dsRNA-Seq: Identification of Viral Infection by Purifying and Sequencing dsRNA

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
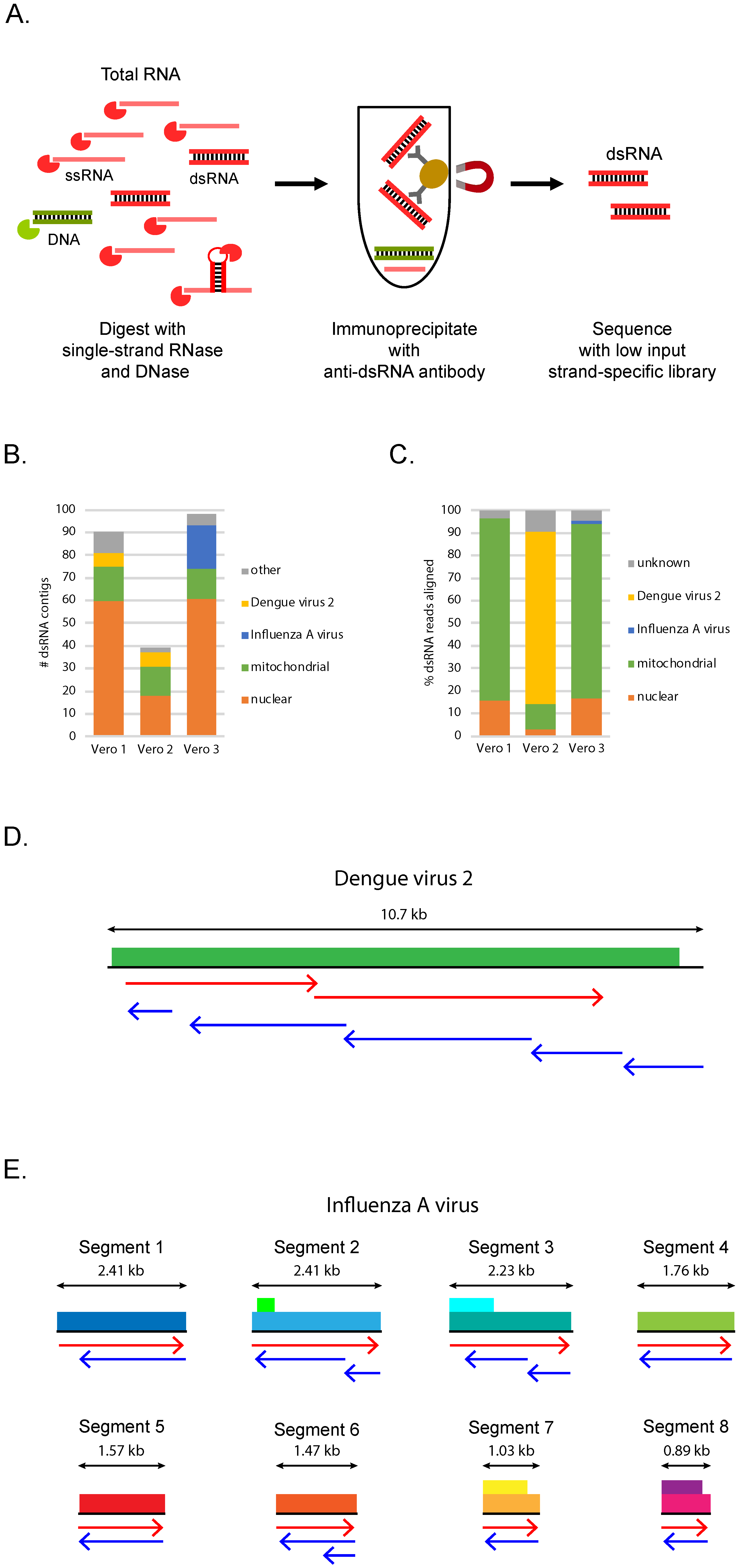
2.1. dsRNA Purification
2.2. RNA Library Construction and Sequencing
2.3. Analysis of dsRNA-Seq Sequences from Vero Cell Samples
2.4. Analysis of dsRNA-Seq and Ribodepleted RNA-Seq Sequences from Mouse Skeletal Muscle Samples
2.5. Analysis of dsRNA-Seq Sequences from Reptile and Deer Tissue Samples
2.6. Sequence Data Availability
3. Results
3.1. dsRNA-Seq Detects Viral Infections of Cultured Mammalian Cells
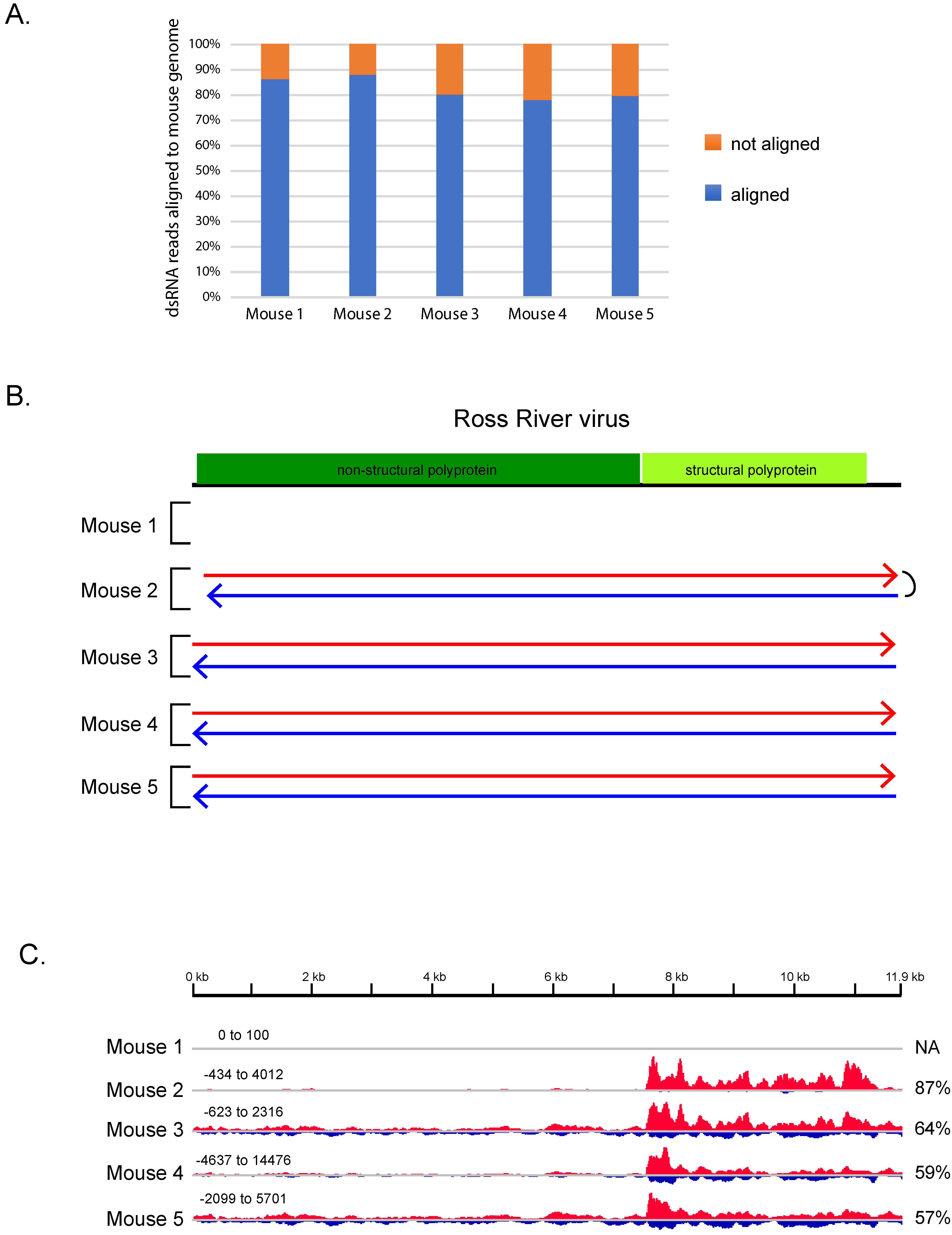
3.2. dsRNA-Seq Correctly Detects Viral Infection in Infected Mice
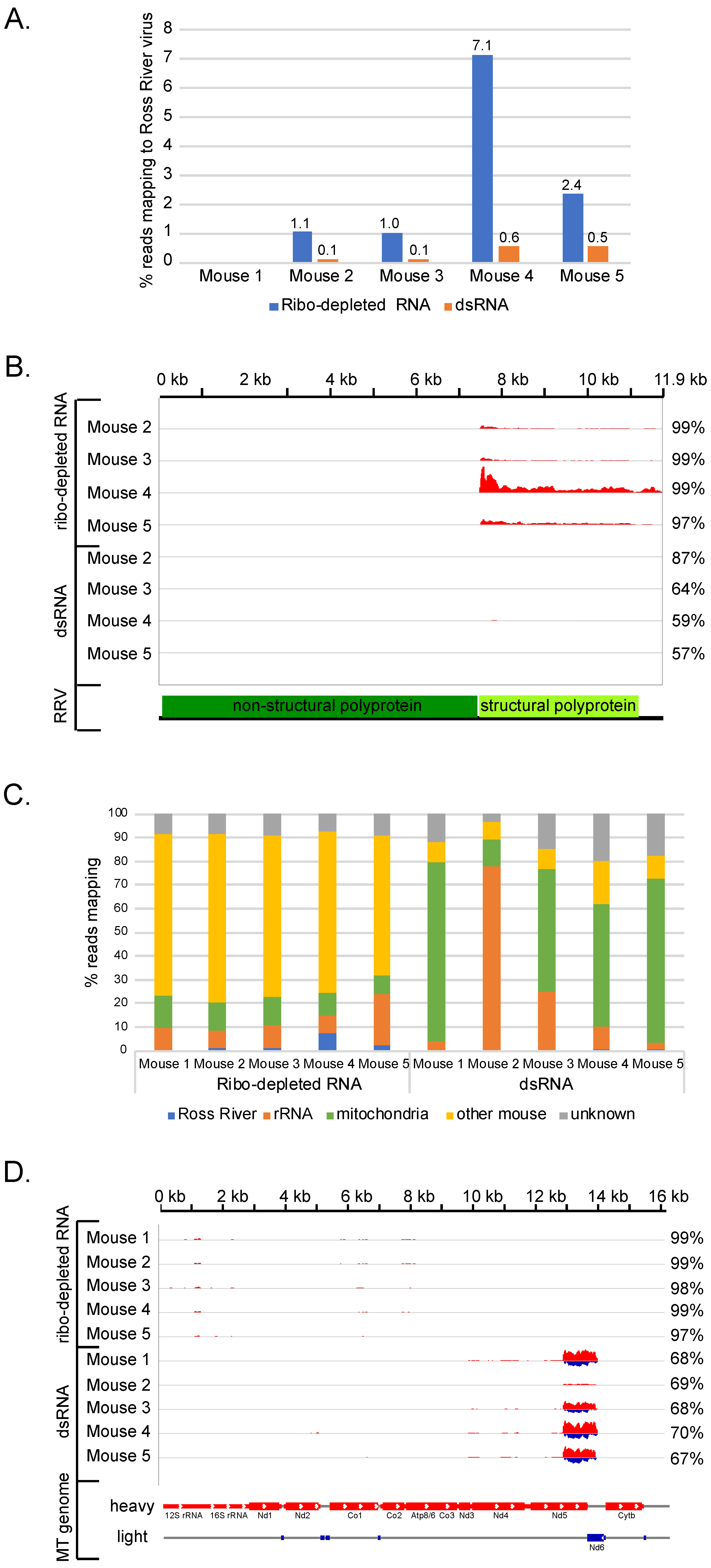
3.3. Impact of dsRNA-Seq on Detecting RNA Viral Infections
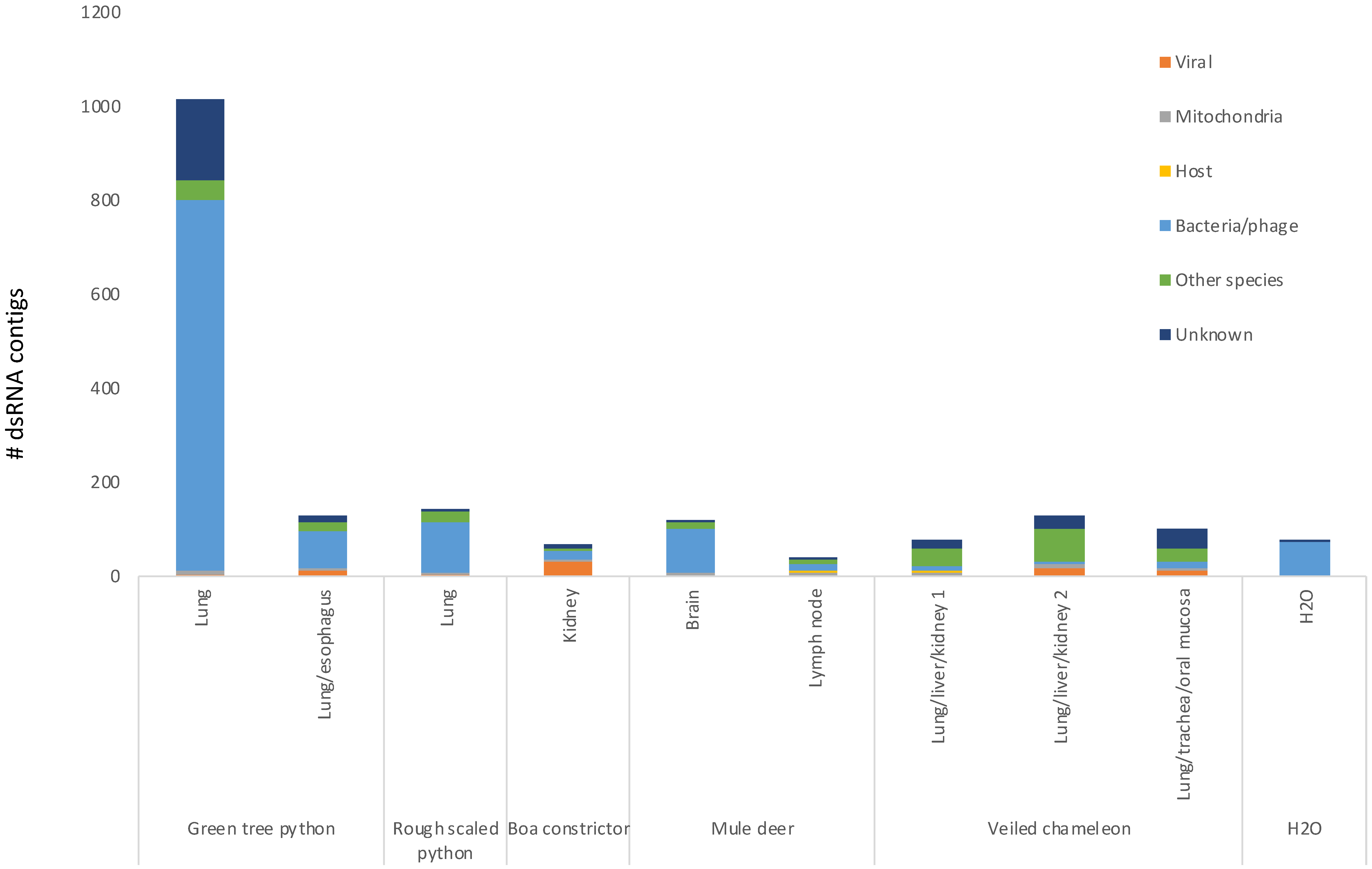
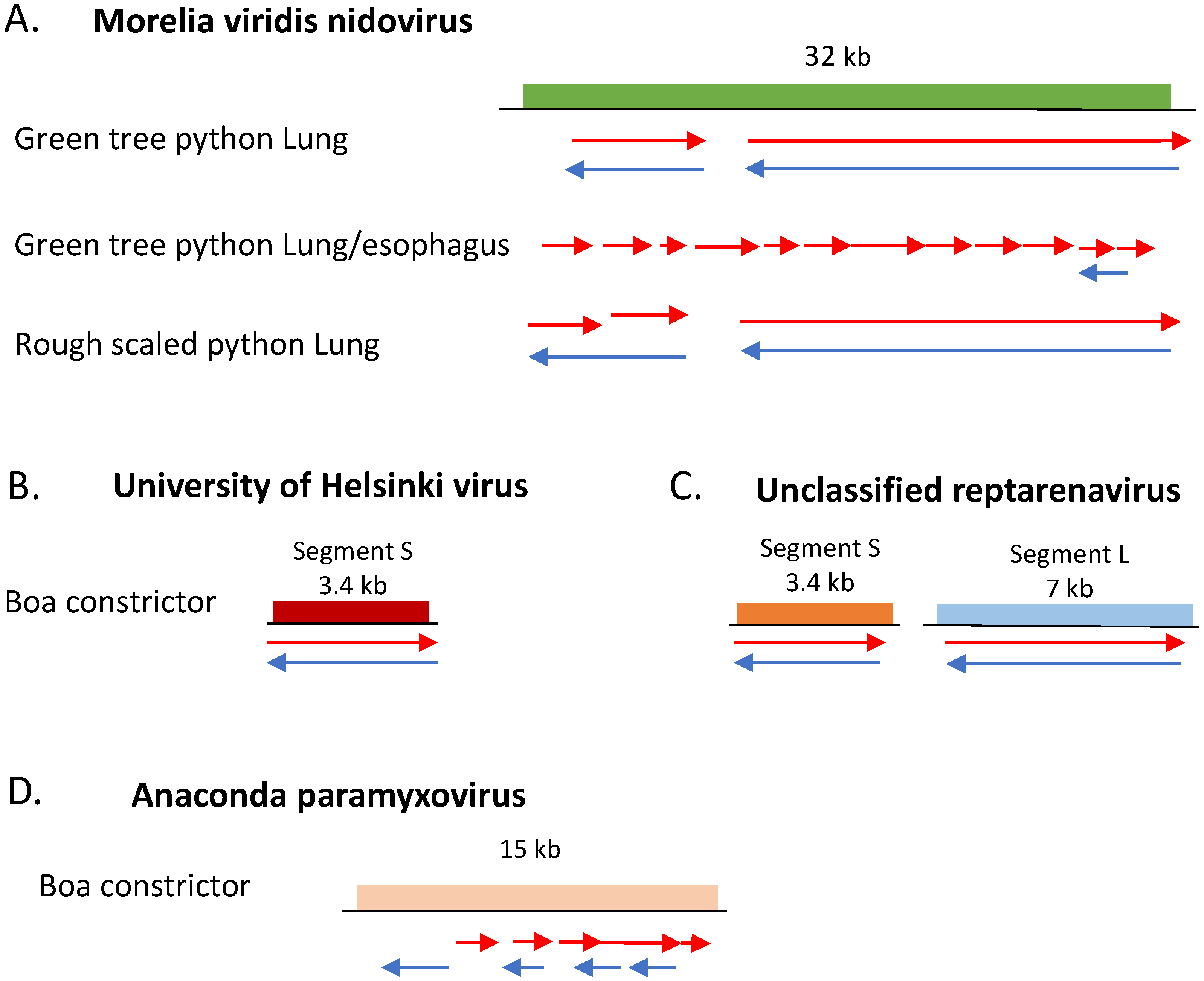
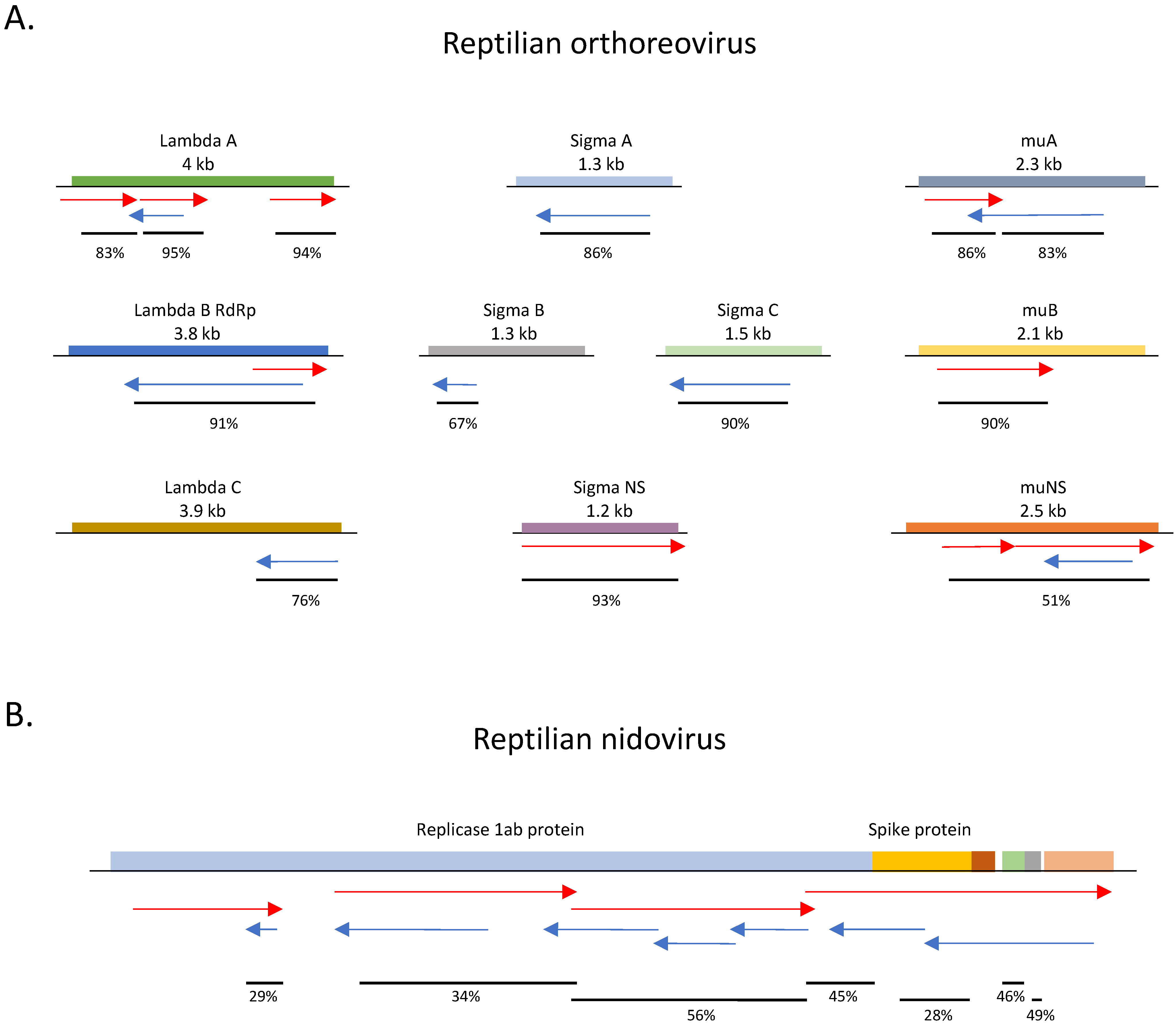
3.4. dsRNA-Seq Detects RNA Viruses of Multiple Genome Types in Infected Animals
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rosenberg, R. Detecting the emergence of novel, zoonotic viruses pathogenic to humans. Cell Mol. Life Sci. 2015, 72, 1115–1125. [Google Scholar] [CrossRef] [PubMed]
- Howard, C.R.; Fletcher, N.F. Emerging virus diseases: Can we ever expect the unexpected? Emerg. Microbes Infect. 2012, 1, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Marston, H.D.; Felkers, G.K.; Morens, D.M.; Fauci, A.S. Emerging Viral Diseases: Confronting Threats with New Technologies. Sci. Transl. Med. 2014, 6, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Rojas, M.R.; Gilbertson, R.L. Emerging Plant Viruses: A Diversity of Mechanisms and Opportunities. In Plant Virus Evolution; Roossinck, M.J., Ed.; Springer: Berlin/Heidelberg, Germany, 2008; pp. 27–51. [Google Scholar]
- Nicaise, V. Crop immunity against viruses: Outcomes and future challenges. Front. Plant Sci. 2014, 5, 660. [Google Scholar] [CrossRef] [PubMed]
- Bodewes, R. Novel viruses in birds: Flying through the roof or is a cage needed? Vet. J. 2018, 233, 55–62. [Google Scholar] [CrossRef]
- National Academies of Sciences Engineering and Medicine. Biodefense in the Age of Synthetic Biology; The National Academies Press: Washington, DC, USA, 2018. [Google Scholar]
- Moore, R.A.; Warren, R.L.; Freeman, J.D.; Gustavsen, J.A.; Chénard, C.; Friedman, J.M.; Suttle, C.A.; Zhao, Y.; Holt, R.A. The sensitivity of massively parallel sequencing for detecting candidate infectious agents associated with human tissue. PLoS ONE 2011, 6, e19838. [Google Scholar] [CrossRef]
- Matranga, C.B.; Andersen, K.G.; Winnicki, S.; Busby, M.; Gladden, A.D.; Tewhey, R.; Stremlau, M.; Berlin, A.; Gire, S.K.; England, E.; et al. Enhanced methods for unbiased deep sequencing of Lassa and Ebola RNA viruses from clinical and biological samples. Genome Biol. 2014, 15, 519. [Google Scholar] [CrossRef]
- Furuta, R.A.; Sakamoto, H.; Kuroishi, A.; Yasiui, K.; Matsukura, H.; Hirayama, F. Metagenomic profiling of the viromes of plasma collected from blood donors with elevated serum alanine aminotransferase levels. Transfusion 2015, 55, 1889–1899. [Google Scholar] [CrossRef]
- Thorburn, F.; Bennett, S.; Modha, S.; Murdoch, D.; Gunson, R.; Murcia, P.R. The use of next generation sequencing in the diagnosis and typing of respiratory infections. J. Clin. Virol. 2015, 69, 96–100. [Google Scholar] [CrossRef] [Green Version]
- Rosseel, T.; Ozhelvaci, O.; Freimanis, G.; Van Borm, S. Evaluation of convenient pretreatment protocols for RNA virus metagenomics in serum and tissue samples. J. Virol. Methods 2015, 222, 72–80. [Google Scholar] [CrossRef]
- Li, L.; Delwart, E. From orphan virus to pathogen: The path to the clinical lab. Curr. Opin. Virol. 2011, 1, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Urisman, A.; Liu, Y.-T.; Springer, M.; Ksiazek, T.G.; Erdman, D.D.; Mardis, E.R.; Hickenbotham, M.; Magrini, V.; Eldred, J.; et al. Viral discovery and sequence recovery using DNA microarrays. PLoS Biol. 2003, 1, e2. [Google Scholar] [CrossRef] [PubMed]
- Briese, T.; Kapoor, A.; Mishra, N.; Jain, K.; Kumar, A.; Jabado, O.J.; Lipkin, W.I. Virome Capture Sequencing Enables Sensitive Viral Diagnosis and Comprehensive Virome Analysis. MBio 2015, 6, e01491-15. [Google Scholar] [CrossRef] [PubMed]
- Wylie, T.N.; Wylie, K.M.; Herter, B.N.; Storch, G.A. Enhanced virome sequencing through solution-based capture enrichment. Genome Res. 2015, 25, 1910–1920. [Google Scholar] [CrossRef]
- Roossinck, M.J.; Saha, P.; Wiley, G.B.; Quan, J.; White, J.D.; Lai, H.; Chavarría, F.; Shen, G.; Roe, B.A. Ecogenomics: Using massively parallel pyrosequencing to understand virus ecology. Mol. Ecol. 2010, 19, 81–88. [Google Scholar] [CrossRef]
- Blouin, A.G.; Ross, H.A.; Hobson-Peters, J.; O’Brien, C.A.; Warren, B.; MacDiarmid, R. A new virus discovered by immunocapture of double-stranded RNA, a rapid method for virus enrichment in metagenomic studies. Mol. Ecol. Resour. 2016, 16, 1255–1263. [Google Scholar] [CrossRef]
- Yanagisawa, H.; Tomita, R.; Katsu, K.; Uehara, T.; Atsumi, G.; Tateda, C. Combined DECS Analysis and Next-Generation Sequencing Enable Efficient Detection of Novel Plant RNA Viruses. Viruses 2016, 8, 70. [Google Scholar] [CrossRef]
- Marais, A.; Faure, C.; Bergey, B.; Candresse, T. Viral Double-Stranded RNAs (dsRNAs) from Plants: Alternative Nucleic Acid Substrates for High-Throughput Sequencing. In Viral Metagenomics. Methods in Molecular Biology; Pantaleo, V., Chiumenti, M., Eds.; Humana Press: New York, NY, USA, 2018; Volume 1746, pp. 45–53. [Google Scholar]
- Crabtree, A.M.; Kizer, E.A.; Hunter, S.S.; Van Leuven, J.T.; New, D.D.; Fagnan, M.W.; Rowley, P.A. A Rapid Method for Sequencing Double-Stranded RNAs Purified from Yeasts and the Identification of a Potent K1 Killer Toxin Isolated from Saccharomyces cerevisiae. Viruses 2019, 11, 70. [Google Scholar] [CrossRef]
- Decker, C.J.; Parker, R. Analysis of Double-Stranded RNA from Microbial Communities Identifies Double-Stranded RNA Virus-like Elements. Cell Rep. 2014, 7, 898–906. [Google Scholar] [CrossRef] [Green Version]
- Urayama, S.; Takaki, Y.; Nishi, S.; Yoshida-Takashima, Y.; Deguchi, S.; Takai, K.; Nunoura, T. Unveiling the RNA virosphere associated with marine microorganisms. Mol. Ecol. Resour. 2018, 18, 1444–1456. [Google Scholar] [CrossRef]
- Urayama, S.; Takaki, Y.; Nunoura, T.; Miyamoto, N. Complete Genome Sequence of a Novel RNA Virus Identified from a Deep-Sea Animal, Osedax japonicus. Microbes Environ. 2018, 33, 446–449. [Google Scholar] [CrossRef] [Green Version]
- Schönborn, J.; Oberstrass, J.; Breyel, E.; Tittgen, J.; Schumacher, J.; Lukacs, N. Monoclonal antibodies to double-stranded RNA as probes of RNA structure in crude nucleic acid extracts. Nucleic Acids Res. 1991, 19, 2993–3000. [Google Scholar] [CrossRef]
- Bonin, M.; Oberstrass, J.; Lukacs, N.; Ewert, K.; Oesterschulze, E.; Kassing, R.; Nellen, W. Determination of preferential binding sites for anti-dsRNA antibodies on double-stranded RNA by scanning force microscopy. RNA 2000, 6, 563–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chomczynski, P. A reagent for the single-step simultaneous isolation of RNA, DNA and proteins from cell and tissue samples. BioTechniques 1993, 15, 532–537. [Google Scholar] [PubMed]
- Haist, K.C.; Burrack, K.S.; Davenport, B.J.; Morrison, T.E. Inflammatory monocytes mediate control of acute alphavirus infection in mice. PLoS Pathog. 2017, 13, e10006748. [Google Scholar] [CrossRef] [PubMed]
- Hoon-Hanks, L.L.; Layton, M.L.; Ossiboff, R.J.; Parker, J.S.L.; Dubovi, E.J.; Stenglein, M.D. Respiratory disease in ball pythons (Python regius) experimentally infected with ball python nidovirus. Virology 2018, 517, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [Green Version]
- Li, H. Aligning Sequence Reads, Clone Sequences and Assembly Contigs with BWA-MEM. arXiv 2013, arXiv:13033997v2. [Google Scholar]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bushnell, B. BBMap Short-Read Aligner, and other Bioinformatics Tools. 2016. Available online: https://sourceforge.net/projects/bbmap/ (accessed on 6 June 2018).
- Quinlan, A.R.; Hall, I.M. BEDtools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed]
- Bradnam, K.R.; Fass, J.N.; Alexandrov, A.; Baranay, P.; Bechner, M.; Birol, I.; Boisvert, S.; Chapman, J.A.; Chapuis, G.; Chikhi, R.; et al. Assemblathon 2: Evaluating de novo methods of genome assembly in three vertebrate species. Gigascience 2013, 2, 10. [Google Scholar] [CrossRef] [PubMed]
- Kircher, M.; Sawyer, S.; Meyer, M. Double indexing overcomes inaccuracies in multiplex sequencing on the Illumina platform. Nucleic Acids Res. 2012, 40, e3. [Google Scholar] [CrossRef] [PubMed]
- Jose, J.; Snyder, J.E.; Kuhn, R.J. A structural and functional perspective of alphavirus replication and assembly. Future Microbiol. 2009, 4, 37–56. [Google Scholar] [CrossRef]
- Pietilä, M.K.; Hellström, K.; Ahola, T. Alphavirus polymerase and RNA replication. Virus Res. 2017, 234, 44–57. [Google Scholar] [CrossRef] [Green Version]
- Strauss, E.G.; Rice, C.M.; Strauss, J.H. Complete nucleotide sequence of the genomic RNA of Sindbis virus. Virology 1984, 133, 92–110. [Google Scholar] [CrossRef]
- Rupp, J.C.; Sokoloski, K.J.; Gebhart, N.N.; Hardy, R.W. Alphavirus RNA synthesis and non-structural protein functions. J. Gen. Virol. 2015, 96, 2483–2500. [Google Scholar] [CrossRef]
- Salter, S.J.; Cox, M.J.; Turek, E.M.; Calus, S.T.; Cookson, W.O.; Moffatt, M.F.; Turner, P.; Parkhill, J.L.; Nicholas, J.; Walker, A.W. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biology 2014, 12, 87. [Google Scholar] [CrossRef]
- De Goffau, M.C.; Lager, S.; Salter, S.J.; Wagner, J.; Kronbichler, A.; Charnock-Jones, D.S.; Peacock, S.J.; Smith, G.C.S.; Parkhill, J. Recognizing the reagent microbiome. Nat. Microbiol. 2018, 3, 851–853. [Google Scholar] [CrossRef] [PubMed]
- Dervas, E.; Hepojoki, J.; Laimbacher, A.; Romero-Palomo, F.; Jelinek, C.; Keller, S.; Smura, T.; Hepojoki, S.; Kipar, A.; Hetzel, U. Nidovirus-Associated Proliferative Pneumonia in the Green Tree Python (Morelia viridis). J. Virol. 2017, 91, e00718-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hepojoki, J.; Salmenperä, P.; Sironen, T.; Hetzel, U.; Korzyukov, Y.; Kipar, A.; Vapalahti, O. Arenavirus Coinfections Are Common in Snakes with Boid Inclusion Body Disease. J. Virol. 2015, 89, 8657–8660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stenglein, M.D.; Jacobson, E.R.; Chang, L.-W.; Sanders, C.; Hawkins, M.G.; Guzman, D.S.-M.; Drazenovich, T.; Dunker, F.; Kamaka, E.K.; Fisher, D.; et al. Widespread recombination, reassortment, and transmission of unbalanced compound viral genotypes in natural arenavirus infections. PLoS Pathog. 2015, 11, e1004900. [Google Scholar] [CrossRef] [PubMed]
- Hyndman, T.H.; Shilton, C.M.; Marschang, R.E. Paramyxoviruses in reptiles: A review. Vet. Microbiol. 2013, 165, 200–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoon-Hanks, L.L.; McGrath, S.; Tyler, K.L.; Owen, C.; Stenglein, M.D. Metagenomic Investigation of Idiopathic Meningoencephalomyelitis in Dogs. J. Vet. Intern. Med. 2018, 32, 324–330. [Google Scholar] [CrossRef]
- Dodds, J.A.; Morris, T.J.; Jordan, R.L. Plant Viral Double-Stranded RNA. Annu. Rev. Phytopathol. 1984, 22, 151–168. [Google Scholar] [CrossRef]
- Kobayashi, K.; Tomita, R.; Sakamoto, M. Recombinant plant dsRNA-binding protein as an effective tool for the isolation of viral replicative form dsRNA and universal detection of RNA viruses. J. Gen. Plant Pathol. 2009, 75, 87–91. [Google Scholar] [CrossRef]
- Weber, F.; Wagner, V.; Rasmussen, S.B.; Hartmann, R.; Paludan, S.R. Double-Stranded RNA Is Produced by Positive-Strand RNA Viruses and DNA Viruses but Not in Detectable Amounts by Negative-Strand RNA Viruses. J. Virol. 2006, 80, 5059–5064. [Google Scholar] [CrossRef] [Green Version]
- Son, K.-N.; Liang, Z.; Lipton, H.L. Double-Stranded RNA Is Detected by Immunofluorescence Analysis in RNA and DNA Virus Infections, Including Those by Negative-Stranded RNA Viruses. J. Virol. 2015, 89, 9383–9392. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Tissue Type | Virus | Genome Type | Average % Identity | |
|---|---|---|---|---|---|
| Nucleotide a | Protein b | ||||
| Green Tree Python | lung | Morelia viridis nidovirus | ssRNA (+) | 95 | 93 |
| lung/esophagus | Morelia viridis nidovirus | ssRNA (+) | 94 | 93 | |
| Rough Scaled Python | lung | Morelia viridis nidovirus | ssRNA (+) | 94 | 97 |
| Boa Constrictor | kidney | University of Helsinki virus reptarenavirus | Segmented ssRNA (−) | 98 | 98 |
| Unidentified reptarenavirus | Segmented ssRNA (−) | 97 | 98 | ||
| Reptilian paramixovirus | ssRNA (−) | 89 | 93 | ||
| Veiled Chameleon | lung/liver/kidney 1 | none | |||
| lung/liver/kidney 2 | Reptilian orthoreovirus | Segmented dsRNA | 76 | 85 | |
| lung/trachea/oral mucosa | Reptilian nidoviruses | ssRNA (+) | ND c | 43 | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Decker, C.J.; Steiner, H.R.; Hoon-Hanks, L.L.; Morrison, J.H.; Haist, K.C.; Stabell, A.C.; Poeschla, E.M.; Morrison, T.E.; Stenglein, M.D.; Sawyer, S.L.; et al. dsRNA-Seq: Identification of Viral Infection by Purifying and Sequencing dsRNA. Viruses 2019, 11, 943. https://doi.org/10.3390/v11100943
Decker CJ, Steiner HR, Hoon-Hanks LL, Morrison JH, Haist KC, Stabell AC, Poeschla EM, Morrison TE, Stenglein MD, Sawyer SL, et al. dsRNA-Seq: Identification of Viral Infection by Purifying and Sequencing dsRNA. Viruses. 2019; 11(10):943. https://doi.org/10.3390/v11100943
Chicago/Turabian StyleDecker, Carolyn J., Halley R. Steiner, Laura L. Hoon-Hanks, James H. Morrison, Kelsey C. Haist, Alex C. Stabell, Eric M. Poeschla, Thomas E. Morrison, Mark D. Stenglein, Sara L. Sawyer, and et al. 2019. "dsRNA-Seq: Identification of Viral Infection by Purifying and Sequencing dsRNA" Viruses 11, no. 10: 943. https://doi.org/10.3390/v11100943
APA StyleDecker, C. J., Steiner, H. R., Hoon-Hanks, L. L., Morrison, J. H., Haist, K. C., Stabell, A. C., Poeschla, E. M., Morrison, T. E., Stenglein, M. D., Sawyer, S. L., & Parker, R. (2019). dsRNA-Seq: Identification of Viral Infection by Purifying and Sequencing dsRNA. Viruses, 11(10), 943. https://doi.org/10.3390/v11100943