, K.O. Broad-spectrum antivirals against 3c or 3c-like proteases of picornaviruses, noroviruses, and coronaviruses. J. Virol. 2012, 86, 11754–11762. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Shivanna, V.; Narayanan, S.; Prior, A.M.; Weerasekara, S.; Hua, D.H.; Kankanamalage, A.C.; Groutas, W.C.; Chang, K.O. Broad-spectrum inhibitors against 3c-like proteases of feline coronaviruses and feline caliciviruses. J. Virol. 2015, 89, 4942–4950. [Google Scholar] [CrossRef] [PubMed]
- De Clercq, E. The nucleoside reverse transcriptase inhibitors, nonnucleoside reverse transcriptase inhibitors, and protease inhibitors in the treatment of HIV infections (aids). Adv. Pharmacol. 2013, 67, 317–358. [Google Scholar] [PubMed]
- De Clercq, E. Current race in the development of daas (direct-acting antivirals) against HCV. Biochem. Pharmacol. 2014, 89, 441–452. [Google Scholar] [CrossRef] [PubMed]
- Netzler, N.E.; Enosi Tuipulotu, D.; Eltahla, A.A.; Lun, J.H.; Ferla, S.; Brancale, A.; Urakova, N.; Frese, M.; Strive, T.; Mackenzie, J.M.; et al. Broad-spectrum non-nucleoside inhibitors for caliciviruses. Antivir. Res. 2017, 146, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Urakova, N.; Netzler, N.; Kelly, A.G.; Frese, M.; White, P.A.; Strive, T. Purification and biochemical characterisation of rabbit calicivirus RNA-dependent RNA polymerases and identification of non-nucleoside inhibitors. Viruses 2016, 8, 100. [Google Scholar] [CrossRef] [PubMed]
- Eltahla, A.A.; Lackovic, K.; Marquis, C.; Eden, J.S.; White, P.A. A fluorescence-based high-throughput screen to identify small compound inhibitors of the genotype 3a hepatitis C virus RNA polymerase. J. Biomol. Screen. 2013, 18, 1027–1034. [Google Scholar] [CrossRef] [PubMed]
- Eltahla, A.A.; Lim, K.L.; Eden, J.S.; Kelly, A.G.; Mackenzie, J.M.; White, P.A. Nonnucleoside inhibitors of norovirus RNA polymerase: Scaffolds for rational drug design. Antimicrob. Agents Chemother. 2014, 58, 3115–3123. [Google Scholar] [CrossRef] [PubMed]
- Bidawid, S.; Malik, N.; Adegbunrin, O.; Sattar, S.A.; Farber, J.M. A feline kidney cell line-based plaque assay for feline calicivirus, a surrogate for norwalk virus. J. Virol. Methods 2003, 107, 163–167. [Google Scholar] [CrossRef]
- Ianevski, A.; He, L.; Aittokallio, T.; Tang, J. Synergyfinder: A web application for analyzing drug combination dose-response matrix data. Bioinformatics 2017, 33, 2413–2415. [Google Scholar] [CrossRef] [PubMed]
- Yadav, B.; Wennerberg, K.; Aittokallio, T.; Tang, J. Searching for drug synergy in complex dose-response landscapes using an interaction potency model. Comput. Struct. Biotechnol. J. 2015, 13, 504–513. [Google Scholar] [CrossRef] [PubMed]
- Helps, C.; Lait, P.; Tasker, S.; Harbour, D. Melting curve analysis of feline calicivirus isolates detected by real-time reverse transcription PCR. J. Virol. Methods 2002, 106, 241–244. [Google Scholar] [CrossRef]
- Cotin, S.; Calliste, C.A.; Mazeron, M.C.; Hantz, S.; Duroux, J.L.; Rawlinson, W.D.; Ploy, M.C.; Alain, S. Eight flavonoids and their potential as inhibitors of human cytomegalovirus replication. Antivir. Res. 2012, 96, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Tarantino, D.; Pezzullo, M.; Mastrangelo, E.; Croci, R.; Rohayem, J.; Robel, I.; Bolognesi, M.; Milani, M. Naphthalene-sulfonate inhibitors of human norovirus RNA-dependent RNA-polymerase. Antivir. Res. 2014, 102, 23–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferla, S.; Netzler, N.E.; Ferla, S.; Veronese, S.; Tuipulotu, D.E.; Guccione, S.; Brancale, A.; White, P.A.; Bassetto, M. In silico screening for human norovirus antivirals reveals a novel non-nucleoside inhibitor of the viral polymerase. Sci. Rep. 2018, 8, 4129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gentles, R.G.; Ding, M.; Bender, J.A.; Bergstrom, C.P.; Grant-Young, K.; Hewawasam, P.; Hudyma, T.; Martin, S.; Nickel, A.; Regueiro-Ren, A.; et al. Discovery and preclinical characterization of the cyclopropylindolobenzazepine BMS-791325, a potent allosteric inhibitor of the hepatitis C virus NS5B polymerase. J. Med. Chem. 2014, 57, 1855–1879. [Google Scholar] [CrossRef] [PubMed]
- Devogelaere, B.; Berke, J.M.; Vijgen, L.; Dehertogh, P.; Fransen, E.; Cleiren, E.; van der Helm, L.; Nyanguile, O.; Tahri, A.; Amssoms, K.; et al. TMC647055, a potent nonnucleoside hepatitis C virus NS5B polymerase inhibitor with cross-genotypic coverage. Antimicrob. Agents Chemother. 2012, 56, 4676–4684. [Google Scholar] [CrossRef] [PubMed]
- Hirashima, S.; Suzuki, T.; Ishida, T.; Noji, S.; Yata, S.; Ando, I.; Komatsu, M.; Ikeda, S.; Hashimoto, H. Benzimidazole derivatives bearing substituted biphenyls as hepatitis C virus NS5B RNA-dependent RNA polymerase inhibitors: Structure-activity relationship studies and identification of a potent and highly selective inhibitor JTK-109. J. Med. Chem. 2006, 49, 4721–4736. [Google Scholar] [CrossRef] [PubMed]
- Rocha-Pereira, J.; Jochmans, D.; Dallmeier, K.; Leyssen, P.; Cunha, R.; Costa, I.; Nascimento, M.S.; Neyts, J. Inhibition of norovirus replication by the nucleoside analogue 2′-c-methylcytidine. Biochem. Biophys. Res. Commun. 2012, 427, 796–800. [Google Scholar] [CrossRef] [PubMed]
- Lam, A.M.; Murakami, E.; Espiritu, C.; Steuer, H.M.; Niu, C.; Keilman, M.; Bao, H.; Zennou, V.; Bourne, N.; Julander, J.G.; et al. Psi-7851, a pronucleotide of beta-d-2′-deoxy-2′-fluoro-2′-c-methyluridine monophosphate, is a potent and pan-genotype inhibitor of hepatitis C virus replication. Antimicrob. Agents Chemother. 2010, 54, 3187–3196. [Google Scholar] [CrossRef] [PubMed]
- Furuta, Y.; Gowen, B.B.; Takahashi, K.; Shiraki, K.; Smee, D.F.; Barnard, D.L. Favipiravir (T-705), a novel viral RNA polymerase inhibitor. Antivir. Res. 2013, 100, 446–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olsen, D.B.; Eldrup, A.B.; Bartholomew, L.; Bhat, B.; Bosserman, M.R.; Ceccacci, A.; Colwell, L.F.; Fay, J.F.; Flores, O.A.; Getty, K.L.; et al. A 7-deaza-adenosine analog is a potent and selective inhibitor of hepatitis C virus replication with excellent pharmacokinetic properties. Antimicrob. Agents Chemother. 2004, 48, 3944–3953. [Google Scholar] [CrossRef] [PubMed]
- Boyd, M.R.; Bacon, T.H.; Sutton, D.; Cole, M. Antiherpesvirus activity of 9-(4-hydroxy-3-hydroxy-methylbut-1-yl)guanine (BRL 39123) in cell culture. Antimicrob. Agents Chemother. 1987, 31, 1238–1242. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.O.; Takahashi, D.; Prakash, O.; Kim, Y. Characterization and inhibition of norovirus proteases of genogroups I and II using a fluorescence resonance energy transfer assay. Virology 2012, 423, 125–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dragovich, P.S.; Prins, T.J.; Zhou, R.; Fuhrman, S.A.; Patick, A.K.; Matthews, D.A.; Ford, C.E.; Meador, J.W., 3rd; Ferre, R.A.; Worland, S.T. Structure-based design, synthesis, and biological evaluation of irreversible human rhinovirus 3c protease inhibitors. 3. Structure-activity studies of ketomethylene-containing peptidomimetics. J. Med. Chem. 1999, 42, 1203–1212. [Google Scholar] [CrossRef] [PubMed]
- Rossignol, J.F. Nitazoxanide: A first-in-class broad-spectrum antiviral agent. Antivir. Res. 2014, 110, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Sosnovtseva, S.A.; Sosnovtsev, S.V.; Green, K.Y. Mapping of the feline calicivirus proteinase responsible for autocatalytic processing of the nonstructural polyprotein and identification of a stable proteinase-polymerase precursor protein. J. Virol. 1999, 73, 6626–6633. [Google Scholar] [PubMed]
- Radford, A.D.; Dawson, S.; Gaskell, R.M.; Foley, J.; Hurley, K.; Pedersen, N.C. Haemorrhagic fever, oedema and high mortality associated with FCV infection. Vet. Rec. 2002, 151, 155. [Google Scholar] [PubMed]
- Rocha-Pereira, J.; Jochmans, D.; Debing, Y.; Verbeken, E.; Nascimento, M.S.; Neyts, J. The viral polymerase inhibitor 2′-c-methylcytidine inhibits norwalk virus replication and protects against norovirus-induced diarrhea and mortality in a mouse model. J. Virol. 2013, 87, 11798–11805. [Google Scholar] [CrossRef] [PubMed]
- Rocha-Pereira, J.; Van Dycke, J.; Neyts, J. Treatment with a nucleoside polymerase inhibitor reduces shedding of murine norovirus in stool to undetectable levels without emergence of drug-resistant variants. Antimicrob. Agents Chemother. 2015, 60, 1907–1911. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Tucker, K.; Lin, X.; Kao, C.C.; Shaw, K.; Tan, H.; Symons, J.; Behera, I.; Rajwanshi, V.K.; Dyatkina, N.; et al. Biochemical evaluation of the inhibition properties of favipiravir and 2′-c-methyl-cytidine triphosphates against human and mouse norovirus RNA polymerases. Antimicrob. Agents Chemother. 2015, 59, 7504–7516. [Google Scholar] [CrossRef] [PubMed]
- Kolawole, A.O.; Rocha-Pereira, J.; Elftman, M.D.; Neyts, J.; Wobus, C.E. Inhibition of human norovirus by a viral polymerase inhibitor in the b cell culture system and in the mouse model. Antivir. Res. 2016, 132, 46–49. [Google Scholar] [CrossRef] [PubMed]
- Rossignol, J.F.; Ayoub, A.; Ayers, M.S. Treatment of diarrhea caused by cryptosporidium parvum: A prospective randomized, double-blind, placebo-controlled study of nitazoxanide. J. Infect. Dis. 2001, 184, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Fox, L.M.; Saravolatz, L.D. Nitazoxanide: A new thiazolide antiparasitic agent. Clin. Infect. Dis. 2005, 40, 1173–1180. [Google Scholar] [CrossRef] [PubMed]
- Rossignol, J.F.; El-Gohary, Y.M. Nitazoxanide in the treatment of viral gastroenteritis: A randomized double-blind placebo-controlled clinical trial. Aliment. Pharmacol. Ther. 2006, 24, 1423–1430. [Google Scholar] [CrossRef] [PubMed]
- Dang, W.; Yin, Y.; Peppelenbosch, M.P.; Pan, Q. Opposing effects of nitazoxanide on murine and human norovirus. J. Infect. Dis. 2017, 216, 780–782. [Google Scholar] [CrossRef] [PubMed]
- Waddington, C.S.; McLeod, C.; Morris, P.; Bowen, A.; Naunton, M.; Carapetis, J.; Grimwood, K.; Robins-Browne, R.; Kirkwood, C.D.; Baird, R.; et al. The nice-gut trial protocol: A randomised, placebo controlled trial of oral nitazoxanide for the empiric treatment of acute gastroenteritis among australian aboriginal children. BMJ Open 2018, 8, e019632. [Google Scholar] [PubMed]
- Haubrich, K.; Gantt, S.; Blydt-Hansen, T. Successful treatment of chronic norovirus gastroenteritis with nitazoxanide in a pediatric kidney transplant recipient. Pediatr. Transplant. 2018, 22, e13186. [Google Scholar] [CrossRef] [PubMed]
- Siddiq, D.M.; Koo, H.L.; Adachi, J.A.; Viola, G.M. Norovirus gastroenteritis successfully treated with nitazoxanide. J. Infect. 2011, 63, 394–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gookin, J.L.; Levy, M.G.; Law, J.M.; Papich, M.G.; Poore, M.F.; Breitschwerdt, E.B. Experimental infection of cats with tritrichomonas foetus. Am. J. Vet. Res. 2001, 62, 1690–1697. [Google Scholar] [CrossRef] [PubMed]
- Moron-Soto, M.; Gutierrez, L.; Sumano, H.; Tapia, G.; Alcala-Canto, Y. Efficacy of nitazoxanide to treat natural giardia infections in dogs. Parasit. Vectors 2017, 10, 52. [Google Scholar] [CrossRef] [PubMed]
- Sosnovtsev, S.V.; Sosnovtseva, S.A.; Green, K.Y. Cleavage of the feline calicivirus capsid precursor is mediated by a virus-encoded proteinase. J. Virol. 1998, 72, 3051–3059. [Google Scholar] [PubMed]
- Croci, R.; Tarantino, D.; Milani, M.; Pezzullo, M.; Rohayem, J.; Bolognesi, M.; Mastrangelo, E. Ppnds inhibits murine norovirus RNA-dependent RNA-polymerase mimicking two RNA stacking bases. FEBS Lett. 2014, 588, 1720–1725. [Google Scholar] [CrossRef] [PubMed]
- Croci, R.; Pezzullo, M.; Tarantino, D.; Milani, M.; Tsay, S.C.; Sureshbabu, R.; Tsai, Y.J.; Mastrangelo, E.; Rohayem, J.; Bolognesi, M.; et al. Structural bases of norovirus RNA dependent RNA polymerase inhibition by novel suramin-related compounds. PLoS ONE 2014, 9, e91765. [Google Scholar] [CrossRef] [PubMed]
- Klinger, M.; Bofill-Cardona, E.; Mayer, B.; Nanoff, C.; Freissmuth, M.; Hohenegger, M. Suramin and the suramin analogue NF307 discriminate among calmodulin-binding sites. Biochem. J. 2001, 355, 827–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed-Belkacem, A.; Guichou, J.F.; Brillet, R.; Ahnou, N.; Hernandez, E.; Pallier, C.; Pawlotsky, J.M. Inhibition of RNA binding to hepatitis C virus RNA-dependent RNA polymerase: A new mechanism for antiviral intervention. Nucleic Acids Res. 2014, 42, 9399–9409. [Google Scholar] [CrossRef] [PubMed]
- Lani, R.; Hassandarvish, P.; Shu, M.H.; Phoon, W.H.; Chu, J.J.; Higgs, S.; Vanlandingham, D.; Abu Bakar, S.; Zandi, K. Antiviral activity of selected flavonoids against chikungunya virus. Antivir. Res. 2016, 133, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Xi, K.; Grum-Tokars, V.; Xu, X.; Ratia, K.; Fu, W.; Houser, K.V.; Baker, S.C.; Johnson, M.E.; Mesecar, A.D. Structure-based design, synthesis, and biological evaluation of peptidomimetic SARS-CoV 3CLpro inhibitors. Bioorg. Med. Chem. Lett. 2007, 17, 5876–5880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wegzyn, C.M.; Wyles, D.L. Antiviral drug advances in the treatment of human immunodeficiency virus (HIV) and chronic hepatitis C virus (HCV). Curr. Opin. Pharmacol. 2012, 12, 556–561. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, N.C.; Kim, Y.; Liu, H.; Galasiti Kankanamalage, A.C.; Eckstrand, C.; Groutas, W.C.; Bannasch, M.; Meadows, J.M.; Chang, K.O. Efficacy of a 3c-like protease inhibitor in treating various forms of acquired feline infectious peritonitis. J. Feline Med. Surg. 2018, 20, 378–392. [Google Scholar] [CrossRef] [PubMed]
- Thomasy, S.M.; Lim, C.C.; Reilly, C.M.; Kass, P.H.; Lappin, M.R.; Maggs, D.J. Evaluation of orally administered famciclovir in cats experimentally infected with feline herpesvirus type-1. Am. J. Vet. Res. 2011, 72, 85–95. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Molecular Mass (g/mol) | Stage of Antiviral Development | Original Target Viral Family | Antiviral Class | Cell Viability CC50 (µM) | Plaque Reduction Assay EC50 (µM) | RdRp Activity [% of Mock at 10 µM] (IC50 µM) | In vitro Protease IC50 (µM) | Reference |
|---|---|---|---|---|---|---|---|---|---|
| Quercetagetin | 318.2 | Research | Herpesviridae | NNI | >100 | >10 | 11.7 (2.8) | ND | Cotin et al. [41] |
| PPNDS | 694.3 | Research | Caliciviridae | NNI | >100 | >10 | 7.4 (2.7) | ND | Tarantino et al. [42] |
| Compound 54 | 485.5 | Research | Caliciviridae | NNI | 55.8 | ND | 65.7 | ND | Ferla et al. [43] |
| Beclabuvir | 659.8 | Phase 2 | Flaviviridae | NNI | 27.5 | ND | 92.2 | ND | Gentles et al. [44] |
| TMC-647055 | 606.7 | Phase 2 | Flaviviridae | NNI | 27.1 | ND | 93.5 | ND | Devogelaere et al. [45] |
| JTK-109 | 638.1 | Phase 2 | Flaviviridae | NNI | 11.9 | ND | 89.8 | ND | Hirashima et al. [46] |
| 2CMC | 257.2 | Pre-clinical | Flaviviridae | NA | >100 | 2.6 | ND | ND | Rocha-Pereira et al. [47] |
| Sofosbuvir | 529.5 | Approved | Flaviviridae | NA | >100 | >10 | ND | ND | Lam et al. [48] |
| T-705 | 157.1 | Phase 3 | Orthomyxoviridae | NA | >100 | >10 | ND | ND | Furuta et al. [49] |
| 7D2M | 280.3 | Research | Flaviviridae | NA | >100 | >10 | ND | ND | Olsen et al. [50] |
| Famciclovir | 321.3 | Approved | Herpesviridae | NA | >100 | >50 | ND | ND | Boyd et al. [51] |
| GC376 | 507.5 | Research | Broad spectrum | PI | >100 | >10 | ND | 18.7 | Kim et al. [29] |
| Chymostatin | 607.7 | Research | Caliciviridae | PI | ND | ND | ND | >50 | Chang et al. [52] |
| Rupintrivir | 598.7 | Research | Picornaviridae | PI | >100 | ND | ND | >50 | Dragovich et al. [53] |
| Nitazoxanide | 307.3 | Approved | Broad spectrum | Unknown | 12.7 | 0.6 | >10 | >10 | Rossignol [54] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fumian, T.M.; Tuipulotu, D.E.; Netzler, N.E.; Lun, J.H.; Russo, A.G.; Yan, G.J.H.; White, P.A. Potential Therapeutic Agents for Feline Calicivirus Infection. Viruses 2018, 10, 433. https://doi.org/10.3390/v10080433
Fumian TM, Tuipulotu DE, Netzler NE, Lun JH, Russo AG, Yan GJH, White PA. Potential Therapeutic Agents for Feline Calicivirus Infection. Viruses. 2018; 10(8):433. https://doi.org/10.3390/v10080433
Chicago/Turabian StyleFumian, Tulio M., Daniel Enosi Tuipulotu, Natalie E. Netzler, Jennifer H. Lun, Alice G. Russo, Grace J. H. Yan, and Peter A. White. 2018. "Potential Therapeutic Agents for Feline Calicivirus Infection" Viruses 10, no. 8: 433. https://doi.org/10.3390/v10080433
APA StyleFumian, T. M., Tuipulotu, D. E., Netzler, N. E., Lun, J. H., Russo, A. G., Yan, G. J. H., & White, P. A. (2018). Potential Therapeutic Agents for Feline Calicivirus Infection. Viruses, 10(8), 433. https://doi.org/10.3390/v10080433