Neuroinflammation, Microglia, and Cell-Association during Prion Disease

Abstract
:
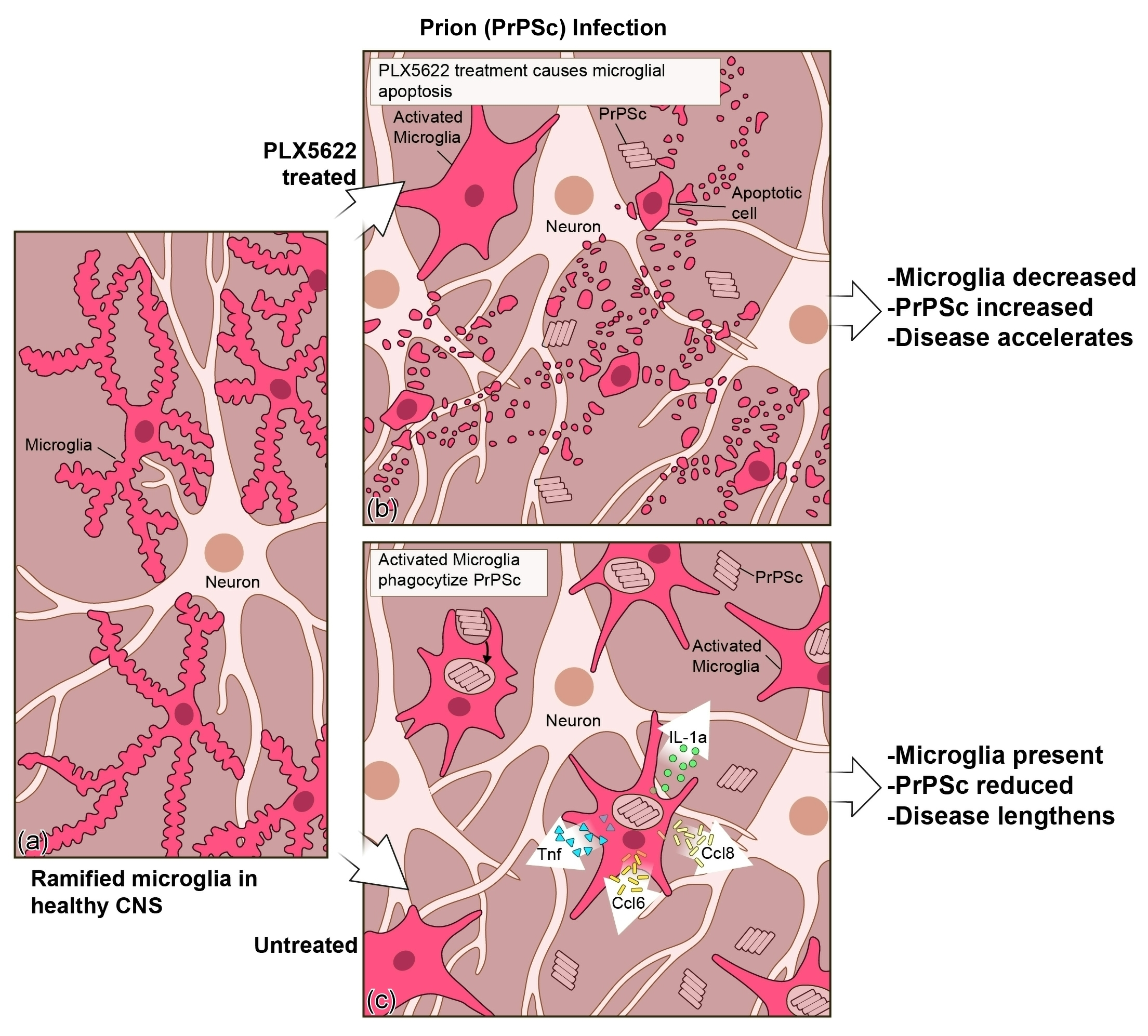{kind=link}
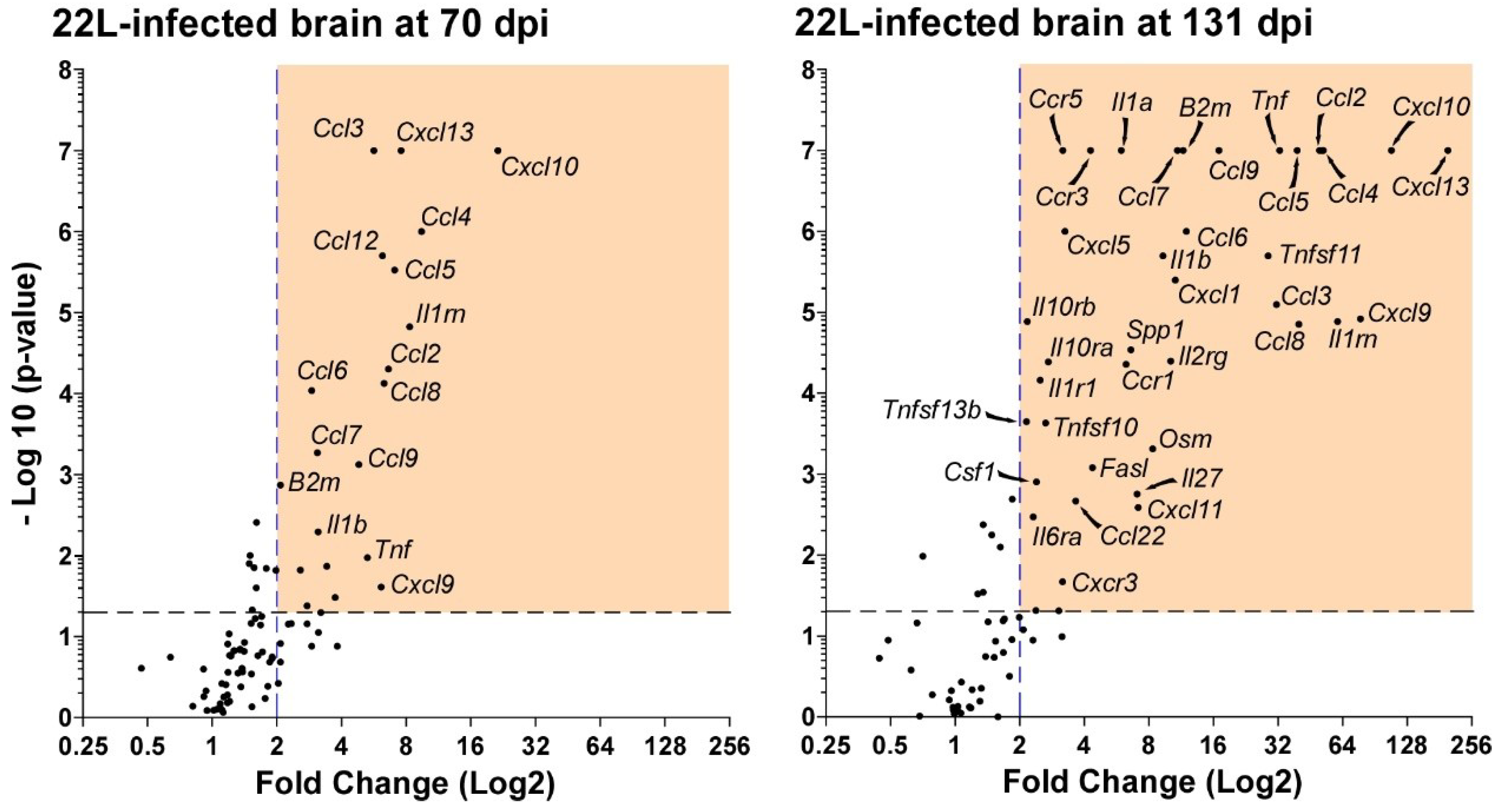{kind=link}
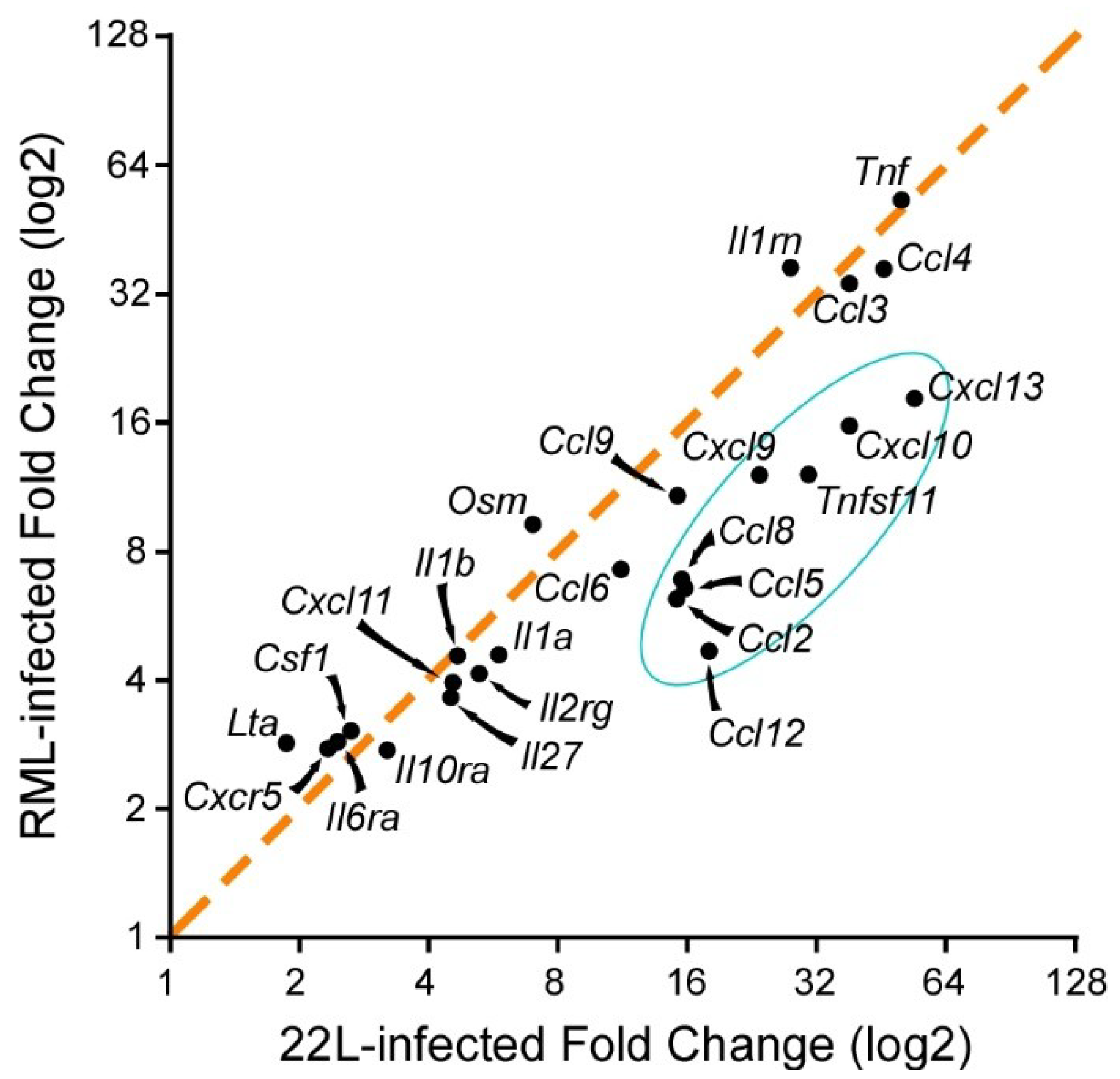{kind=link}
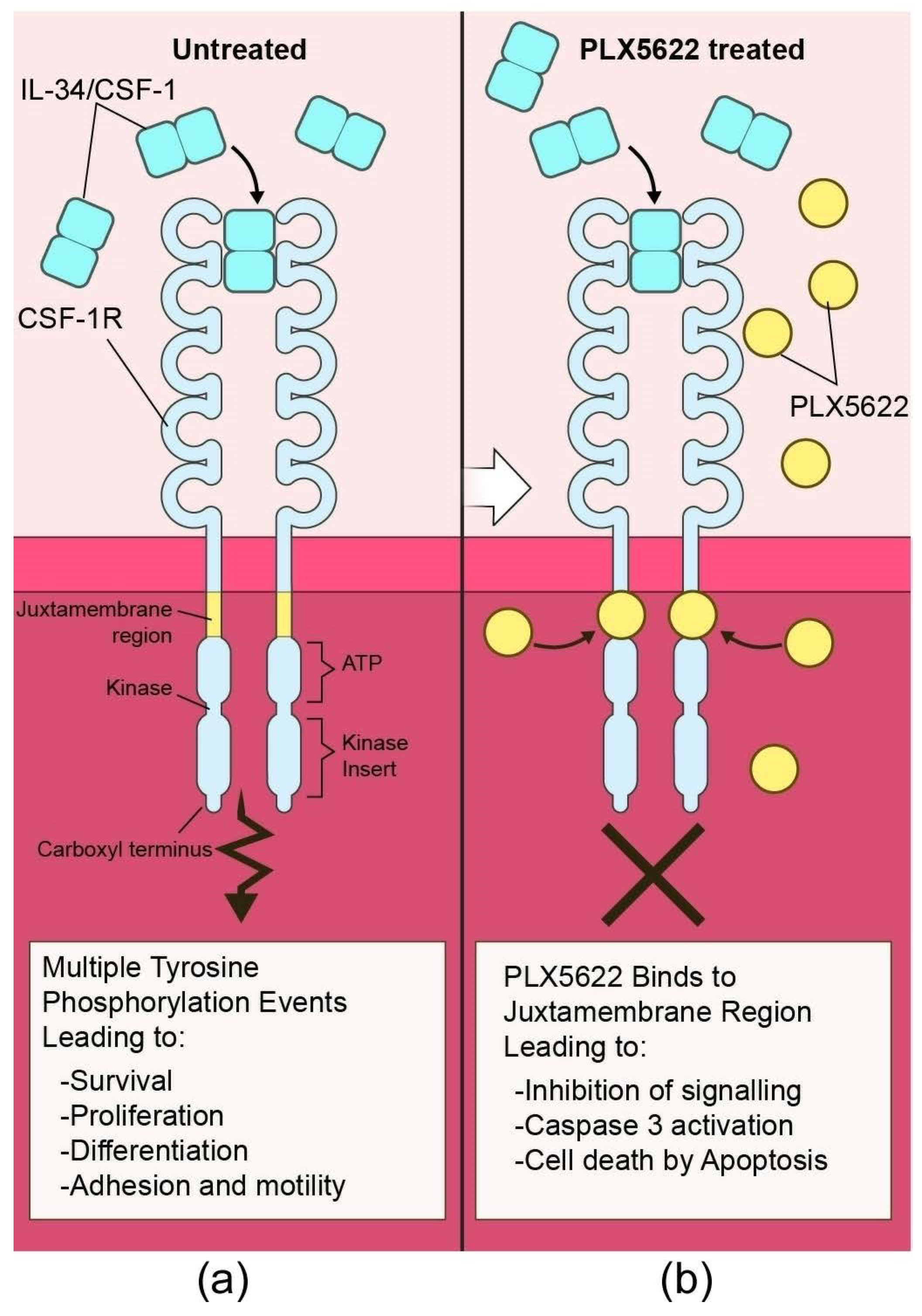{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Prions and Disease
2. Neuroinflammation in Prion Disease
3. In Vivo Assessment of Microglia in Prion Disease
4. Cell-Association Studies
5. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Caughey, B.; Baron, G.S.; Chesebro, B.; Jeffrey, M. Getting a grip on prions: Oligomers, amyloids, and pathological membrane interactions. Annu. Rev. Biochem. 2009, 78, 177–204. [Google Scholar] [CrossRef]
- Tribouillard-Tanvier, D.; Race, B.; Striebel, J.F.; Carroll, J.A.; Phillips, K.; Chesebro, B. Early cytokine elevation, PrPres deposition, and gliosis in mouse scrapie: No effect on disease by deletion of cytokine genes IL-12p40 and IL-12p35. J. Virol. 2012, 86, 10377–10383. [Google Scholar] [CrossRef]
- Scallet, A.C.; Ye, X. Excitotoxic mechanisms of neurodegeneration in transmissible spongiform encephalopathies. Ann. N. Y. Acad. Sci. 1997, 825, 194–205. [Google Scholar] [CrossRef]
- Black, S.A.; Stys, P.K.; Zamponi, G.W.; Tsutsui, S. Cellular prion protein and NMDA receptor modulation: Protecting against excitotoxicity. Front. Cell Dev. Biol. 2014, 2, 45. [Google Scholar] [CrossRef]
- Carroll, J.A.; Striebel, J.F.; Race, B.; Phillips, K.; Chesebro, B. Prion infection of mouse brain reveals multiple new upregulated genes involved in neuroinflammation or signal transduction. J. Virol. 2015, 89, 2388–2404. [Google Scholar] [CrossRef]
- Carroll, J.A.; Striebel, J.F.; Rangel, A.; Woods, T.; Phillips, K.; Peterson, K.E.; Race, B.; Chesebro, B. Prion Strain Differences in Accumulation of PrPSc on Neurons and Glia Are Associated with Similar Expression Profiles of Neuroinflammatory Genes: Comparison of Three Prion Strains. PLoS Pathog. 2016, 12, e1005551. [Google Scholar] [CrossRef]
- Tribouillard-Tanvier, D.; Striebel, J.F.; Peterson, K.E.; Chesebro, B. Analysis of protein levels of 24 cytokines in scrapie agent-infected brain and glial cell cultures from mice differing in prion protein expression levels. J. Virol. 2009, 83, 11244–11253. [Google Scholar] [CrossRef]
- Aiken, J.M.; Williamson, J.L.; Marsh, R.F. Evidence of mitochondrial involvement in scrapie infection. J. Virol. 1989, 63, 1686–1694. [Google Scholar]
- Choi, S.I.; Ju, W.K.; Choi, E.K.; Kim, J.; Lea, H.Z.; Carp, R.I.; Wisniewski, H.M.; Kim, Y.S. Mitochondrial dysfunction induced by oxidative stress in the brains of hamsters infected with the 263 K scrapie agent. Acta Neuropathol. 1998, 96, 279–286. [Google Scholar] [CrossRef]
- Jeffrey, M.; McGovern, G.; Chambers, E.V.; King, D.; Gonzalez, L.; Manson, J.C.; Ghetti, B.; Piccardo, P.; Barron, R.M. Mechanism of PrP-amyloid formation in mice without transmissible spongiform encephalopathy. Brain Pathol. 2012, 22, 58–66. [Google Scholar] [CrossRef]
- Siskova, Z.; Mahad, D.J.; Pudney, C.; Campbell, G.; Cadogan, M.; Asuni, A.; O’Connor, V.; Perry, V.H. Morphological and functional abnormalities in mitochondria associated with synaptic degeneration in prion disease. Am. J. Pathol. 2010, 177, 1411–1421. [Google Scholar] [CrossRef]
- Choi, H.S.; Choi, Y.G.; Shin, H.Y.; Oh, J.M.; Park, J.H.; Kim, J.I.; Carp, R.I.; Choi, E.K.; Kim, Y.S. Dysfunction of mitochondrial dynamics in the brains of scrapie-infected mice. Biochem. Biophys. Res. Commun. 2014, 448, 157–162. [Google Scholar] [CrossRef]
- Forloni, G.; Angeretti, N.; Chiesa, R.; Monzani, E.; Salmona, M.; Bugiani, O.; Tagliavini, F. Neurotoxicity of a prion protein fragment. Nature 1993, 362, 543–546. [Google Scholar] [CrossRef]
- Liberski, P.P.; Sikorska, B.; Bratosiewicz-Wasik, J.; Gajdusek, D.C.; Brown, P. Neuronal cell death in transmissible spongiform encephalopathies (prion diseases) revisited: From apoptosis to autophagy. Int. J. Biochem. Cell Biol. 2004, 36, 2473–2490. [Google Scholar] [CrossRef]
- Hope, J.; Shearman, M.S.; Baxter, H.C.; Chong, A.; Kelly, S.M.; Price, N.C. Cytotoxicity of prion protein peptide (PrP106–126) differs in mechanism from the cytotoxic activity of the Alzheimer’s disease amyloid peptide, A beta 25–35. Neurodegeneration 1996, 5, 1–11. [Google Scholar] [CrossRef]
- Gonzalez, L.; Martin, S.; Begara-McGorum, I.; Hunter, N.; Houston, F.; Simmons, M.; Jeffrey, M. Effects of agent strain and host genotype on PrP accumulation in the brain of sheep naturally and experimentally affected with scrapie. J. Comp. Pathol. 2002, 126, 17–29. [Google Scholar] [CrossRef]
- Jeffrey, M.; Martin, S.; Barr, J.; Chong, A.; Fraser, J.R. Onset of accumulation of PrPres in murine ME7 scrapie in relation to pathological and PrP immunohistochemical changes. J. Comp. Pathol. 2001, 124, 20–28. [Google Scholar] [CrossRef]
- Thal, D.R.; von Arnim, C.A.; Griffin, W.S.; Mrak, R.E.; Walker, L.; Attems, J.; Arzberger, T. Frontotemporal lobar degeneration FTLD-tau: Preclinical lesions, vascular, and Alzheimer-related co-pathologies. J. Neural Transm. 2015, 122, 1007–1018. [Google Scholar] [CrossRef]
- Baron, G.S.; Hughson, A.G.; Raymond, G.J.; Offerdahl, D.K.; Barton, K.A.; Raymond, L.D.; Dorward, D.W.; Caughey, B. Effect of glycans and the glycophosphatidylinositol anchor on strain dependent conformations of scrapie prion protein: Improved purifications and infrared spectra. Biochemistry 2011, 50, 4479–4490. [Google Scholar] [CrossRef]
- Harper, J.D.; Lansbury, P.T., Jr. Models of amyloid seeding in Alzheimer’s disease and scrapie: Mechanistic truths and physiological consequences of the time-dependent solubility of amyloid proteins. Annu. Rev. Biochem. 1997, 66, 385–407. [Google Scholar] [CrossRef]
- Aguzzi, A.; Polymenidou, M. Mammalian prion biology: One century of evolving concepts. Cell 2004, 116, 313–327. [Google Scholar] [CrossRef]
- Kraus, A.; Groveman, B.R.; Caughey, B. Prions and the potential transmissibility of protein misfolding diseases. Annu. Rev. Microbiol. 2013, 67, 543–564. [Google Scholar] [CrossRef]
- Riesner, D. The Scrapie Isoform of the Prion Protein PrPSc Compared to the Cellular Isoform PrPC. In Prions in Humans and Animals; Hörnlimann, B., Riesner, D., Kretzschmar, H.A., Eds.; Walter de Gruyter: Berlin, German; New York, NY, USA, 2007; pp. 104–118. [Google Scholar]
- Williams, A.E.; Ryder, S.; Blakemore, W.F. Monocyte recruitment into the scrapie-affected brain. Acta. Neuropathol. 1995, 90, 164–169. [Google Scholar] [CrossRef]
- Lewicki, H.; Tishon, A.; Homann, D.; Mazarguil, H.; Laval, F.; Asensio, V.C.; Campbell, I.L.; DeArmond, S.; Coon, B.; Teng, C.; et al. T cells infiltrate the brain in murine and human transmissible spongiform encephalopathies. J. Virol. 2003, 77, 3799–3808. [Google Scholar] [CrossRef]
- Baker, C.A.; Manuelidis, L. Unique inflammatory RNA profiles of microglia in Creutzfeldt-Jakob disease. Proc. Natl. Acad. Sci. USA 2003, 100, 675–679. [Google Scholar] [CrossRef] [Green Version]
- Booth, S.; Bowman, C.; Baumgartner, R.; Sorensen, G.; Robertson, C.; Coulthart, M.; Phillipson, C.; Somorjai, R.L. Identification of central nervous system genes involved in the host response to the scrapie agent during preclinical and clinical infection. J. Gen. Virol. 2004, 85, 3459–3471. [Google Scholar] [CrossRef] [Green Version]
- Brown, A.R.; Webb, J.; Rebus, S.; Williams, A.; Fazakerley, J.K. Identification of up-regulated genes by array analysis in scrapie-infected mouse brains. Neuropathol. Appl. Neurobiol. 2004, 30, 555–567. [Google Scholar] [CrossRef]
- Hwang, D.; Lee, I.Y.; Yoo, H.; Gehlenborg, N.; Cho, J.H.; Petritis, B.; Baxter, D.; Pitstick, R.; Young, R.; Spicer, D.; et al. A systems approach to prion disease. Mol. Syst. Biol. 2009, 5, 252. [Google Scholar] [CrossRef] [Green Version]
- Moody, L.R.; Herbst, A.J.; Aiken, J.M. Upregulation of interferon-gamma-induced genes during prion infection. J. Toxicol. Environ. Health A 2011, 74, 146–153. [Google Scholar] [CrossRef]
- Riemer, C.; Neidhold, S.; Burwinkel, M.; Schwarz, A.; Schultz, J.; Kratzschmar, J.; Monning, U.; Baier, M. Gene expression profiling of scrapie-infected brain tissue. Biochem. Biophys. Res. Commun. 2004, 323, 556–564. [Google Scholar] [CrossRef]
- Skinner, P.J.; Abbassi, H.; Chesebro, B.; Race, R.E.; Reilly, C.; Haase, A.T. Gene expression alterations in brains of mice infected with three strains of scrapie. BMC Genomics 2006, 7, 114. [Google Scholar] [CrossRef]
- Sorensen, G.; Medina, S.; Parchaliuk, D.; Phillipson, C.; Robertson, C.; Booth, S.A. Comprehensive transcriptional profiling of prion infection in mouse models reveals networks of responsive genes. BMC Genomics 2008, 9, 114. [Google Scholar] [CrossRef]
- Xiang, W.; Windl, O.; Wunsch, G.; Dugas, M.; Kohlmann, A.; Dierkes, N.; Westner, I.M.; Kretzschmar, H.A. Identification of differentially expressed genes in scrapie-infected mouse brains by using global gene expression technology. J. Virol. 2004, 78, 11051–11060. [Google Scholar] [CrossRef]
- Crespo, I.; Roomp, K.; Jurkowski, W.; Kitano, H.; del Sol, A. Gene regulatory network analysis supports inflammation as a key neurodegeneration process in prion disease. BMC Syst. Biol. 2012, 6, 132. [Google Scholar] [CrossRef]
- Castelli, J.C.; Hassel, B.A.; Maran, A.; Paranjape, J.; Hewitt, J.A.; Li, X.L.; Hsu, Y.T.; Silverman, R.H.; Youle, R.J. The role of 2′-5′ oligoadenylate-activated ribonuclease L in apoptosis. Cell Death Differ. 1998, 5, 313–320. [Google Scholar] [CrossRef] [Green Version]
- Potu, H.; Sgorbissa, A.; Brancolini, C. Identification of USP18 as an important regulator of the susceptibility to IFN-alpha and drug-induced apoptosis. Cancer Res. 2010, 70, 655–665. [Google Scholar] [CrossRef]
- Wong, B.R.; Rho, J.; Arron, J.; Robinson, E.; Orlinick, J.; Chao, M.; Kalachikov, S.; Cayani, E.; Bartlett, F.S., 3rd; Frankel, W.N.; et al. TRANCE is a novel ligand of the tumor necrosis factor receptor family that activates c-Jun N-terminal kinase in T cells. J. Biol. Chem. 1997, 272, 25190–25194. [Google Scholar] [CrossRef]
- Oka, K.; Sawamura, T.; Kikuta, K.; Itokawa, S.; Kume, N.; Kita, T.; Masaki, T. Lectin-like oxidized low-density lipoprotein receptor 1 mediates phagocytosis of aged/apoptotic cells in endothelial cells. Proc. Natl. Acad. Sci. USA 1998, 95, 9535–9540. [Google Scholar] [CrossRef] [Green Version]
- Valerio, A.; Ferrario, M.; Martinez, F.O.; Locati, M.; Ghisi, V.; Bresciani, L.G.; Mantovani, A.; Spano, P. Gene expression profile activated by the chemokine CCL5/RANTES in human neuronal cells. J. Neurosci. Res. 2004, 78, 371–382. [Google Scholar] [CrossRef]
- Li, Y.; Duan, Z.; Gao, D.; Huang, S.; Yuan, H.; Niu, X. The new role of LOX-1 in hypertension induced neuronal apoptosis. Biochem. Biophys. Res. Commun. 2012, 425, 735–740. [Google Scholar] [CrossRef]
- Van Marle, G.; Henry, S.; Todoruk, T.; Sullivan, A.; Silva, C.; Rourke, S.B.; Holden, J.; McArthur, J.C.; Gill, M.J.; Power, C. Human immunodeficiency virus type 1 Nef protein mediates neural cell death: A neurotoxic role for IP-10. Virology 2004, 329, 302–318. [Google Scholar] [CrossRef]
- Sui, Y.; Stehno-Bittel, L.; Li, S.; Loganathan, R.; Dhillon, N.K.; Pinson, D.; Nath, A.; Kolson, D.; Narayan, O.; Buch, S. CXCL10-induced cell death in neurons: Role of calcium dysregulation. Eur. J. Neurosci. 2006, 23, 957–964. [Google Scholar] [CrossRef]
- Severini, C.; Passeri, P.P.; Ciotti, M.; Florenzano, F.; Possenti, R.; Zona, C.; Di Matteo, A.; Guglielmotti, A.; Calissano, P.; Pachter, J.; et al. Bindarit, inhibitor of CCL2 synthesis, protects neurons against amyloid-beta-induced toxicity. J. Alzheimers Dis. 2014, 38, 281–293. [Google Scholar] [CrossRef]
- Fabrizi, C.; Businaro, R.; Lauro, G.M.; Starace, G.; Fumagalli, L. Activated alpha2macroglobulin increases beta-amyloid (25–35)-induced toxicity in LAN5 human neuroblastoma cells. Exp. Neurol. 1999, 155, 252–259. [Google Scholar] [CrossRef]
- Kovacs, D.M. Alpha2-macroglobulin in late-onset Alzheimer’s disease. Exp. Gerontol. 2000, 35, 473–479. [Google Scholar] [CrossRef]
- Gelbard, H.A.; Dzenko, K.A.; DiLoreto, D.; del Cerro, C.; del Cerro, M.; Epstein, L.G. Neurotoxic effects of tumor necrosis factor alpha in primary human neuronal cultures are mediated by activation of the glutamate AMPA receptor subtype: Implications for AIDS neuropathogenesis. Dev. Neurosci. 1993, 15, 417–422. [Google Scholar] [CrossRef]
- Na, Y.J.; Jin, J.K.; Kim, J.I.; Choi, E.K.; Carp, R.I.; Kim, Y.S. JAK-STAT signaling pathway mediates astrogliosis in brains of scrapie-infected mice. J. Neurochem. 2007, 103, 637–649. [Google Scholar] [CrossRef] [Green Version]
- Fan, Y.; Mao, R.; Yang, J. NF-kappaB and STAT3 signaling pathways collaboratively link inflammation to cancer. Protein Cell 2013, 4, 176–185. [Google Scholar] [CrossRef]
- Quinton, L.J.; Mizgerd, J.P. NF-kappaB and STAT3 signaling hubs for lung innate immunity. Cell Tissue Res. 2011, 343, 153–165. [Google Scholar] [CrossRef]
- Yang, J.; Liao, X.; Agarwal, M.K.; Barnes, L.; Auron, P.E.; Stark, G.R. Unphosphorylated STAT3 accumulates in response to IL-6 and activates transcription by binding to NFkappaB. Genes Dev. 2007, 21, 1396–1408. [Google Scholar] [CrossRef]
- Uskokovic, A.; Dinic, S.; Mihailovic, M.; Grigorov, I.; Ivanovic-Matic, S.; Bogojevic, D.; Grdovic, N.; Arambasic, J.; Vidakovic, M.; Martinovic, V.; et al. STAT3/NFkappaB interplay in the regulation of alpha2-macroglobulin gene expression during rat liver development and the acute phase response. IUBMB Life 2007, 59, 170–178. [Google Scholar] [CrossRef]
- Hosokawa, Y.; Hosokawa, I.; Ozaki, K.; Nakae, H.; Matsuo, T. Oncostatin M synergistically induces CXCL10 and ICAM-1 expression in IL-1beta-stimulated-human gingival fibroblasts. J. Cell. Biochem. 2010, 111, 40–48. [Google Scholar] [CrossRef]
- Bode, J.G.; Albrecht, U.; Haussinger, D.; Heinrich, P.C.; Schaper, F. Hepatic acute phase proteins--regulation by IL-6- and IL-1-type cytokines involving STAT3 and its crosstalk with NF-kappaB-dependent signaling. Eur. J. Cell. Biol. 2012, 91, 496–505. [Google Scholar] [CrossRef]
- Meling, S.; Bardsen, K.; Ulvund, M.J. Presence of an acute phase response in sheep with clinical classical scrapie. BMC Vet. Res. 2012, 8, 113. [Google Scholar] [CrossRef]
- Campbell, I.L.; Eddleston, M.; Kemper, P.; Oldstone, M.B.; Hobbs, M.V. Activation of cerebral cytokine gene expression and its correlation with onset of reactive astrocyte and acute-phase response gene expression in scrapie. J. Virol. 1994, 68, 2383–2387. [Google Scholar]
- Cunningham, C.; Wilcockson, D.C.; Boche, D.; Perry, V.H. Comparison of inflammatory and acute-phase responses in the brain and peripheral organs of the ME7 model of prion disease. J. Virol. 2005, 79, 5174–5184. [Google Scholar] [CrossRef]
- Yu, Z.; Kone, B.C. The STAT3 DNA-binding domain mediates interaction with NF-kappaB p65 and inducible nitric oxide synthase transrepression in mesangial cells. J. Am. Soc. Nephrol. 2004, 15, 585–591. [Google Scholar] [CrossRef]
- Yu, Z.; Zhang, W.; Kone, B.C. Signal transducers and activators of transcription 3 (STAT3) inhibits transcription of the inducible nitric oxide synthase gene by interacting with nuclear factor kappaB. Biochem. J. 2002, 367, 97–105. [Google Scholar] [CrossRef]
- Lee, H.; Herrmann, A.; Deng, J.H.; Kujawski, M.; Niu, G.; Li, Z.; Forman, S.; Jove, R.; Pardoll, D.M.; Yu, H. Persistently activated Stat3 maintains constitutive NF-kappaB activity in tumors. Cancer Cell 2009, 15, 283–293. [Google Scholar] [CrossRef]
- Hiroi, M.; Ohmori, Y. The transcriptional coactivator CREB-binding protein cooperates with STAT1 and NF-kappa B for synergistic transcriptional activation of the CXC ligand 9/monokine induced by interferon-gamma gene. J. Biol. Chem. 2003, 278, 651–660. [Google Scholar] [CrossRef]
- Jahnke, A.; Johnson, J.P. Synergistic activation of intercellular adhesion molecule 1 (ICAM-1) by TNF-alpha and IFN-gamma is mediated by p65/p50 and p65/c-Rel and interferon-responsive factor Stat1 alpha (p91) that can be activated by both IFN-gamma and IFN-alpha. FEBS Lett. 1994, 354, 220–226. [Google Scholar] [CrossRef]
- Ohmori, Y.; Schreiber, R.D.; Hamilton, T.A. Synergy between interferon-gamma and tumor necrosis factor-alpha in transcriptional activation is mediated by cooperation between signal transducer and activator of transcription 1 and nuclear factor kappaB. J. Biol. Chem. 1997, 272, 14899–14907. [Google Scholar] [CrossRef]
- Sekine, N.; Ishikawa, T.; Okazaki, T.; Hayashi, M.; Wollheim, C.B.; Fujita, T. Synergistic activation of NF-kappab and inducible isoform of nitric oxide synthase induction by interferon-gamma and tumor necrosis factor-alpha in INS-1 cells. J. Cell. Physiol. 2000, 184, 46–57. [Google Scholar] [CrossRef]
- Kim, M.O.; Suh, H.S.; Brosnan, C.F.; Lee, S.C. Regulation of RANTES/CCL5 expression in human astrocytes by interleukin-1 and interferon-beta. J. Neurochem. 2004, 90, 297–308. [Google Scholar] [CrossRef] [Green Version]
- Striebel, J.F.; Race, B.; Carroll, J.A.; Phillips, K.; Chesebro, B. Knockout of fractalkine receptor Cx3cr1 does not alter disease or microglial activation in prion-infected mice. J. Gen. Virol. 2016, 97, 1481–1487. [Google Scholar] [CrossRef] [Green Version]
- Carroll, J.A.; Race, B.; Williams, K.; Chesebro, B. Toll-like receptor 2 confers partial neuroprotection during prion disease. PLoS ONE 2019, in press. [Google Scholar] [CrossRef]
- Mabbott, N.A.; Williams, A.; Farquhar, C.F.; Pasparakis, M.; Kollias, G.; Bruce, M.E. Tumor necrosis factor alpha-deficient, but not interleukin-6-deficient, mice resist peripheral infection with scrapie. J. Virol. 2000, 74, 3338–3344. [Google Scholar] [CrossRef]
- Prinz, M.; Montrasio, F.; Klein, M.A.; Schwarz, P.; Priller, J.; Odermatt, B.; Pfeffer, K.; Aguzzi, A. Lymph nodal prion replication and neuroinvasion in mice devoid of follicular dendritic cells. Proc. Natl. Acad. Sci. USA 2002, 99, 919–924. [Google Scholar] [CrossRef] [Green Version]
- Tamguney, G.; Giles, K.; Glidden, D.V.; Lessard, P.; Wille, H.; Tremblay, P.; Groth, D.F.; Yehiely, F.; Korth, C.; Moore, R.C.; et al. Genes contributing to prion pathogenesis. J. Gen. Virol. 2008, 89, 1777–1788. [Google Scholar] [CrossRef] [Green Version]
- Klein, M.A.; Frigg, R.; Flechsig, E.; Raeber, A.J.; Kalinke, U.; Bluethmann, H.; Bootz, F.; Suter, M.; Zinkernagel, R.M.; Aguzzi, A. A crucial role for B cells in neuroinvasive scrapie. Nature 1997, 390, 687–690. [Google Scholar] [CrossRef]
- Prinz, M.; Heikenwalder, M.; Junt, T.; Schwarz, P.; Glatzel, M.; Heppner, F.L.; Fu, Y.X.; Lipp, M.; Aguzzi, A. Positioning of follicular dendritic cells within the spleen controls prion neuroinvasion. Nature 2003, 425, 957–962. [Google Scholar] [CrossRef]
- Felton, L.M.; Cunningham, C.; Rankine, E.L.; Waters, S.; Boche, D.; Perry, V.H. MCP-1 and murine prion disease: Separation of early behavioural dysfunction from overt clinical disease. Neurobiol. Dis. 2005, 20, 283–295. [Google Scholar] [CrossRef]
- O’Shea, M.; Maytham, E.G.; Linehan, J.M.; Brandner, S.; Collinge, J.; Lloyd, S.E. Investigation of mcp1 as a quantitative trait gene for prion disease incubation time in mouse. Genetics 2008, 180, 559–566. [Google Scholar] [CrossRef]
- Thackray, A.M.; McKenzie, A.N.; Klein, M.A.; Lauder, A.; Bujdoso, R. Accelerated prion disease in the absence of interleukin-10. J. Virol. 2004, 78, 13697–13707. [Google Scholar] [CrossRef]
- Schultz, J.; Schwarz, A.; Neidhold, S.; Burwinkel, M.; Riemer, C.; Simon, D.; Kopf, M.; Otto, M.; Baier, M. Role of interleukin-1 in prion disease-associated astrocyte activation. Am. J. Pathol. 2004, 165, 671–678. [Google Scholar] [CrossRef]
- Riemer, C.; Schultz, J.; Burwinkel, M.; Schwarz, A.; Mok, S.W.; Gultner, S.; Bamme, T.; Norley, S.; van Landeghem, F.; Lu, B.; et al. Accelerated prion replication in, but prolonged survival times of, prion-infected CXCR3−/− mice. J. Virol. 2008, 82, 12464–12471. [Google Scholar] [CrossRef]
- Spinner, D.S.; Cho, I.S.; Park, S.Y.; Kim, J.I.; Meeker, H.C.; Ye, X.; Lafauci, G.; Kerr, D.J.; Flory, M.J.; Kim, B.S.; et al. Accelerated prion disease pathogenesis in Toll-like receptor 4 signaling-mutant mice. J. Virol. 2008, 82, 10701–10708. [Google Scholar] [CrossRef]
- Wong, Y.C.; Krainc, D. alpha-synuclein toxicity in neurodegeneration: Mechanism and therapeutic strategies. Nat. Med. 2017, 23, 1–13. [Google Scholar] [CrossRef]
- Zilka, N.; Kazmerova, Z.; Jadhav, S.; Neradil, P.; Madari, A.; Obetkova, D.; Bugos, O.; Novak, M. Who fans the flames of Alzheimer’s disease brains? Misfolded tau on the crossroad of neurodegenerative and inflammatory pathways. J. Neuroinflammation. 2012, 9, 47. [Google Scholar]
- Zilka, N.; Korenova, M.; Novak, M. Misfolded tau protein and disease modifying pathways in transgenic rodent models of human tauopathies. Acta Neuropathol. 2009, 118, 71–86. [Google Scholar] [CrossRef]
- Valera, E.; Spencer, B.; Masliah, E. Immunotherapeutic Approaches Targeting Amyloid-beta, alpha-Synuclein, and Tau for the Treatment of Neurodegenerative Disorders. Neurotherapeutics 2016, 13, 179–189. [Google Scholar] [CrossRef]
- Ransohoff, R.M. How neuroinflammation contributes to neurodegeneration. Science 2016, 353, 777–783. [Google Scholar] [CrossRef]
- Amor, S.; Peferoen, L.A.; Vogel, D.Y.; Breur, M.; van der Valk, P.; Baker, D.; van Noort, J.M. Inflammation in neurodegenerative diseases—An update. Immunology 2014, 142, 151–166. [Google Scholar] [CrossRef]
- Outram, G.W.; Dickinson, A.G.; Fraser, H. Reduced susceptibility to scrapie in mice after steroid administration. Nature 1974, 249, 855–856. [Google Scholar] [CrossRef]
- Outram, G.W.; Dickinson, A.G.; Fraser, H. Slow encephalopathies, inflammatory responses and arachis oil. Lancet 1975, 1, 198–200. [Google Scholar] [CrossRef]
- Manuelidis, L.; Fritch, W.; Zaitsev, I. Dapsone to delay symptoms in Creutzfeldt-Jakob disease. Lancet 1998, 352, 456. [Google Scholar] [CrossRef]
- Riemer, C.; Burwinkel, M.; Schwarz, A.; Gultner, S.; Mok, S.W.; Heise, I.; Holtkamp, N.; Baier, M. Evaluation of drugs for treatment of prion infections of the central nervous system. J. Gen. Virol. 2008, 89, 594–597. [Google Scholar] [CrossRef] [Green Version]
- Reiss, A.B.; Wirkowski, E. Statins in neurological disorders: Mechanisms and therapeutic value. Sci. World J. 2009, 9, 1242–1259. [Google Scholar] [CrossRef]
- Wang, Q.; Yan, J.; Chen, X.; Li, J.; Yang, Y.; Weng, J.; Deng, C.; Yenari, M.A. Statins: Multiple neuroprotective mechanisms in neurodegenerative diseases. Exp. Neurol. 2011, 230, 27–34. [Google Scholar] [CrossRef]
- Kumar, A.; Sharma, N.; Gupta, A.; Kalonia, H.; Mishra, J. Neuroprotective potential of atorvastatin and simvastatin (HMG-CoA reductase inhibitors) against 6-hydroxydopamine (6-OHDA) induced Parkinson-like symptoms. Brain. Res. 2012, 1471, 13–22. [Google Scholar] [CrossRef]
- Xu, Y.Q.; Long, L.; Yan, J.Q.; Wei, L.; Pan, M.Q.; Gao, H.M.; Zhou, P.; Liu, M.; Zhu, C.S.; Tang, B.S.; et al. Simvastatin induces neuroprotection in 6-OHDA-lesioned PC12 via the PI3K/AKT/caspase 3 pathway and anti-inflammatory responses. CNS Neurosci. Ther. 2013, 19, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Roy, A.; Matras, J.; Brahmachari, S.; Gendelman, H.E.; Pahan, K. Simvastatin inhibits the activation of p21ras and prevents the loss of dopaminergic neurons in a mouse model of Parkinson’s disease. J. Neurosci. 2009, 29, 13543–13556. [Google Scholar] [CrossRef]
- Selley, M.L. Simvastatin prevents 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced striatal dopamine depletion and protein tyrosine nitration in mice. Brain Res. 2005, 1037, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Chen, T.; Wang, C.; Li, G.; Zhi, W.; Yin, J.; Wan, Q.; Chen, L. Atorvastatin in improvement of cognitive impairments caused by amyloid beta in mice: Involvement of inflammatory reaction. BMC Neurol. 2016, 16, 18. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.Y.; Fan, Y.C.; Wang, M.; Wang, D.; Li, X.H. Atorvastatin attenuates the production of IL-1beta, IL-6, and TNF-alpha in the hippocampus of an amyloid beta1-42-induced rat model of Alzheimer’s disease. Clin. Interv. Aging 2013, 8, 103–110. [Google Scholar] [PubMed]
- Youssef, S.; Stuve, O.; Patarroyo, J.C.; Ruiz, P.J.; Radosevich, J.L.; Hur, E.M.; Bravo, M.; Mitchell, D.J.; Sobel, R.A.; Steinman, L.; et al. The HMG-CoA reductase inhibitor, atorvastatin, promotes a Th2 bias and reverses paralysis in central nervous system autoimmune disease. Nature 2002, 420, 78–84. [Google Scholar] [CrossRef]
- Stanislaus, R.; Singh, A.K.; Singh, I. Lovastatin treatment decreases mononuclear cell infiltration into the CNS of Lewis rats with experimental allergic encephalomyelitis. J. Neurosci. Res. 2001, 66, 155–162. [Google Scholar] [CrossRef]
- Greenwood, J.; Walters, C.E.; Pryce, G.; Kanuga, N.; Beraud, E.; Baker, D.; Adamson, P. Lovastatin inhibits brain endothelial cell Rho-mediated lymphocyte migration and attenuates experimental autoimmune encephalomyelitis. FASEB J. 2003, 17, 905–907. [Google Scholar] [CrossRef] [Green Version]
- Stanislaus, R.; Pahan, K.; Singh, A.K.; Singh, I. Amelioration of experimental allergic encephalomyelitis in Lewis rats by lovastatin. Neurosci. Lett. 1999, 269, 71–74. [Google Scholar] [CrossRef]
- Undela, K.; Gudala, K.; Malla, S.; Bansal, D. Statin use and risk of Parkinson’s disease: A meta-analysis of observational studies. J. Neurol. 2013, 260, 158–165. [Google Scholar] [CrossRef]
- Friedman, B.; Lahad, A.; Dresner, Y.; Vinker, S. Long-term statin use and the risk of Parkinson’s disease. Am. J. Manag. Care 2013, 19, 626–632. [Google Scholar] [PubMed]
- Gao, X.; Simon, K.C.; Schwarzschild, M.A.; Ascherio, A. Prospective study of statin use and risk of Parkinson disease. Arch. Neurol. 2012, 69, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Alonso, A.; Guo, X.; Umbach, D.M.; Lichtenstein, M.L.; Ballantyne, C.M.; Mailman, R.B.; Mosley, T.H.; Chen, H. Statins, plasma cholesterol, and risk of Parkinson’s disease: A prospective study. Mov. Disord. 2015, 30, 552–559. [Google Scholar] [CrossRef] [PubMed]
- Tison, F.; Negre-Pages, L.; Meissner, W.G.; Dupouy, S.; Li, Q.; Thiolat, M.L.; Thiollier, T.; Galitzky, M.; Ory-Magne, F.; Milhet, A.; et al. Simvastatin decreases levodopa-induced dyskinesia in monkeys, but not in a randomized, placebo-controlled, multiple cross-over (“n-of-1”) exploratory trial of simvastatin against levodopa-induced dyskinesia in Parkinson’s disease patients. Parkinsonism Relat. Disord. 2013, 19, 416–421. [Google Scholar] [CrossRef] [PubMed]
- Sparks, D.L.; Sabbagh, M.; Connor, D.; Soares, H.; Lopez, J.; Stankovic, G.; Johnson-Traver, S.; Ziolkowski, C.; Browne, P. Statin therapy in Alzheimer’s disease. Acta Neurol. Scand. Suppl. 2006, 185, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Yiannopoulou, K.G.; Papageorgiou, S.G. Current and future treatments for Alzheimer’s disease. Ther. Adv. Neurol. Disord. 2013, 6, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Bedi, O.; Dhawan, V.; Sharma, P.L.; Kumar, P. Pleiotropic effects of statins: New therapeutic targets in drug design. Naunyn. Schmiedebergs Arch. Pharmacol. 2016, 389, 695–712. [Google Scholar] [CrossRef] [PubMed]
- Feldman, H.H.; Doody, R.S.; Kivipelto, M.; Sparks, D.L.; Waters, D.D.; Jones, R.W.; Schwam, E.; Schindler, R.; Hey-Hadavi, J.; DeMicco, D.A.; et al. Randomized controlled trial of atorvastatin in mild to moderate Alzheimer disease: LEADe. Neurology 2010, 74, 956–964. [Google Scholar] [CrossRef]
- Simons, M.; Schwarzler, F.; Lutjohann, D.; von Bergmann, K.; Beyreuther, K.; Dichgans, J.; Wormstall, H.; Hartmann, T.; Schulz, J.B. Treatment with simvastatin in normocholesterolemic patients with Alzheimer’s disease: A 26-week randomized, placebo-controlled, double-blind trial. Ann. Neurol. 2002, 52, 346–350. [Google Scholar] [CrossRef]
- Trompet, S.; van Vliet, P.; de Craen, A.J.; Jolles, J.; Buckley, B.M.; Murphy, M.B.; Ford, I.; Macfarlane, P.W.; Sattar, N.; Packard, C.J.; et al. Pravastatin and cognitive function in the elderly. Results of the PROSPER study. J. Neurol. 2010, 257, 85–90. [Google Scholar]
- Pihl-Jensen, G.; Tsakiri, A.; Frederiksen, J.L. Statin treatment in multiple sclerosis: A systematic review and meta-analysis. CNS Drugs 2015, 29, 277–291. [Google Scholar] [CrossRef] [PubMed]
- Birnbaum, G.; Cree, B.; Altafullah, I.; Zinser, M.; Reder, A.T. Combining beta interferon and atorvastatin may increase disease activity in multiple sclerosis. Neurology 2008, 71, 1390–1395. [Google Scholar] [CrossRef] [PubMed]
- Lanzillo, R.; Orefice, G.; Quarantelli, M.; Rinaldi, C.; Prinster, A.; Ventrella, G.; Spitaleri, D.; Lus, G.; Vacca, G.; Carotenuto, B.; et al. Atorvastatin combined to interferon to verify the efficacy (ACTIVE) in relapsing-remitting active multiple sclerosis patients: A longitudinal controlled trial of combination therapy. Mult. Scler. 2010, 16, 450–454. [Google Scholar] [CrossRef] [PubMed]
- Togha, M.; Karvigh, S.A.; Nabavi, M.; Moghadam, N.B.; Harirchian, M.H.; Sahraian, M.A.; Enzevaei, A.; Nourian, A.; Ghanaati, H.; Firouznia, K.; et al. Simvastatin treatment in patients with relapsing-remitting multiple sclerosis receiving interferon beta 1a: A double-blind randomized controlled trial. Mult. Scler. 2010, 16, 848–854. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, P.S.; Lycke, J.; Eralinna, J.P.; Edland, A.; Wu, X.; Frederiksen, J.L.; Oturai, A.; Malmestrom, C.; Stenager, E.; Sellebjerg, F.; et al. Simvastatin as add-on therapy to interferon beta-1a for relapsing-remitting multiple sclerosis (SIMCOMBIN study): A placebo-controlled randomised phase 4 trial. Lancet Neurol. 2011, 10, 691–701. [Google Scholar] [CrossRef]
- Haviv, Y.; Avrahami, D.; Ovadia, H.; Ben-Hur, T.; Gabizon, R.; Sharon, R. Induced neuroprotection independently from PrPSc accumulation in a mouse model for prion disease treated with simvastatin. Arch. Neurol. 2008, 65, 762–775. [Google Scholar] [CrossRef] [PubMed]
- Kempster, S.; Bate, C.; Williams, A. Simvastatin treatment prolongs the survival of scrapie-infected mice. Neuroreport 2007, 18, 479–482. [Google Scholar] [CrossRef]
- Mok, S.W.; Thelen, K.M.; Riemer, C.; Bamme, T.; Gultner, S.; Lutjohann, D.; Baier, M. Simvastatin prolongs survival times in prion infections of the central nervous system. Biochem. Biophys. Res. Commun. 2006, 348, 697–702. [Google Scholar] [CrossRef]
- Vetrugno, V.; Di Bari, M.A.; Nonno, R.; Puopolo, M.; D’Agostino, C.; Pirisinu, L.; Pocchiari, M.; Agrimi, U. Oral pravastatin prolongs survival time of scrapie-infected mice. J. Gen. Virol. 2009, 90, 1775–1780. [Google Scholar] [CrossRef] [Green Version]
- Carroll, J.A.; Race, B.; Phillips, K.; Striebel, J.F.; Chesebro, B. Statins are ineffective at reducing neuroinflammation or prolonging survival in scrapie-infected mice. J. Gen. Virol. 2017, 98, 2190–2199. [Google Scholar] [CrossRef] [Green Version]
- Hong, S.; Dissing-Olesen, L.; Stevens, B. New insights on the role of microglia in synaptic pruning in health and disease. Curr. Opin. Neurobiol. 2016, 36, 128–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bilimoria, P.M.; Stevens, B. Microglia function during brain development: New insights from animal models. Brain. Res. 2015, 1617, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Schafer, D.P.; Stevens, B. Phagocytic glial cells: Sculpting synaptic circuits in the developing nervous system. Curr. Opin. Neurobiol. 2013, 23, 1034–1040. [Google Scholar] [CrossRef] [PubMed]
- Schafer, D.P.; Stevens, B. Microglia Function in Central Nervous System Development and Plasticity. Cold Spring Harb. Perspect. Biol. 2015, 7, a020545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salter, M.W.; Stevens, B. Microglia emerge as central players in brain disease. Nat. Med. 2017, 23, 1018–1027. [Google Scholar] [CrossRef] [PubMed]
- Mandrekar-Colucci, S.; Landreth, G.E. Microglia and inflammation in Alzheimer’s disease. CNS Neurol. Disord. Drug Targets 2010, 9, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Muzio, L.; Martino, G.; Furlan, R. Multifaceted aspects of inflammation in multiple sclerosis: The role of microglia. J. Neuroimmunol. 2007, 191, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Rogers, J.; Mastroeni, D.; Leonard, B.; Joyce, J.; Grover, A. Neuroinflammation in Alzheimer’s disease and Parkinson’s disease: Are microglia pathogenic in either disorder? Int. Rev. Neurobiol. 2007, 82, 235–246. [Google Scholar] [PubMed]
- Garden, G.A. Microglia in human immunodeficiency virus-associated neurodegeneration. Glia 2002, 40, 240–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Betmouni, S.; Perry, V.H.; Gordon, J.L. Evidence for an early inflammatory response in the central nervous system of mice with scrapie. Neuroscience 1996, 74, 1–5. [Google Scholar] [CrossRef]
- Williams, A.; Lucassen, P.J.; Ritchie, D.; Bruce, M. PrP deposition, microglial activation, and neuronal apoptosis in murine scrapie. Exp. Neurol. 1997, 144, 433–438. [Google Scholar] [CrossRef]
- Vincenti, J.E.; Murphy, L.; Grabert, K.; McColl, B.W.; Cancellotti, E.; Freeman, T.C.; Manson, J.C. Defining the Microglia Response during the Time Course of Chronic Neurodegeneration. J. Virol. 2015, 90, 3003–3017. [Google Scholar] [CrossRef] [Green Version]
- Gomez Perdiguero, E.; Klapproth, K.; Schulz, C.; Busch, K.; Azzoni, E.; Crozet, L.; Garner, H.; Trouillet, C.; de Bruijn, M.F.; Geissmann, F.; et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature 2015, 518, 547–551. [Google Scholar] [CrossRef]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef] [PubMed]
- Ajami, B.; Bennett, J.L.; Krieger, C.; Tetzlaff, W.; Rossi, F.M. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat. Neurosci. 2007, 10, 1538–1543. [Google Scholar] [CrossRef] [PubMed]
- Elmore, M.R.; Lee, R.J.; West, B.L.; Green, K.N. Characterizing newly repopulated microglia in the adult mouse: Impacts on animal behavior, cell morphology, and neuroinflammation. PLoS ONE 2015, 10, e0122912. [Google Scholar] [CrossRef] [PubMed]
- Elmore, M.R.; Najafi, A.R.; Koike, M.A.; Dagher, N.N.; Spangenberg, E.E.; Rice, R.A.; Kitazawa, M.; Matusow, B.; Nguyen, H.; West, B.L.; et al. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron 2014, 82, 380–397. [Google Scholar] [CrossRef]
- Erblich, B.; Zhu, L.; Etgen, A.M.; Dobrenis, K.; Pollard, J.W. Absence of colony stimulation factor-1 receptor results in loss of microglia, disrupted brain development and olfactory deficits. PLoS ONE 2011, 6, e26317. [Google Scholar] [CrossRef]
- Chitu, V.; Gokhan, S.; Nandi, S.; Mehler, M.F.; Stanley, E.R. Emerging Roles for CSF-1 Receptor and its Ligands in the Nervous System. Trends Neurosci. 2016, 39, 378–393. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Szretter, K.J.; Vermi, W.; Gilfillan, S.; Rossini, C.; Cella, M.; Barrow, A.D.; Diamond, M.S.; Colonna, M. IL-34 is a tissue-restricted ligand of CSF1R required for the development of Langerhans cells and microglia. Nat. Immunol. 2012, 13, 753–760. [Google Scholar] [CrossRef]
- Wegiel, J.; Wisniewski, H.M.; Dziewiatkowski, J.; Tarnawski, M.; Kozielski, R.; Trenkner, E.; Wiktor-Jedrzejczak, W. Reduced number and altered morphology of microglial cells in colony stimulating factor-1-deficient osteopetrotic op/op mice. Brain Res. 1998, 804, 135–139. [Google Scholar] [CrossRef]
- Gomez-Nicola, D.; Fransen, N.L.; Suzzi, S.; Perry, V.H. Regulation of microglial proliferation during chronic neurodegeneration. J. Neurosci. 2013, 33, 2481–2493. [Google Scholar] [CrossRef]
- Zhu, C.; Herrmann, U.S.; Falsig, J.; Abakumova, I.; Nuvolone, M.; Schwarz, P.; Frauenknecht, K.; Rushing, E.J.; Aguzzi, A. A neuroprotective role for microglia in prion diseases. J. Exp. Med. 2016, 213, 1047–1059. [Google Scholar] [CrossRef] [Green Version]
- Carroll, J.A.; Race, B.; Williams, K.; Striebel, J.; Chesebro, B. Microglia Are Critical in Host Defense Against Prion Disease. J. Virol. 2018, 92, 1–17. [Google Scholar]
- Chhor, V.; Le Charpentier, T.; Lebon, S.; Ore, M.V.; Celador, I.L.; Josserand, J.; Degos, V.; Jacotot, E.; Hagberg, H.; Savman, K.; et al. Characterization of phenotype markers and neuronotoxic potential of polarised primary microglia in vitro. Brain Behav. Immun. 2013, 32, 70–85. [Google Scholar] [CrossRef] [Green Version]
- Butovsky, O.; Jedrychowski, M.P.; Moore, C.S.; Cialic, R.; Lanser, A.J.; Gabriely, G.; Koeglsperger, T.; Dake, B.; Wu, P.M.; Doykan, C.E.; et al. Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nat. Neurosci. 2014, 17, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M.; Perry, V.H. Microglial physiology: Unique stimuli, specialized responses. Annu. Rev. Immunol. 2009, 27, 119–145. [Google Scholar] [CrossRef] [PubMed]
- Streit, W.J.; Walter, S.A.; Pennell, N.A. Reactive microgliosis. Prog. Neurobiol. 1999, 57, 563–581. [Google Scholar] [CrossRef]
- Nelson, P.T.; Soma, L.A.; Lavi, E. Microglia in diseases of the central nervous system. Ann. Med. 2002, 34, 491–500. [Google Scholar] [CrossRef] [PubMed]
- Mathys, H.; Adaikkan, C.; Gao, F.; Young, J.Z.; Manet, E.; Hemberg, M.; De Jager, P.L.; Ransohoff, R.M.; Regev, A.; Tsai, L.H. Temporal Tracking of Microglia Activation in Neurodegeneration at Single-Cell Resolution. Cell. Rep. 2017, 21, 366–380. [Google Scholar] [CrossRef] [Green Version]
- Bruce, M.E.; McConnell, I.; Fraser, H.; Dickinson, A.G. The disease characteristics of different strains of scrapie in Sinc congenic mouse lines: Implications for the nature of the agent and host control of pathogenesis. J. Gen. Virol. 1991, 72, 595–603. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, A.G.; Meikle, V.M. Host-genotype and agent effects in scrapie incubation: Change in allelic interaction with different strains of agent. Mol. Gen. Genet. 1971, 112, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Bruce, M.E.; Dickinson, A.G. Biological evidence that scrapie agent has an independent genome. J. Gen. Virol. 1987, 68, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Fraser, H. The pathology of a natural and experimental scrapie. Front. Biol. 1976, 44, 267–305. [Google Scholar] [PubMed]
- Carp, R.I.; Callahan, S.M.; Sersen, E.A.; Moretz, R.C. Preclinical changes in weight of scrapie-infected mice as a function of scrapie agent-mouse strain combination. Intervirology 1984, 21, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Kimberlin, R.H.; Walker, C.A.; Millson, G.C.; Taylor, D.M.; Robertson, P.A.; Tomlinson, A.H.; Dickinson, A.G. Disinfection studies with two strains of mouse-passaged scrapie agent. Guidelines for Creutzfeldt-Jakob and related agents. J. Neurol. Sci. 1983, 59, 355–369. [Google Scholar] [CrossRef]
- Caughey, B.; Raymond, G.J.; Bessen, R.A. Strain-dependent differences in beta-sheet conformations of abnormal prion protein. J. Biol. Chem. 1998, 273, 32230–32235. [Google Scholar] [CrossRef] [PubMed]
- Pattison, I.H.; Millson, G.C. Scrapie produced experimentally in goats with special reference to the clinical syndrome. J. Comp. Pathol. 1961, 71, 101–109. [Google Scholar] [CrossRef]
- Foster, J.D.; Dickinson, A.G. The unusual properties of CH1641, a sheep-passaged isolate of scrapie. Vet. Rec. 1988, 123, 5–8. [Google Scholar] [CrossRef]
- Kimberlin, R.H.; Walker, C.A. Evidence that the transmission of one source of scrapie agent to hamsters involves separation of agent strains from a mixture. J. Gen. Virol. 1978, 39, 487–496. [Google Scholar] [CrossRef]
- Kimberlin, R.H.; Walker, C.A. Pathogenesis of mouse scrapie: Dynamics of agent replication in spleen, spinal cord and brain after infection by different routes. J. Comp. Pathol. 1979, 89, 551–562. [Google Scholar] [CrossRef]
- Van Keulen, L.J.M.; Langeveld, J.P.; Dolstra, C.H.; Jacobs, J.; Bossers, A.; van Zijderveld, F.G. TSE strain differentiation in mice by immunohistochemical PrPSc profiles and triplex Western blot. Neuropathol. Appl. Neurobiol. 2015, 41, 756–779. [Google Scholar] [CrossRef] [PubMed]
- Siso, S.; Chianini, F.; Eaton, S.L.; Witz, J.; Hamilton, S.; Martin, S.; Finlayson, J.; Pang, Y.; Stewart, P.; Steele, P.; et al. Disease phenotype in sheep after infection with cloned murine scrapie strains. Prion 2012, 6, 174–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Chen, K.; Sloan, S.A.; Bennett, M.L.; Scholze, A.R.; O’Keeffe, S.; Phatnani, H.P.; Guarnieri, P.; Caneda, C.; Ruderisch, N.; et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 2014, 34, 11929–11947. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sloan, S.A.; Clarke, L.E.; Caneda, C.; Plaza, C.A.; Blumenthal, P.D.; Vogel, H.; Steinberg, G.K.; Edwards, M.S.; Li, G.; et al. Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with Mouse. Neuron 2016, 89, 37–53. [Google Scholar] [CrossRef] [PubMed]
- Race, R.E.; Priola, S.A.; Bessen, R.A.; Ernst, D.; Dockter, J.; Rall, G.F.; Mucke, L.; Chesebro, B.; Oldstone, M.B. Neuron-specific expression of a hamster prion protein minigene in transgenic mice induces susceptibility to hamster scrapie agent. Neuron 1995, 15, 1183–1191. [Google Scholar] [CrossRef] [Green Version]
- Raeber, A.J.; Race, R.E.; Brandner, S.; Priola, S.A.; Sailer, A.; Bessen, R.A.; Mucke, L.; Manson, J.; Aguzzi, A.; Oldstone, M.B.; et al. Astrocyte-specific expression of hamster prion protein (PrP) renders PrP knockout mice susceptible to hamster scrapie. EMBO J. 1997, 16, 6057–6065. [Google Scholar] [CrossRef] [Green Version]
- Kercher, L.; Favara, C.; Striebel, J.F.; LaCasse, R.; Chesebro, B. Prion protein expression differences in microglia and astroglia influence scrapie-induced neurodegeneration in the retina and brain of transgenic mice. J. Virol. 2007, 81, 10340–10351. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Munch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [Green Version]
- Liddelow, S.A.; Barres, B.A. Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity 2017, 46, 957–967. [Google Scholar] [CrossRef]
- Jeffrey, M.; Goodsir, C.M.; Race, R.E.; Chesebro, B. Scrapie-specific neuronal lesions are independent of neuronal PrP expression. Ann. Neurol. 2004, 55, 781–792. [Google Scholar] [CrossRef] [PubMed]
- Deleault, N.R.; Kascsak, R.; Geoghegan, J.C.; Supattapone, S. Species-dependent differences in cofactor utilization for formation of the protease-resistant prion protein in vitro. Biochemistry 2010, 49, 3928–3934. [Google Scholar] [CrossRef] [PubMed]
- Supattapone, S. Elucidating the role of cofactors in mammalian prion propagation. Prion 2014, 8, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, L.; Martin, S.; Jeffrey, M. Distinct profiles of PrP(d) immunoreactivity in the brain of scrapie- and BSE-infected sheep: Implications for differential cell targeting and PrP processing. J. Gen. Virol. 2003, 84, 1339–1350. [Google Scholar] [CrossRef] [PubMed]
- Hughes, M.M.; Field, R.H.; Perry, V.H.; Murray, C.L.; Cunningham, C. Microglia in the degenerating brain are capable of phagocytosis of beads and of apoptotic cells, but do not efficiently remove PrPSc, even upon LPS stimulation. Glia 2010, 58, 2017–2030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguzzi, A.; Zhu, C. Microglia in prion diseases. J. Clin. Investig. 2017, 127, 3230–3239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mok, S.W.; Riemer, C.; Madela, K.; Hsu, D.K.; Liu, F.T.; Gultner, S.; Heise, I.; Baier, M. Role of galectin-3 in prion infections of the CNS. Biochem. Biophys. Res. Commun. 2007, 359, 672–678. [Google Scholar] [CrossRef]
- Hilton, K.J.; Cunningham, C.; Reynolds, R.A.; Perry, V.H. Early Hippocampal Synaptic Loss Precedes Neuronal Loss and Associates with Early Behavioural Deficits in Three Distinct Strains of Prion Disease. PLoS ONE 2013, 8, e68062. [Google Scholar] [CrossRef]
- Cronier, S.; Laude, H.; Peyrin, J.M. Prions can infect primary cultured neurons and astrocytes and promote neuronal cell death. Proc. Natl. Acad. Sci. USA 2004, 101, 12271–12276. [Google Scholar] [CrossRef] [Green Version]
- Cronier, S.; Carimalo, J.; Schaeffer, B.; Jaumain, E.; Beringue, V.; Miquel, M.C.; Laude, H.; Peyrin, J.M. Endogenous prion protein conversion is required for prion-induced neuritic alterations and neuronal death. FASEB J. 2012, 26, 3854–3861. [Google Scholar] [CrossRef]
- Hannaoui, S.; Maatouk, L.; Privat, N.; Levavasseur, E.; Faucheux, B.A.; Haik, S. Prion propagation and toxicity occur in vitro with two-phase kinetics specific to strain and neuronal type. J. Virol. 2013, 87, 2535–2548. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carroll, J.A.; Chesebro, B. Neuroinflammation, Microglia, and Cell-Association during Prion Disease. Viruses 2019, 11, 65. https://doi.org/10.3390/v11010065
Carroll JA, Chesebro B. Neuroinflammation, Microglia, and Cell-Association during Prion Disease. Viruses. 2019; 11(1):65. https://doi.org/10.3390/v11010065
Chicago/Turabian StyleCarroll, James A., and Bruce Chesebro. 2019. "Neuroinflammation, Microglia, and Cell-Association during Prion Disease" Viruses 11, no. 1: 65. https://doi.org/10.3390/v11010065
APA StyleCarroll, J. A., & Chesebro, B. (2019). Neuroinflammation, Microglia, and Cell-Association during Prion Disease. Viruses, 11(1), 65. https://doi.org/10.3390/v11010065