Host and Viral Proteins Modulating Ebola and Marburg Virus Egress


 ,
, 
Abstract
:1. Introduction
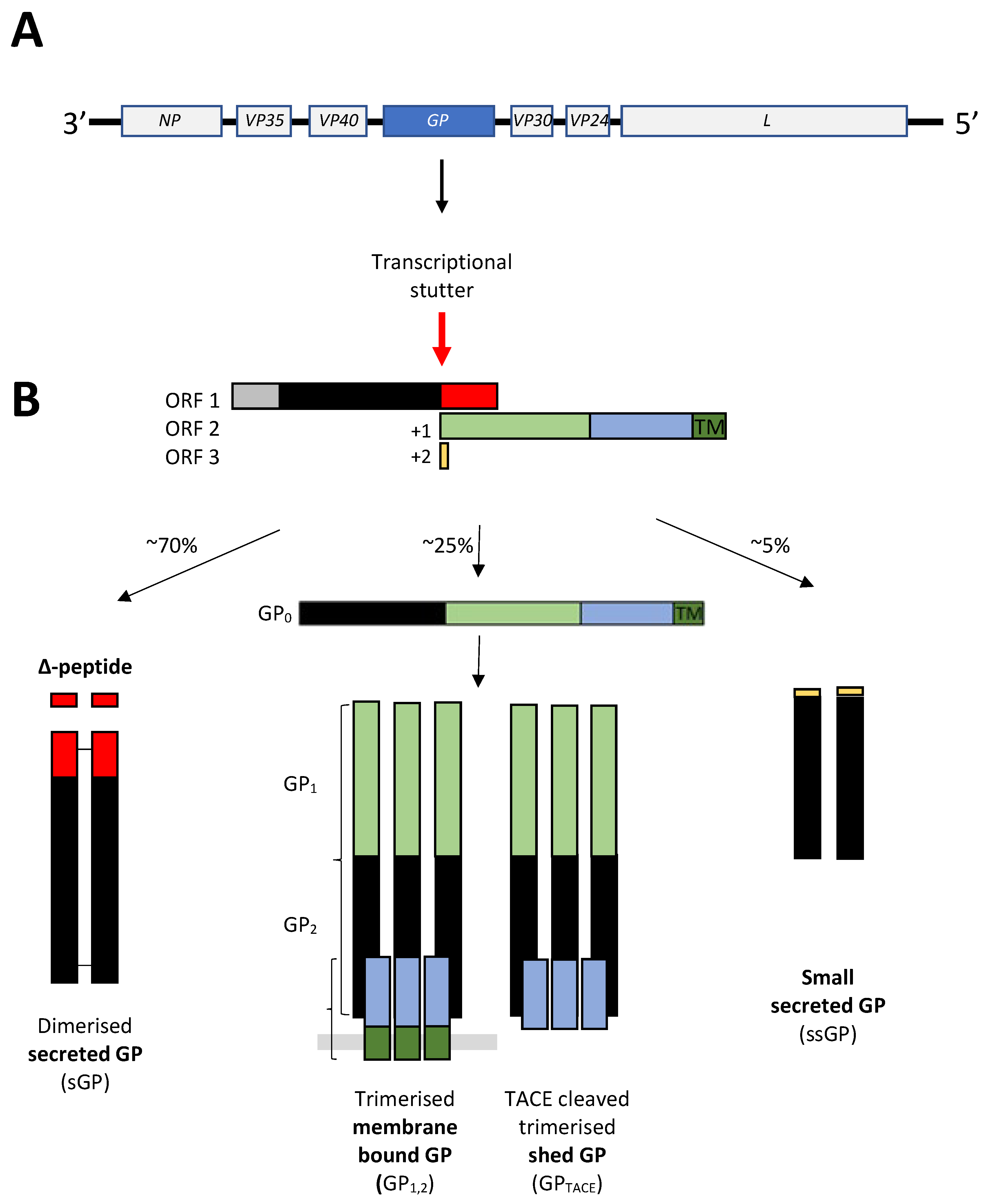
1.1. Filoviral Particle and Genome Structure: A Brief Overview
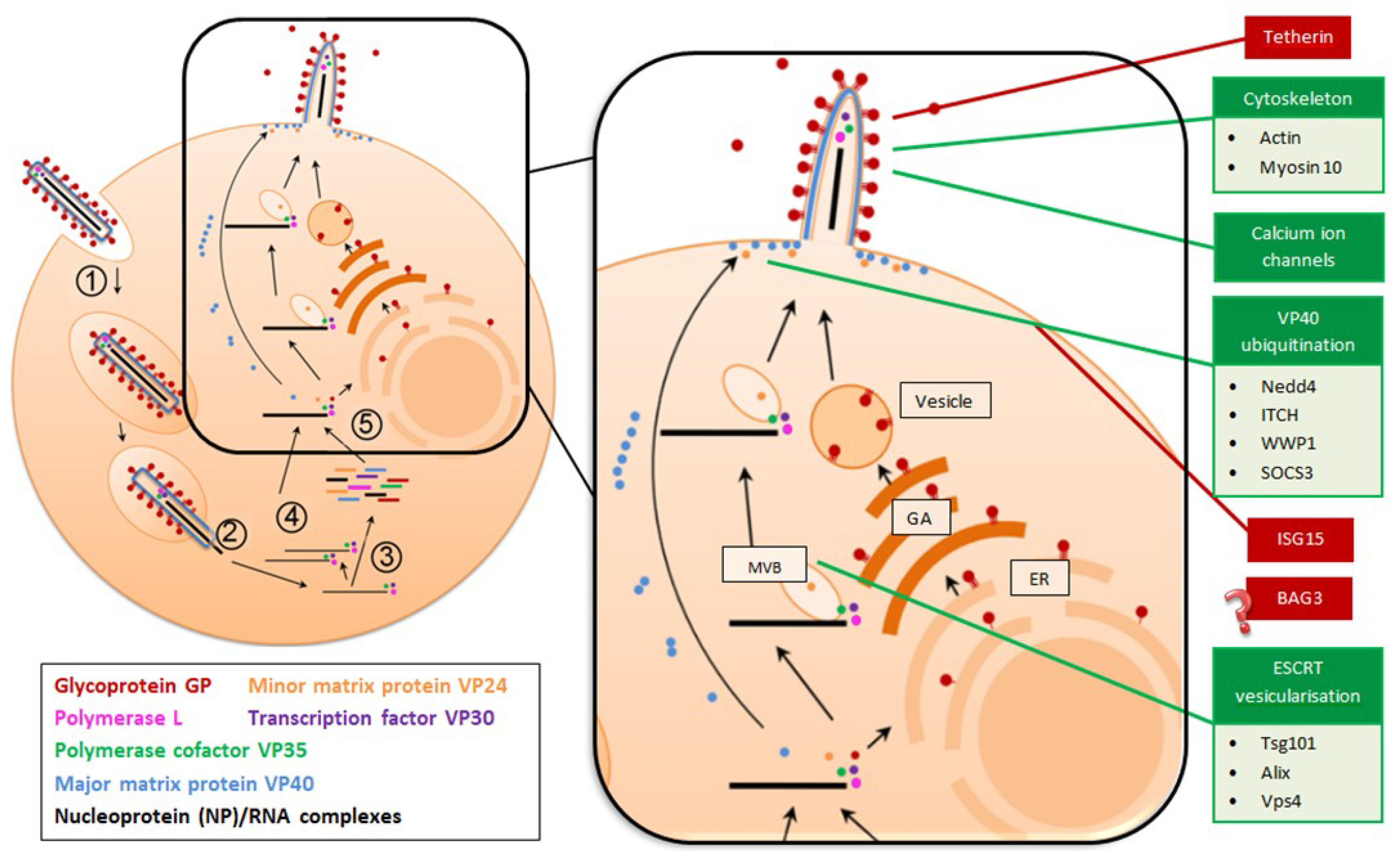
1.2. Filoviruses Bud from the Host Cell to Complete the Replication Cycle
2. Viral Proteins that Promote Filovirus Budding
2.1. Central Role of the VP40 Matrix Protein in Filoviruses Egress
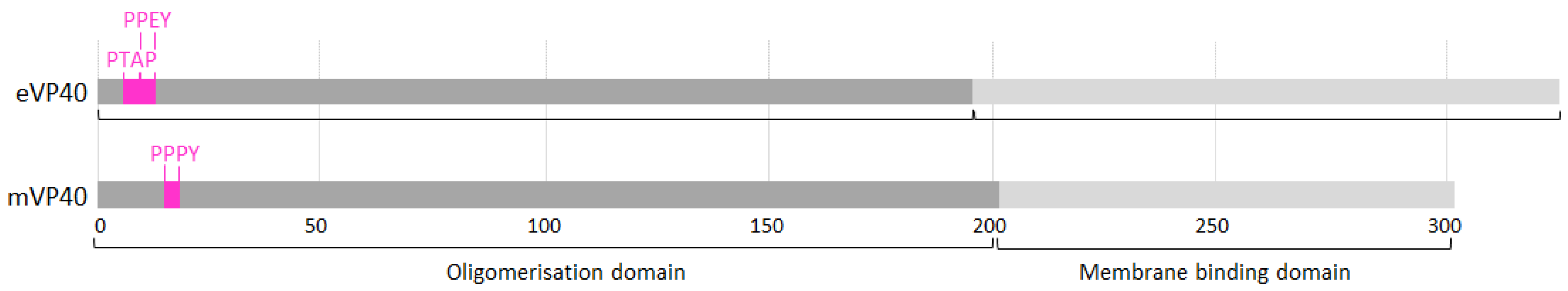
2.1.1. VP40 Late Domain
2.1.2. VP40 Oligomerisation and Membrane Binding Motifs
2.2. GP and NP are Promoters of VP40 Egress
3. Host Cell Proteins that Promote Filovirus Budding
3.1. Hijacking of the Host Cytoskeleton Facilitates Filovirus Budding
3.2. VP40 Ubiquitination is Necessary for Filovirus Budding
3.2.1. Nedd4
3.2.2. ITCH
3.2.3. WWP1
3.2.4. SOCS3
3.3. The Host ESCRT Pathway Drives Filovirus Egress
3.3.1. Tsg101
3.3.2. Alix
3.3.3. Vps4
3.4. Free Calcium Ions Promote Efficient Budding of Filoviruses
4. Host Inhibitors of Budding
4.1. Tetherin Antagonises Virion Release from the Host Cell Surface
4.2. ISG15 Interferes with Nedd4 Ubiquitination of VP40
4.3. Bag3, A Newly Identified Inhibitor of Filovirus Budding
4.4. Application of Egress Inhibitors in Drug Development
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Brainard, J.; Hooper, L.; Pond, K.; Edmunds, K.; Hunter, P.R. Risk factors for transmission of Ebola or Marburg virus disease: A systematic review and meta-analysis. Int. J. Epidemiol. 2016, 45, 102–116. [Google Scholar] [CrossRef] [PubMed]
- Towner, J.S.; Amman, B.R.; Sealy, T.K.; Reeder Carroll, S.A.; Comer, J.A.; Kemp, A.; Swanepoel, R.; Paddock, C.D.; Balinandi, S.; Khristova, M.L.; et al. Isolation of genetically diverse Marburg viruses from Egyptian fruit bats. PLoS Pathog. 2009, 5, e1000536. [Google Scholar] [CrossRef] [PubMed]
- Leroy, E.M.; Kumulungui, B.; Pourrut, X.; Rouquet, P.; Hassanin, A.; Yaba, P.; Délicat, A.; Paweska, J.T.; Gonzalez, J.P.; Swanepoel, R. Fruit bats as reservoirs of Ebola virus. Nature 2005, 438, 575–576. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, T.; Anthony, S.J.; Gbakima, A.; Bird, B.H.; Bangura, J.; Tremeau-Bravard, A.; Belaganahalli, M.N.; Wells, H.L.; Dhanota, J.K.; Liang, E.; et al. The discovery of Bombali virus adds further support for bats as hosts of ebolaviruses. Nat. Microbiol. 2018, 3, 1084–1089. [Google Scholar] [CrossRef] [PubMed]
- Towner, J.S.; Khristova, M.L.; Sealy, T.K.; Vincent, M.J.; Erickson, B.R.; Bawiec, D.A.; Hartman, A.L.; Comer, J.A.; Zaki, S.R.; Stroher, U.; et al. Marburgvirus genomics and association with a large hemorrhagic fever outbreak in Angola. J. Virol. 2006, 80, 6497–6516. [Google Scholar] [CrossRef] [PubMed]
- De Clercq, E. Ebola virus (EBOV) infection: Therapeutic strategies. Biochem. Pharmacol. 2015, 93, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Henao-Restrepo, A.M.; Camacho, A.; Longini, I.M.; Watson, C.H.; Edmunds, W.J.; Egger, M.; Carroll, M.W.; Dean, N.E.; Diatta, I.; Doumbia, M.; et al. Efficacy and effectiveness of an rVSV-vectored vaccine in preventing Ebola virus disease: Final results from the Guinea ring vaccination, open-label, cluster-randomised trial (Ebola Ca Suffit!). Lancet 2017, 389, 505–518. [Google Scholar] [CrossRef]
- The PREVAIL II Writing Group, for the Multi-National PREVAIL II Study Team. A randomized, controlled trial of ZMapp for Ebola virus infection. N. Engl. J. Med. 2016, 375, 1448–1456. [Google Scholar] [CrossRef]
- Pavot, V. Ebola virus vaccines: Where do we stand? Clin. Immunol. 2016, 173, 44–49. [Google Scholar] [CrossRef]
- Feldmann, H.; Klenk, H.D.; Sanchez, A. Molecular biology and evolution of filoviruses. Arch. Virol. Suppl. 1993, 7, 81–100. [Google Scholar]
- Martin, B.; Hoenen, T.; Canard, B.; Decroly, E. Filovirus proteins for antiviral drug discovery: A structure/function analysis of surface glycoproteins and virus entry. Antiviral Res. 2016, 135, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Whelan, S.P.J.; Barr, J.N.; Wertz, G.W. Transcription and replication of nonsegmented negative-strand RNA viruses. Curr. Top. Microbiol. Immunol. 2004, 283, 61–119. [Google Scholar] [PubMed]
- Licata, J.M.; Johnson, R.F.; Han, Z.; Harty, R.N. Contribution of Ebola virus glycoprotein, nucleoprotein, and VP24 to budding of VP40 virus-like particles. J. Virol. 2004, 78, 7344–7351. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, A.; Trappier, S.G.; Mahy, B.W.J.; Peters, C.J.; Nichol, S.T. The virion glycoproteins of Ebola viruses are encoded in two reading frames and are expressed through transcriptional editing. Proc. Natl. Acad. Sci. USA 1996, 93, 3602–3607. [Google Scholar] [CrossRef] [PubMed]
- Volchkov, V.E.; Feldmann, H.; Volchkova, V.A.; Klenk, H.D. Processing of the Ebola virus glycoprotein by the proprotein convertase furin. Proc. Natl. Acad. Sci. USA 1998, 95, 5762–5767. [Google Scholar] [CrossRef] [Green Version]
- Wool-Lewis, R.J.; Bates, P. Characterization of Ebola virus entry by using pseudotyped viruses: Identification of receptor-deficient cell lines. J. Virol. 1998, 72, 3155–3160. [Google Scholar] [PubMed]
- Dolnik, O.; Volchkova, V.; Garten, W.; Carbonnelle, C.; Becker, S.; Kahnt, J.; Stroher, U.; Klenk, H.D.; Volchkov, V. Ectodomain shedding of the glycoprotein GP of Ebola virus. EMBO J. 2004, 23, 2175–2184. [Google Scholar] [CrossRef] [Green Version]
- Mehedi, M.; Falzarano, D.; Seebach, J.; Hu, X.; Carpenter, M.S.; Schnittler, H.-J.; Feldmann, H. A new Ebola virus nonstructural glycoprotein expressed through RNA editing. J. Virol. 2011, 85, 5406–5414. [Google Scholar] [CrossRef]
- Cook, J.D.; Lee, J.E. The secret life of viral entry glycoproteins: Moonlighting in immune evasion. PLoS Pathog. 2013, 9, e1003258. [Google Scholar] [CrossRef]
- Davey, R.A.; Shtanko, O.; Anantpadma, M.; Sakurai, Y.; Chandran, K.; Maury, W. Mechanisms of filovirus entry. Curr. Top. Microbiol. Immunol. 2017, 411, 323–352. [Google Scholar]
- Herbert, A.S.; Davidson, C.; Kuehne, A.I.; Bakken, R.; Braigen, S.Z.; Gunn, K.E.; Whelan, S.P.; Brummelkamp, T.R.; Twenhafel, N.A.; Chandran, K.; et al. Niemann-Pick C1 is essential for Ebolavirus replication and pathogenesis. mBio 2015, 6, e00565-15. [Google Scholar] [CrossRef] [PubMed]
- Nanbo, A.; Imai, M.; Watanabe, S.; Noda, T.; Takahashi, K.; Neumann, G.; Halfmann, P.; Kawaoka, Y. Ebolavirus is internalized into host cells via macropinocytosis in a viral glycoprotein-dependent manner. PLoS Pathog. 2010, 6, e1001121. [Google Scholar] [CrossRef] [PubMed]
- Hood, C.L.; Abraham, J.; Boyington, J.C.; Leung, K.; Kwong, P.D.; Nabel, G.J. Biochemical and structural characterization of cathepsin L-processed Ebola virus glycoprotein: implications for viral entry and immunogenicity. J. Virol. 2010, 84, 2972–2982. [Google Scholar] [CrossRef] [PubMed]
- Chandran, K.; Sullivan, N.J.; Felbor, U.; Whelan, S.P.; Cunningham, J.M. Endosomal proteolysis of the Ebola virus glycoprotein is necessary for infection. Science 2005, 308, 1643–1645. [Google Scholar] [CrossRef] [PubMed]
- Kaletsky, R.L.; Simmons, G.; Bates, P. Proteolysis of the Ebola virus glycoproteins enhances virus binding and infectivity. J. Virol. 2007, 81, 13378–13384. [Google Scholar] [CrossRef]
- Schornberg, K.; Matsuyama, S.; Kabsch, K.; Delos, S.; Bouton, A.; White, J. Role of endosomal cathepsins in entry mediated by the Ebola virus glycoprotein. J. Virol. 2006, 80, 4174–4178. [Google Scholar] [CrossRef]
- Brecher, M.; Schornberg, K.L.; Delos, S.E.; Fusco, M.L.; Saphire, E.O.; White, J.M. Cathepsin cleavage potentiates the ebola virus glycoprotein to undergo a subsequent fusion-relevant conformational change. J. Virol. 2012, 86, 364–372. [Google Scholar] [CrossRef]
- Weik, M.; Modrof, J.; Klenk, H.; Becker, S.; Mühlberger, E. Ebola virus VP30-mediated transcription is regulated by RNA secondary structure formation. J. Virol. 2002, 76, 8532–8539. [Google Scholar] [CrossRef]
- Muhlberger, E.; Lotfering, B.; Klenk, H.; Becker, S. Three of the Four Nucleocapsid Proteins of Marburg Virus, NP, VP35, and L, Are Sufficient To Mediate Replication and Transcription of Marburg Virus-Specific Monocistronic Minigenomes. J. Virol. 1998, 72, 8756–8764. [Google Scholar]
- Hoenen, T.; Shabman, R.S.; Groseth, A.; Herwig, A.; Weber, M.; Schudt, G.; Dolnik, O.; Basler, C.F.; Becker, S.; Feldmann, H. Inclusion bodies are a site of ebolavirus replication. J. Virol. 2012, 86, 11779–11788. [Google Scholar] [CrossRef]
- Nanbo, A.; Watanabe, S.; Halfmann, P.; Kawaoka, Y. The spatio-temporal distribution dynamics of Ebola virus proteins and RNA in infected cells. Sci. Rep. 2013, 3, 1206. [Google Scholar] [CrossRef] [PubMed]
- Bavari, S.; Bosio, C.M.; Wiegand, E.; Ruthel, G.; Will, A.B.; Geisbert, T.W.; Hevey, M.; Schmaljohn, C.; Schmaljohn, A.; Aman, M.J. Lipid raft microdomains: A gateway for compartmentalized trafficking of Ebola and Marburg viruses. J. Exp. Med. 2002, 195, 593–602. [Google Scholar] [CrossRef] [PubMed]
- Elliott, L.H.; Kiley, M.P.; McCormick, J.B. Descriptive analysis of Ebola virus proteins. Virology 1985, 147, 169–176. [Google Scholar] [CrossRef]
- Geisbert, T.W.; Jahrling, P.B. Differentiation of filoviruses by electron microscopy. Virus Res. 1995, 39, 129–150. [Google Scholar] [CrossRef] [Green Version]
- Noda, T.; Sagara, H.; Suzuki, E.; Takada, A.; Kida, H.; Kawaoka, Y. Ebola virus VP40 drives the formation of virus-like filamentous particles along with GP. J. Virol. 2002, 76, 4855–4865. [Google Scholar] [CrossRef] [PubMed]
- Noda, T.; Watanabe, S.; Sagara, H.; Kawaoka, Y. Mapping of the VP40-binding regions of the nucleoprotein of Ebola virus. J. Virol. 2007, 81, 3554–3562. [Google Scholar] [CrossRef]
- Timmins, J.; Scianimanico, S.; Schoehn, G.; Weissenhorn, W. Vesicular release of Ebola virus matrix protein VP40. Virology 2001, 283, 1–6. [Google Scholar] [CrossRef]
- Wenigenrath, J.; Kolesnikova, L.; Hoenen, T.; Mittler, E.; Becker, S. Establishment and application of an infectious virus-like particle system for Marburg virus. J. Gen. Virol. 2010, 91, 1325–1334. [Google Scholar] [CrossRef] [Green Version]
- Oda, S.; Noda, T.; Wijesinghe, K.J.; Halfmann, P.; Bornholdt, Z.A.; Abelson, D.M.; Armbrust, T.; Stahelin, R.V.; Kawaoka, Y.; Saphire, E.O. Crystal structure of Marburg virus VP40 reveals a broad, basic patch for matrix assembly and a requirement of the N-terminal domain for immunosuppression. J. Virol. 2015, 90, 1839–1848. [Google Scholar] [CrossRef]
- McCarthy, S.E.; Johnson, R.F.; Zhang, Y.A.; Sunyer, J.O.; Harty, R.N. Role for amino acids 212KLR214 of Ebola virus VP40 in assembly and budding. J. Virol. 2007, 81, 11452–11460. [Google Scholar] [CrossRef]
- Licata, J.M.; Simpson-Holley, M.; Wright, N.T.; Han, Z.; Paragas, J.; Harty, R.N. Overlapping motifs (PTAP and PPEY) within the Ebola virus VP40 protein function independently as late budding domains: Involvement of host proteins TSG101 and VPS-4. J. Virol. 2003, 77, 1812–1819. [Google Scholar] [CrossRef] [PubMed]
- Martin-Serrano, J.; Perez-Caballero, D.; Bieniasz, P.D. Context-dependent effects of L domains and ubiquitination on viral budding. J. Virol. 2004, 78, 5554–5563. [Google Scholar] [CrossRef] [PubMed]
- Jasenosky, L.D.; Neumann, G.; Lukashevich, I.; Kawaoka, Y. Ebola virus VP40-induced particle formation and association with the lipid bilayer. J. Virol. 2001, 75, 5205–5214. [Google Scholar] [CrossRef]
- Neumann, G.; Ebihara, H.; Takada, A.; Noda, T.; Kobasa, D.; Jasenosky, L.D.; Watanabe, S.; Kim, J.H.; Feldmann, H.; Kawaoka, Y. Ebola virus VP40 late domains are not essential for viral replication in cell culture. J. Virol. 2005, 79, 10300–10307. [Google Scholar] [CrossRef]
- Urata, S.; Yasuda, J. Regulation of Marburg virus (MARV) budding by Nedd4.1: A different WW domain of Nedd4.1 is critical for binding to MARV and Ebola virus VP40. J. Gen. Virol. 2010, 91, 228–234. [Google Scholar] [CrossRef]
- Makino, A.; Yamayoshi, S.; Shinya, K.; Noda, T.; Kawaoka, Y. Identification of amino acids in Marburg virus VP40 that are important for virus-like particle budding. J. Infect. Dis. 2011, 204 (Suppl. 3), S871–S877. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.I.; Sudol, M. The WW domain of Yes-associated protein binds a proline-rich ligand that differs from the consensus established for Src homology 3-binding modules. Proc. Natl. Acad. Sci. USA 1995, 92, 7819–7823. [Google Scholar] [CrossRef]
- Harvey, K.F.; Kumar, S. Nedd4-like proteins: An emerging family of ubiquitin-protein ligases implicated in diverse cellular functions. Trends Cell Biol. 1999, 9, 166–169. [Google Scholar] [CrossRef]
- Han, Z.; Sagum, C.A.; Bedford, M.T.; Sidhu, S.S.; Sudol, M.; Harty, R.N. ITCH E3 ubiquitin ligase interacts with Ebola virus VP40 to regulate budding. J. Virol. 2016, 90, 9163–9171. [Google Scholar] [CrossRef]
- Liang, J.; Sagum, C.A.; Bedford, M.T.; Sidhu, S.S.; Sudol, M.; Han, Z.; Harty, R.N. Chaperone-mediated autophagy protein BAG3 negatively regulates Ebola and Marburg VP40-mediated egress. PLoS Pathog. 2017, 13, e1006132. [Google Scholar]
- Bornholdt, Z.A.; Noda, T.; Abelson, D.M.; Halfmann, P.; Wood, M.R.; Kawaoka, Y.; Saphire, E.O. Structural rearrangement of Ebola virus VP40 begets multiple functions in the virus life cycle. Cell 2013, 154, 763–774. [Google Scholar] [CrossRef] [PubMed]
- Timmins, J.; Schoehn, G.; Kohlhaas, C.; Klenk, H.D.; Ruigrok, R.W.; Weissenhorn, W. Oligomerization and polymerization of the filovirus matrix protein VP40. Virology 2003, 312, 359–368. [Google Scholar] [CrossRef] [Green Version]
- Hoenen, T.; Volchkov, V.; Kolesnikova, L.; Mittler, E.; Timmins, J.; Ottmann, M.; Reynard, O.; Becker, S.; Weissenhorn, W. VP40 octamers are essential for Ebola virus replication. J. Virol. 2005, 79, 1898–1905. [Google Scholar] [CrossRef] [PubMed]
- Gomis-Rüth, F.X.; Dessen, A.; Timmins, J.; Bracher, A.; Kolesnikowa, L.; Becker, S.; Klenk, H.-D.; Weissenhorn, W. The matrix protein VP40 from Ebola virus octamerizes into pore-like structures with specific RNA binding properties. Structure 2003, 11, 423–433. [Google Scholar] [CrossRef]
- Hoenen, T.; Biedenkopf, N.; Zielecki, F.; Jung, S.; Groseth, A.; Feldmann, H.; Becker, S. Oligomerization of Ebola virus VP40 is essential for particle morphogenesis and regulation of viral transcription. J. Virol. 2010, 84, 7053–7063. [Google Scholar] [CrossRef] [PubMed]
- Gc, J.B.; Gerstman, B.S.; Stahelin, R.V.; Chapagain, P.P. The Ebola virus protein VP40 hexamer enhances the clustering of PI(4,5)P2 lipids in the plasma membrane. Phys. Chem. Chem. Phys. 2016, 18, 28409–28417. [Google Scholar] [CrossRef]
- Yamayoshi, S.; Kawaoka, Y. Mapping of a region of Ebola virus VP40 that is important in the production of virus-like particles. J. Infect. Dis. 2007, 196 (Suppl. 2), S291–S295. [Google Scholar] [CrossRef]
- Adu-Gyamfi, E.; Soni, S.P.; Xue, Y.; Digman, M.A.; Gratton, E.; Stahelin, R.V. The Ebola virus matrix protein penetrates into the plasma membrane: A key step in viral protein 40 (VP40) oligomerization and viral egress. J. Biol. Chem. 2013, 288, 5779–5789. [Google Scholar] [CrossRef]
- Silvestri, L.S.; Ruthel, G.; Kallstrom, G.; Warfield, K.L.; Swenson, D.L.; Nelle, T.; Iversen, P.L.; Bavari, S.; Aman, M.J. Involvement of vacuolar protein sorting pathway in Ebola virus release independent of TSG101 interaction. J. Infect. Dis. 2007, 196 (Suppl. 2), S264–S270. [Google Scholar] [CrossRef]
- Adu-Gyamfi, E.; Soni, S.P.; Jee, C.S.; Digman, M.A.; Gratton, E.; Stahelin, R.V. A loop region in the N-terminal domain of Ebola virus VP40 is important in viral assembly, budding, and egress. Viruses 2014, 6, 3837–3854. [Google Scholar] [CrossRef]
- Liu, Y.; Cocka, L.; Okumura, A.; Zhang, Y.A.; Sunyer, J.O.; Harty, R.N. Conserved motifs within Ebola and Marburg virus VP40 proteins are important for stability, localization, and subsequent budding of virus-like particles. J. Virol. 2010, 84, 2294–2303. [Google Scholar] [CrossRef]
- Liu, Y.; Harty, R.N. Viral and host proteins that modulate filovirus budding. Future Virol. 2010, 5, 481–491. [Google Scholar] [CrossRef] [Green Version]
- Ruthel, G.; Demmin, G.L.; Kallstrom, G.; Javid, M.P.; Badie, S.S.; Will, A.B.; Nelle, T.; Schokman, R.; Nguyen, T.L.; Carra, J.H.; et al. Association of ebola virus matrix protein VP40 with microtubules. J. Virol. 2005, 79, 4709–4719. [Google Scholar] [CrossRef]
- Panchal, R.G.; Ruthel, G.; Kenny, T.A.; Kallstrom, G.H.; Lane, D.; Badie, S.S.; Li, L.; Bavari, S.; Aman, M.J. In vivo oligomerization and raft localization of Ebola virus protein VP40 during vesicular budding. Proc. Natl. Acad. Sci. USA 2003, 100, 15936–15941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soni, S.P.; Adu-Gyamfi, E.; Yong, S.S.; Jee, C.S.; Stahelin, R.V. The Ebola virus matrix protein deeply penetrates the plasma membrane: An important step in viral egress. Biophys. J. 2013, 104, 1940–1949. [Google Scholar] [CrossRef] [PubMed]
- Yamayoshi, S.; Noda, T.; Ebihara, H.; Goto, H.; Morikawa, Y.; Lukashevich, I.S.; Neumann, G.; Feldmann, H.; Kawaoka, Y. Ebola virus matrix protein VP40 uses the COPII transport system for its intracellular transport. Cell Host Microbe 2008, 3, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Scianimanico, S.; Schoehn, G.; Timmins, J.; Ruigrok, R.H.; Klenk, H.D.; Weissenhorn, W. Membrane association induces a conformational change in the Ebola virus matrix protein. EMBO J. 2000, 19, 6732–6741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soni, S.P.; Stahelin, R.V. The Ebola virus matrix protein VP40 selectively induces vesiculation from phosphatidylserine-enriched membranes. J. Biol. Chem. 2014, 289, 33590–33597. [Google Scholar] [CrossRef]
- Rog, T.; Pasenkiewicz-Gierula, M. Cholesterol effects on the phospholipid condensation and packing in the bilayer: A molecular simulation study. FEBS Lett. 2001, 502, 68–71. [Google Scholar] [CrossRef]
- Wijesinghe, K.J.; Stahelin, R.V. Investigation of the lipid binding properties of the Marburg virus matrix protein VP40. J. Virol. 2015, 90, 3074–3085. [Google Scholar] [CrossRef]
- de Meyer, F.; Smit, B. Effect of cholesterol on the structure of a phospholipid bilayer. Proc. Natl. Acad. Sci. USA 2009, 106, 3654–3658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dick, R.A.; Goh, S.L.; Feigenson, G.W.; Vogt, V.M. HIV-1 Gag protein can sense the cholesterol and acyl chain environment in model membranes. Proc. Natl. Acad. Sci. USA 2012, 109, 18761–18766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adu-Gyamfi, E.; Johnson, K.A.; Fraser, M.E.; Scott, J.L.; Soni, S.P.; Jones, K.R.; Digman, M.A.; Gratton, E.; Tessier, C.R.; Stahelin, R.V. Host cell plasma membrane phosphatidylserine regulates the assembly and budding of Ebola virus. J. Virol. 2015, 89, 9440–9453. [Google Scholar] [CrossRef] [PubMed]
- Dessen, A.; Volchkov, V.; Dolnik, O.; Klenk, H.D.; Weissenhorn, W. Crystal structure of the matrix protein VP40 from Ebola virus. EMBO J. 2000, 19, 4228–4236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Vecchio, K.; Frick, C.T.; Gc, J.B.; Oda, S.-I.; Gerstman, B.S.; Saphire, E.O.; Chapagain, P.P.; Stahelin, R.V. A cationic, C-terminal patch and structural rearrangements in Ebola virus matrix VP40 protein control its interactions with phosphatidylserine. J. Biol. Chem. 2018, 293, 3335–3349. [Google Scholar] [CrossRef] [Green Version]
- Shnyrova, A.V.; Ayllon, J.; Mikhalyov, I.I.; Villar, E.; Zimmerberg, J.; Frolov, V.A. Vesicle formation by self-assembly of membrane-bound matrix proteins into a fluidlike budding domain. J. Cell Biol. 2007, 179, 627–633. [Google Scholar] [CrossRef] [Green Version]
- Johnson, K.A.; Taghon, G.J.; Scott, J.L.; Stahelin, R.V. The Ebola virus matrix protein, VP40, requires phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2) for extensive oligomerization at the plasma membrane and viral egress. Sci. Rep. 2016, 6, 19125. [Google Scholar] [CrossRef]
- Monde, K.; Chukkapalli, V.; Ono, A. Assembly and replication of HIV-1 in T cells with low levels of phosphatidylinositol-(4,5)-bisphosphate. J. Virol. 2011, 85, 3584–3595. [Google Scholar] [CrossRef]
- Swenson, D.L.; Warfield, K.L.; Kuehl, K.; Larsen, T.; Hevey, M.C.; Schmaljohn, A.; Bavari, S.; Aman, M.J. Generation of Marburg virus-like particles by co-expression of glycoprotein and matrix protein. FEMS Immunol. Med. Microbiol. 2004, 40, 27–31. [Google Scholar] [CrossRef] [Green Version]
- Urata, S.; Noda, T.; Kawaoka, Y.; Morikawa, S.; Yokosawa, H.; Yasuda, J. Interaction of Tsg101 with Marburg virus VP40 depends on the PPPY motif, but not the PT/SAP motif as in the case of Ebola virus, and Tsg101 plays a critical role in the budding of Marburg virus-like particles induced by VP40, NP, and GP. J. Virol. 2007, 81, 4895–4899. [Google Scholar] [CrossRef]
- Dolnik, O.; Kolesnikova, L.; Stevermann, L.; Becker, S. Tsg101 is recruited by a late domain of the nucleocapsid protein to support budding of Marburg virus-like particles. J. Virol. 2010, 84, 7847–7856. [Google Scholar] [CrossRef] [PubMed]
- Wahl-Jensen, V.M.; Afanasieva, T.A.; Seebach, J.; Ströher, U.; Feldmann, H.; Schnittler, H.-J. Effects of Ebola virus glycoproteins on endothelial cell activation and barrier function. J. Virol. 2005, 79, 10442–10450. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Harty, R.N. Packaging of actin into Ebola virus VLPs. Virol. J. 2005, 2, 92. [Google Scholar] [CrossRef] [PubMed]
- Kolesnikova, L.; Bohil, A.B.; Cheney, R.E.; Becker, S. Budding of Marburgvirus is associated with filopodia. Cell. Microbiol. 2007, 9, 939–951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adu-Gyamfi, E.; Digman, M.A.; Gratton, E.; Stahelin, R.V. Single-particle tracking demonstrates that actin coordinates the movement of the Ebola virus matrix protein. Biophys. J. 2012, 103, L41–L43. [Google Scholar] [CrossRef]
- Martinez, N.W.; Xue, X.; Berro, R.G.; Kreitzer, G.; Resh, M.D. Kinesin KIF4 regulates intracellular trafficking and stability of the human immunodeficiency virus type 1 Gag polyprotein. J. Virol. 2008, 82, 9937–9950. [Google Scholar] [CrossRef] [PubMed]
- Radtke, K.; Kieneke, D.; Wolfstein, A.; Michael, K.; Steffen, W.; Scholz, T.; Karger, A.; Sodeik, B. Plus- and minus-end directed microtubule motors bind simultaneously to herpes simplex virus capsids using different inner tegument structures. PLoS Pathog. 2010, 6, e1000991. [Google Scholar] [CrossRef]
- Ward, B.M. Visualization and characterization of the intracellular movement of Vaccinia virus intracellular mature virions. J. Virol. 2005, 79, 4755–4763. [Google Scholar] [CrossRef] [PubMed]
- Kolesnikova, L.; Bugany, H.; Klenk, H.D.; Becker, S. VP40, the matrix protein of Marburg virus, is associated with membranes of the late endosomal compartment. J. Virol. 2002, 76, 1825–1838. [Google Scholar] [CrossRef] [PubMed]
- Hicke, L. Protein regulation by monoubiquitin. Nat. Rev. Mol. Cell Biol. 2001, 2, 195–201. [Google Scholar] [CrossRef]
- d’Azzo, A.; Bongiovanni, A.; Nastasi, T. E3 ubiquitin ligases as regulators of membrane protein trafficking and degradation. Traffic 2005, 6, 429–441. [Google Scholar] [CrossRef] [PubMed]
- Anan, T.; Nagata, Y.; Koga, H.; Honda, Y.; Yabuki, N.; Miyamoto, C.; Kuwano, A.; Matsuda, I.; Endo, F.; Saya, H.; et al. Human ubiquitin-protein ligase Nedd4: Expression, subcellular localization and selective interaction with ubiquitin-conjugating enzymes. Genes Cells 1998, 3, 751–763. [Google Scholar] [CrossRef] [PubMed]
- Lafont, F.; Simons, K. Raft-partitioning of the ubiquitin ligases Cbl and Nedd4 upon IgE-triggered cell signaling. Proc. Natl. Acad. Sci. USA 2001, 98, 3180–3184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasuda, J.; Nakao, M.; Kawaoka, Y.; Shida, H. Nedd4 regulates egress of Ebola virus-like particles from host cells. J. Virol. 2003, 77, 9987–9992. [Google Scholar] [CrossRef] [PubMed]
- Angers, A.; Ramjaun, A.R.; McPherson, P.S. The HECT domain ligase Itch ubiquitinates endophilin and localizes to the trans-Golgi network and endosomal system. J. Biol. Chem. 2004, 279, 11471–11479. [Google Scholar] [CrossRef] [PubMed]
- Scheffner, M.; Kumar, S. Mammalian HECT ubiquitin-protein ligases: Biological and pathophysiological aspects. Biochim. Biophys. Acta 2014, 1843, 61–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallagher, E.; Gao, M.; Liu, Y.-C.; Karin, M. Activation of the E3 ubiquitin ligase Itch through a phosphorylation-induced conformational change. Proc. Natl. Acad. Sci. USA 2006, 103, 1717–1722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, Z.; Sagum, C.A.; Takizawa, F.; Ruthel, G.; Berry, C.T.; Kong, J.; Sunyer, J.O.; Freedman, B.D.; Bedford, M.T.; Sidhu, S.S.; et al. Ubiquitin ligase WWP1 interacts with Ebola virus VP40 to regulate egress. J. Virol. 2017, 91, e00812-17. [Google Scholar] [CrossRef] [PubMed]
- Kershaw, N.J.; Laktyushin, A.; Nicola, N.A.; Babon, J.J. Reconstruction of an active SOCS3-based E3 ubiquitin ligase complex in vitro: Identification of the active components and JAK2 and gp130 as substrates. Growth Factors 2014, 32, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Babon, J.J.; Sabo, J.K.; Soetopo, A.; Yao, S.; Bailey, M.F.; Zhang, J.G.; Nicola, N.A.; Norton, R.S. The SOCS box domain of SOCS3: Structure and interaction with the elonginBC-cullin5 ubiquitin ligase. J. Mol. Biol. 2008, 381, 928–940. [Google Scholar] [CrossRef] [PubMed]
- Okumura, A.; Rasmussen, A.L.; Halfmann, P.; Feldmann, F.; Yoshimura, A.; Feldmann, H.; Kawaoka, Y.; Harty, R.N.; Katze, M.G. Suppressor of cytokine signaling 3 is an inducible host factor that regulates virus egress during Ebola virus infection. J. Virol. 2015, 89, 10399–10406. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.; Cao, J.; Wu, M.; Zhang, W.; Jiang, T.; Yoshimura, A.; Gao, H. Suppressor of cytokine signaling 3 inhibits LPS-induced IL-6 expression in osteoblasts by suppressing CCAAT/enhancer-binding protein [102] activity. J. Biol. Chem. 2010, 285, 37227–37239. [Google Scholar] [CrossRef] [PubMed]
- Henne, W.M.; Buchkovich, N.J.; Emr, S.D. The ESCRT Pathway. Dev. Cell 2011, 21, 77–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goff, A.; Ehrlich, L.S.; Cohen, S.N.; Carter, C.A. Tsg101 control of human immunodeficiency virus type 1 Gag trafficking and release. J. Virol. 2003, 77, 9173–9182. [Google Scholar] [CrossRef] [PubMed]
- Patton, G.S.; Morris, S.A.; Chung, W.; Bieniasz, P.D.; McClure, M.O. Identification of domains in Gag important for prototypic foamy virus egress. J. Virol. 2005, 79, 6392–6399. [Google Scholar] [CrossRef] [PubMed]
- Segura-Morales, C.; Pescia, C.; Chatellard-Causse, C.; Sadoul, R.; Bertrand, E.; Basyuk, E. Tsg101 and Alix interact with murine leukemia virus Gag and cooperate with Nedd4 ubiquitin ligases during budding. J. Biol. Chem. 2005, 280, 27004–27012. [Google Scholar] [CrossRef]
- Perez, M.; Craven, R.C.; de la Torre, J.C. The small RING finger protein Z drives arenavirus budding: Implications for antiviral strategies. Proc. Natl. Acad. Sci. USA 2003, 100, 12978–12983. [Google Scholar] [CrossRef] [Green Version]
- Dolnik, O.; Kolesnikova, L.; Welsch, S.; Strecker, T.; Schudt, G.; Becker, S. Interaction with Tsg101 is necessary for the efficient transport and release of nucleocapsids in Marburg virus-infected cells. PLoS Pathog. 2014, 10, e1004463. [Google Scholar] [CrossRef]
- Martin-Serrano, J.; Zang, T.; Bieniasz, P.D. HIV-1 and Ebola virus encode small peptide motifs that recruit Tsg101 to sites of particle assembly to facilitate egress. Nat. Med. 2001, 7, 1313–1319. [Google Scholar] [CrossRef]
- Morita, E.; Sandrin, V.; Chung, H.Y.; Morham, S.G.; Gygi, S.P.; Rodesch, C.K.; Sundquist, W.I. Human ESCRT and ALIX proteins interact with proteins of the midbody and function in cytokinesis. EMBO J. 2007, 26, 4215–4227. [Google Scholar] [CrossRef]
- Razi, M.; Futter, C.E. Distinct Roles for Tsg101 and Hrs in multivesicular body formation and inward vesiculation. Mol. Biol. Cell 2006, 17, 3469–3483. [Google Scholar] [CrossRef] [PubMed]
- Pornillos, O.; Alam, S.L.; Rich, R.L.; Myszka, D.G.; Davis, D.R.; Sundquist, W.I. Structure and functional interactions of the Tsg101 UEV domain. EMBO J. 2002, 21, 2397–2406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irie, T.; Licata, J.M.; Harty, R.N. Functional characterization of Ebola virus L-domains using VSV recombinants. Virology 2005, 336, 291–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, J.; Qu, Y.; Liu, Y.; Jambusaria, R.; Han, Z.; Ruthel, G.; Freedman, B.D.; Harty, R.N. Host IQGAP1 and Ebola virus VP40 interactions facilitate virus-like particle egress. J. Virol. 2013, 87, 7777–7780. [Google Scholar] [CrossRef] [PubMed]
- Christ, L.; Wenzel, E.M.; Liestøl, K.; Raiborg, C.; Campsteijn, C.; Stenmark, H. ALIX and ESCRT-I/II function as parallel ESCRT-III recruiters in cytokinetic abscission. J. Cell Biol. 2016, 212, 499–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dussupt, V.; Javid, M.P.; Abou-Jaoude, G.; Jadwin, J.A.; de La Cruz, J.; Nagashima, K.; Bouamr, F. The nucleocapsid region of HIV-1 Gag cooperates with the PTAP and LYPXnL late domains to recruit the cellular machinery necessary for viral budding. PLoS Pathog. 2009, 5, e1000339. [Google Scholar] [CrossRef]
- Bissig, C.; Gruenberg, J. ALIX and the multivesicular endosome: ALIX in Wonderland. Trends Cell Biol. 2014, 24, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Madara, J.J.; Liu, Y.; Liu, W.; Ruthel, G.; Freedman, B.D.; Harty, R.N. ALIX rescues budding of a double PTAP/PPEY L-domain deletion mutant of Ebola VP40: A role for ALIX in Ebola virus egress. J. Infect. Dis. 2015, 212 (Suppl. 2), S138–S145. [Google Scholar] [CrossRef]
- Zhai, Q.; Landesman, M.B.; Chung, H.-Y.; Dierkers, A.; Jeffries, C.M.; Trewhella, J.; Hill, C.P.; Sundquist, W.I. Activation of the retroviral budding factor ALIX. J. Virol. 2011, 85, 9222–9226. [Google Scholar] [CrossRef]
- Sette, P.; Jadwin, J.A.; Dussupt, V.; Bello, N.F.; Bouamr, F. The ESCRT-associated protein Alix recruits the ubiquitin ligase Nedd4-1 to facilitate HIV-1 release through the LYPXnL L domain motif. J. Virol. 2010, 84, 8181–8192. [Google Scholar] [CrossRef]
- Landsberg, M.J.; Vajjhala, P.R.; Rothnagel, R.; Munn, A.L.; Hankamer, B. Three-dimensional structure of AAA ATPase Vps4: Advancing structural insights into the mechanisms of endosomal sorting and enveloped virus budding. Structure 2009, 17, 427–437. [Google Scholar] [CrossRef] [PubMed]
- Babst, M.; Wendland, B.; Estepa, E.J.; Emr, S.D. The Vps4p AAA ATPase regulates membrane association of a Vps protein complex required for normal endosome function. EMBO J. 1998, 17, 2982–2993. [Google Scholar] [CrossRef] [Green Version]
- Schoeneberg, J.; Yan, S.; Righini, M.; Remec Pavlin, M.; Lee, I.-H.; Carlson, L.-A.; Bahrami, A.H.; Goldman, D.H.; Ren, X.; Hummer, G.; et al. ATP-dependent force generation and membrane scission by ESCRT-III and Vps4. bioRxiv 2018. [Google Scholar] [CrossRef]
- Kolesnikova, L.; Strecker, T.; Morita, E.; Zielecki, F.; Mittler, E.; Crump, C.; Becker, S. Vacuolar protein sorting pathway contributes to the release of Marburg virus. J. Virol. 2009, 83, 2327–2337. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Peng, Q.; Lin, Q.; Childress, C.; Carey, D.; Yang, W. Calcium activates Nedd4 E3 ubiquitin ligases by releasing the C2 domain-mediated auto-inhibition. J. Biol. Chem. 2010, 285, 12279–12288. [Google Scholar] [CrossRef] [PubMed]
- Bissig, C.; Lenoir, M.; Velluz, M.C.; Kufareva, I.; Abagyan, R.; Overduin, M.; Gruenberg, J. Viral infection controlled by a calcium-dependent lipid-binding module in ALIX. Dev. Cell 2013, 25, 364–373. [Google Scholar] [CrossRef] [PubMed]
- Scheffer, L.L.; Sreetama, S.C.; Sharma, N.; Medikayala, S.; Brown, K.J.; Defour, A.; Jaiswal, J.K. Mechanism of Ca(2+)-triggered ESCRT assembly and regulation of cell membrane repair. Nat. Commun. 2014, 5, 5646. [Google Scholar] [CrossRef]
- Ehrlich, L.S.; Medina, G.N.; Carter, C.A. ESCRT machinery potentiates HIV-1 utilization of the PI(4,5)P(2)-PLC-IP3R-Ca(2+) signaling cascade. J. Mol. Biol. 2011, 413, 347–358. [Google Scholar] [CrossRef]
- Ehrlich, L.S.; Carter, C.A. HIV assembly and budding: Ca(2+) signaling and non-ESCRT proteins set the stage. Mol. Biol. Int. 2012, 2012, 851670. [Google Scholar] [CrossRef]
- Han, Z.; Harty, R.N. Influence of calcium/calmodulin on budding of Ebola VLPs: Implications for the involvement of the Ras/Raf/MEK/ERK pathway. Virus Genes 2007, 35, 511–520. [Google Scholar] [CrossRef]
- Muik, M.; Fahrner, M.; Schindl, R.; Stathopulos, P.; Frischauf, I.; Derler, I.; Plenk, P.; Lackner, B.; Groschner, K.; Ikura, M.; et al. STIM1 couples to ORAI1 via an intramolecular transition into an extended conformation. EMBO J. 2011, 30, 1678–1689. [Google Scholar] [CrossRef] [Green Version]
- Han, Z.; Madara, J.J.; Herbert, A.; Prugar, L.I.; Ruthel, G.; Lu, J.; Liu, Y.; Liu, W.; Liu, X.; Wrobel, J.E.; et al. Calcium regulation of hemorrhagic fever virus budding: Mechanistic implications for host-oriented therapeutic ontervention. PLoS Pathog. 2015, 11, e1005220. [Google Scholar] [CrossRef] [PubMed]
- Neil, S.J.D.; Sandrin, V.; Sundquist, W.I.; Bieniasz, P.D. An interferon-α-induced tethering mechanism inhibits HIV-1 and Ebola virus particle release but is counteracted by the HIV-1 Vpu protein. Cell Host Microbe 2007, 2, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Radoshitzky, S.R.; Dong, L.; Chi, X.; Clester, J.C.; Retterer, C.; Spurgers, K.; Kuhn, J.H.; Sandwick, S.; Ruthel, G.; Kota, K.; et al. Infectious Lassa virus, but not filoviruses, is restricted by BST-2/tetherin. J. Virol. 2010, 84, 10569–10580. [Google Scholar] [CrossRef] [PubMed]
- Erikson, E.; Adam, T.; Schmidt, S.; Lehmann-Koch, J.; Over, B.; Goffinet, C.; Harter, C.; Bekeredjian-Ding, I.; Sertel, S.; Lasitschka, F.; et al. In vivo expression profile of the antiviral restriction factor and tumor-targeting antigen CD317/BST-2/HM1.24/tetherin in humans. Proc. Natl. Acad. Sci. USA 2011, 108, 13688–13693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammonds, J.; Wang, J.J.; Yi, H.; Spearman, P. Immunoelectron microscopic evidence for Tetherin/BST2 as the physical bridge between HIV-1 virions and the plasma membrane. PLoS Pathog. 2010, 6, e1000749. [Google Scholar] [CrossRef] [PubMed]
- Neil, S.J.; Zang, T.; Bieniasz, P.D. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 2008, 451, 425–430. [Google Scholar] [CrossRef] [Green Version]
- Klimkait, T.; Strebel, K.; Hoggan, M.D.; Martin, M.A.; Orenstein, J.M. The human immunodeficiency virus type 1-specific protein vpu is required for efficient virus maturation and release. J. Virol. 1990, 64, 621–629. [Google Scholar]
- Lopez, L.A.; Yang, S.J.; Exline, C.M.; Rengarajan, S.; Haworth, K.G.; Cannon, P.M. Anti-tetherin activities of HIV-1 Vpu and ebola virus glycoprotein do not involve removal of tetherin from lipid rafts. J. Virol. 2012, 86, 5467–5480. [Google Scholar] [CrossRef]
- Neil, S.J.D.; Eastman, S.W.; Jouvenet, N.; Bieniasz, P.D. HIV-1 Vpu promotes release and prevents endocytosis of nascent retrovirus particles from the plasma membrane. PLoS Pathog. 2006, 2, e39. [Google Scholar] [CrossRef]
- Kuhl, B.D.; Cheng, V.; Wainberg, M.A.; Liang, C. Tetherin and its viral antagonists. J. Neuroimmune Pharmacol. 2011, 6, 188–201. [Google Scholar] [CrossRef] [PubMed]
- le Tortorec, A.; Willey, S.; Neil, S.J.D. Antiviral inhibition of enveloped virus release by Tetherin/BST-2: Action and counteraction. Viruses 2011, 3, 520–540. [Google Scholar] [CrossRef]
- Kupzig, S.; Korolchuk, V.; Rollason, R.; Sugden, A.; Wilde, A.; Banting, G. Bst-2/HM1.24 is a raft-associated apical membrane protein with an unusual topology. Traffic 2003, 4, 694–709. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, J.; Kaisho, T.; Tomizawa, H.; Lee, B.O.; Kobune, Y.; Inazawa, J.; Oritani, K.; Itoh, M.; Ochi, T.; Ishihara, K.; et al. Molecular cloning and chromosomal mapping of a bone marrow stromal cell surface gene, BST2, that may be involved in pre-B-cell growth. Genomics 1995, 26, 527–534. [Google Scholar] [CrossRef]
- Venkatesh, S.; Bieniasz, P.D. Mechanism of HIV-1 virion entrapment by tetherin. PLoS Pathog. 2013, 9, e1003483. [Google Scholar] [CrossRef]
- Perez-Caballero, D.; Zang, T.; Ebrahimi, A.; McNatt, M.W.; Gregory, D.A.; Johnson, M.C.; Bieniasz, P.D. Tetherin inhibits HIV-1 release by directly tethering virions to cells. Cell 2009, 139, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Hinz, A.; Miguet, N.; Natrajan, G.; Usami, Y.; Yamanaka, H.; Renesto, P.; Hartlieb, B.; McCarthy, A.A.; Simorre, J.P.; Gottlinger, H.; et al. Structural basis of HIV-1 tethering to membranes by the BST-2/tetherin ectodomain. Cell Host Microbe 2010, 7, 314–323. [Google Scholar] [CrossRef]
- Hammonds, J.; Ding, L.; Chu, H.; Geller, K.; Robbins, A.; Wang, J.J.; Yi, H.; Spearman, P. The tetherin/BST-2 coiled-coil ectodomain mediates plasma membrane microdomain localization and restriction of particle release. J. Virol. 2012, 86, 2259–2272. [Google Scholar] [CrossRef]
- Bampi, C.; Rasga, L.; Roux, L. Antagonism to human BST-2/tetherin by Sendai virus glycoproteins. J. Gen. Virol. 2013, 94, 1211–1219. [Google Scholar] [CrossRef] [Green Version]
- Jones, P.H.; Maric, M.; Madison, M.N.; Maury, W.; Roller, R.J.; Okeoma, C.M. BST-2/tetherin-mediated restriction of chikungunya (CHIKV) VLP budding is counteracted by CHIKV non-structural protein 1 (nsP1). Virology 2013, 438, 37–49. [Google Scholar] [CrossRef] [Green Version]
- Hauser, H.; Lopez, L.A.; Yang, S.J.; Oldenburg, J.E.; Exline, C.M.; Guatelli, J.C.; Cannon, P.M. HIV-1 Vpu and HIV-2 Env counteract BST-2/tetherin by sequestration in a perinuclear compartment. Retrovirology 2010, 7, 51. [Google Scholar] [CrossRef] [Green Version]
- Mansouri, M.; Viswanathan, K.; Douglas, J.L.; Hines, J.; Gustin, J.; Moses, A.V.; Früh, K. Molecular mechanism of BST2/tetherin downregulation by K5/MIR2 of Kaposi’s sarcoma-associated herpesvirus. J. Virol. 2009, 83, 9672–9681. [Google Scholar] [CrossRef] [PubMed]
- Bates, P.; Kaletsky, R.L.; Francica, J.R.; Agrawal-Gamse, C. Tetherin-mediated restriction of filovirus budding is antagonized by the Ebola glycoprotein. Proc. Natl. Acad. Sci. USA 2009, 106, 2886–2891. [Google Scholar] [Green Version]
- Gustin, J.K.; Bai, Y.; Moses, A.V.; Douglas, J.L. Ebola virus glycoprotein promotes enhanced viral egress by preventing Ebola VP40 from associating with the host restriction factor BST2/tetherin. J. Infect. Dis. 2015, 212 (Suppl. 2), S181–S190. [Google Scholar] [CrossRef] [PubMed]
- Gnirß, K.; Fiedler, M.; Krämer-Kühl, A.; Bolduan, S.; Mittler, E.; Becker, S.; Schindler, M.; Pöhlmann, S. Analysis of determinants in filovirus glycoproteins required for tetherin antagonism. Viruses 2014, 6, 1654–1671. [Google Scholar] [CrossRef] [PubMed]
- González-Hernández, M.; Hoffmann, M.; Brinkmann, C.; Nehls, J.; Winkler, M.; Schindler, M.; Pöhlmann, S. A GXXXA motif in the transmembrane domain of the Ebola virus glycoprotein is required for tetherin antagonism. J. Virol. 2018, 92, e00403-18. [Google Scholar] [CrossRef]
- Pitha-Rowe, I.F.; Pitha, P.M. Viral defense, carcinogenesis and ISG15: Novel roles for an old ISG. Cytokine Growth Factor Rev. 2007, 18, 409–417. [Google Scholar] [CrossRef] [Green Version]
- Zhao, C.; Denison, C.; Huibregtse, J.M.; Gygi, S.; Krug, R.M. Human ISG15 conjugation targets both IFN-induced and constitutively expressed proteins functioning in diverse cellular pathways. Proc. Natl. Acad. Sci. USA 2005, 102, 10200–10205. [Google Scholar] [CrossRef] [Green Version]
- Okumura, A.; Pitha, P.M.; Harty, R.N. ISG15 inhibits Ebola VP40 VLP budding in an l-domain-dependent manner by blocking Nedd4 ligase activity. Proc. Natl. Acad. Sci. USA 2008, 105, 3974–3979. [Google Scholar] [CrossRef]
- Malakhova, O.A.; Zhang, D.-E. ISG15 inhibits Nedd4 ubiquitin E3 activity and enhances the innate antiviral response. J. Biol. Chem. 2008, 283, 8783–8787. [Google Scholar] [CrossRef]
- Lindner, H.A.; Lytvyn, V.; Qi, H.; Lachance, P.; Ziomek, E.; Menard, R. Selectivity in ISG15 and ubiquitin recognition by the SARS coronavirus papain-like protease. Arch. Biochem. Biophys. 2007, 466, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Yuan, W.; Krug, R.M. Influenza B virus NS1 protein inhibits conjugation of the interferon (IFN)-induced ubiquitin-like ISG15 protein. EMBO J. 2001, 20, 362–371. [Google Scholar] [CrossRef] [Green Version]
- Guerra, S.; Caceres, A.; Knobeloch, K.P.; Horak, I.; Esteban, M. Vaccinia virus E3 protein prevents the antiviral action of ISG15. PLoS Pathog. 2008, 4, e1000096. [Google Scholar] [CrossRef] [PubMed]
- Gamerdinger, M.; Kaya, A.M.; Wolfrum, U.; Clement, A.M.; Behl, C. BAG3 mediates chaperone-based aggresome-targeting and selective autophagy of misfolded proteins. EMBO Rep. 2011, 12, 149–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loughran, H.M.; Han, Z.; Wrobel, J.E.; Decker, S.E.; Ruthel, G.; Freedman, B.D.; Harty, R.N.; Reitz, A.B. Quinoxaline-based inhibitors of Ebola and Marburg VP40 egress. Bioorg. Med. Chem. Lett. 2016, 26, 3429–3435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, Z.; Lu, J.; Liu, Y.; Davis, B.; Lee, M.S.; Olson, M.A.; Ruthel, G.; Freedman, B.D.; Schnell, M.J.; Wrobel, J.E.; et al. Small-molecule probes targeting the viral PPxY-host Nedd4 interface block egress of a broad range of RNA viruses. J. Virol. 2014, 88, 7294–7306. [Google Scholar] [CrossRef] [PubMed]
- Teimoori, S.; Seesuay, W.; Jittavisutthikul, S.; Chaisri, U.; Sookrung, N.; Densumite, J.; Saelim, N.; Chulanetra, M.; Maneewatch, S.; Chaicumpa, W. Human transbodies to VP40 inhibit cellular egress of Ebola virus-like particles. Biochem. Biophys. Res. Commun. 2016, 479, 245–252. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Location | Function |
|---|---|
| P7 | Ubiquitination [42] |
| 10PP11 | Ubiquitination [42] |
| Y13 | Ubiquitination [42] |
| P53 | Membrane localisation [57] |
| A55 | Dimerisation [51] |
| H61 | Dimerisation [51] |
| W95 | Oligomerisation [55] |
| 96LPLGVA101 | Membrane localisation and structural stability [62] |
| P108 | Dimerisation [51] |
| 112TA113 | Dimerisation [51] |
| 116ML117 | Dimerisation [51] |
| F134 | Oligomerisation [57] |
| A149 | Oligomerisation [57] |
| A151 | Oligomerisation [57] |
| E160 | Oligomerisation [57] |
| Q184 | Oligomerisation [57] |
| L203 | Hexamerisation [51] |
| 212KLR214 | Structural stability and oligomerisation [40] |
| 224KK225 | Membrane localisation and membrane binding [51] |
| 226GNSADLTSPE255 | Protection of polymerised microtubules [63] |
| I237 | Hexamerisation [51] |
| M241 | Hexamerisation [51] |
| K270 | Membrane localisation and binding [51] |
| 274KK275 | Membrane localisation and binding [51] |
| P283 | Membrane binding [64] |
| P286 | Membrane binding [64] |
| I293 | Membrane penetration [58] |
| L295 | Membrane penetration [58] |
| V298 | Membrane penetration [58] |
| A299 | Membrane penetration [65] |
| 303LTMVI307 | Hexamerisation [66] |
| 309QDCDTCHSP317 | Membrane binding [65] |
| 320LPAVIEK326 | Hexamerisation [67] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gordon, T.B.; Hayward, J.A.; Marsh, G.A.; Baker, M.L.; Tachedjian, G. Host and Viral Proteins Modulating Ebola and Marburg Virus Egress. Viruses 2019, 11, 25. https://doi.org/10.3390/v11010025
Gordon TB, Hayward JA, Marsh GA, Baker ML, Tachedjian G. Host and Viral Proteins Modulating Ebola and Marburg Virus Egress. Viruses. 2019; 11(1):25. https://doi.org/10.3390/v11010025
Chicago/Turabian StyleGordon, Tamsin B., Joshua A. Hayward, Glenn A. Marsh, Michelle L. Baker, and Gilda Tachedjian. 2019. "Host and Viral Proteins Modulating Ebola and Marburg Virus Egress" Viruses 11, no. 1: 25. https://doi.org/10.3390/v11010025
APA StyleGordon, T. B., Hayward, J. A., Marsh, G. A., Baker, M. L., & Tachedjian, G. (2019). Host and Viral Proteins Modulating Ebola and Marburg Virus Egress. Viruses, 11(1), 25. https://doi.org/10.3390/v11010025