Porcine Circovirus 2 Induction of ROS Is Responsible for Mitophagy in PK-15 Cells via Activation of Drp1 Phosphorylation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Virus
2.2. Chemicals and Antibodies
2.3. Virus Infection and Chemical Treatments
2.4. Protein Extraction and SDS-PAGE/Western Blotting
2.5. Detection of Mitophagy
2.6. Transmission Electron Microscopy
2.7. Immunofluorescence
2.8. Measurement of the Mitochondrial Mass, ROS, and Mitochondrial Membrane Potential
2.9. Measurement of Apoptosis
2.10. Statistical Analysis
3. Results
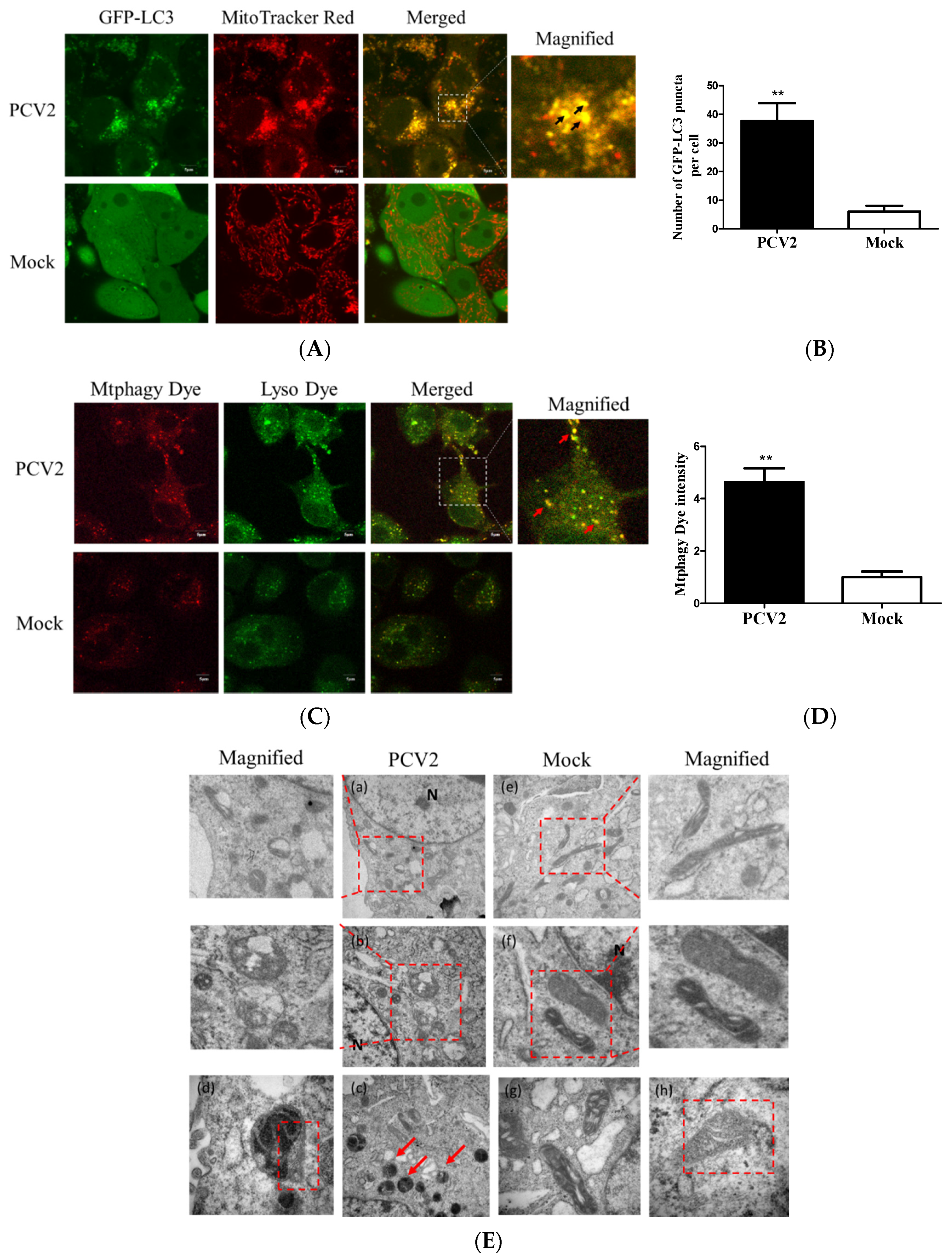
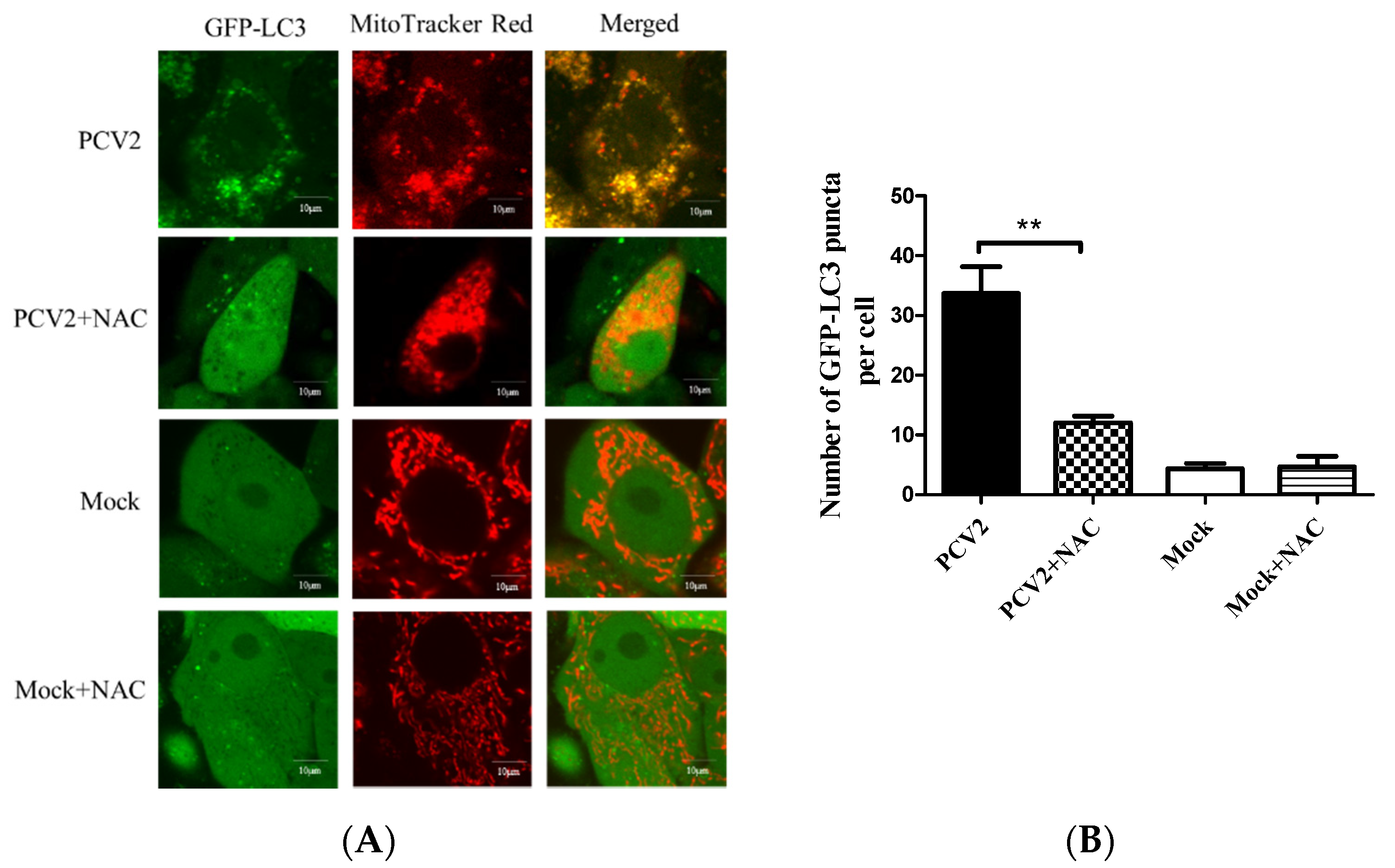
3.1. PCV2 Induced Mitophagy with the Accumulation of Mitophagosomes
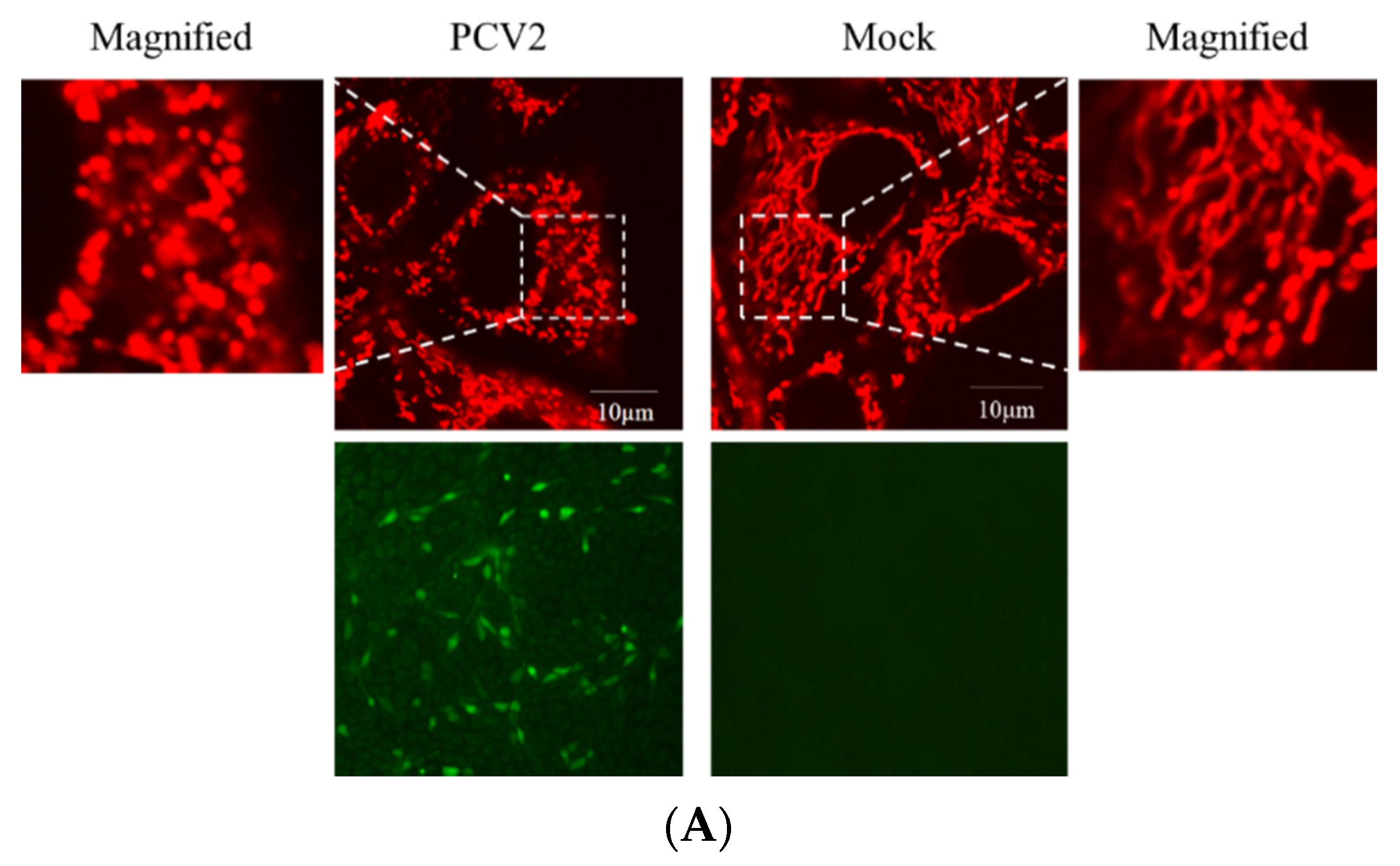
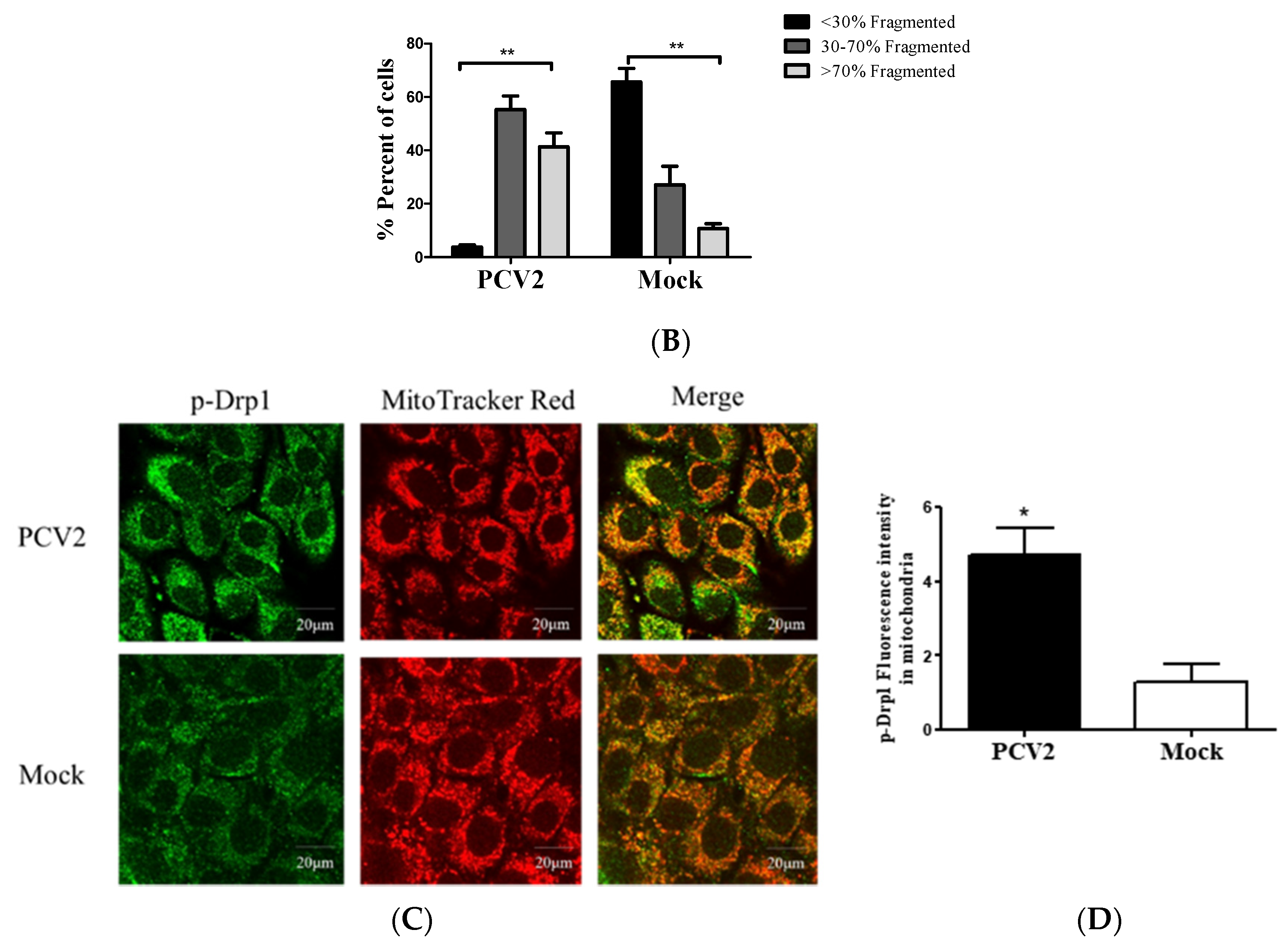
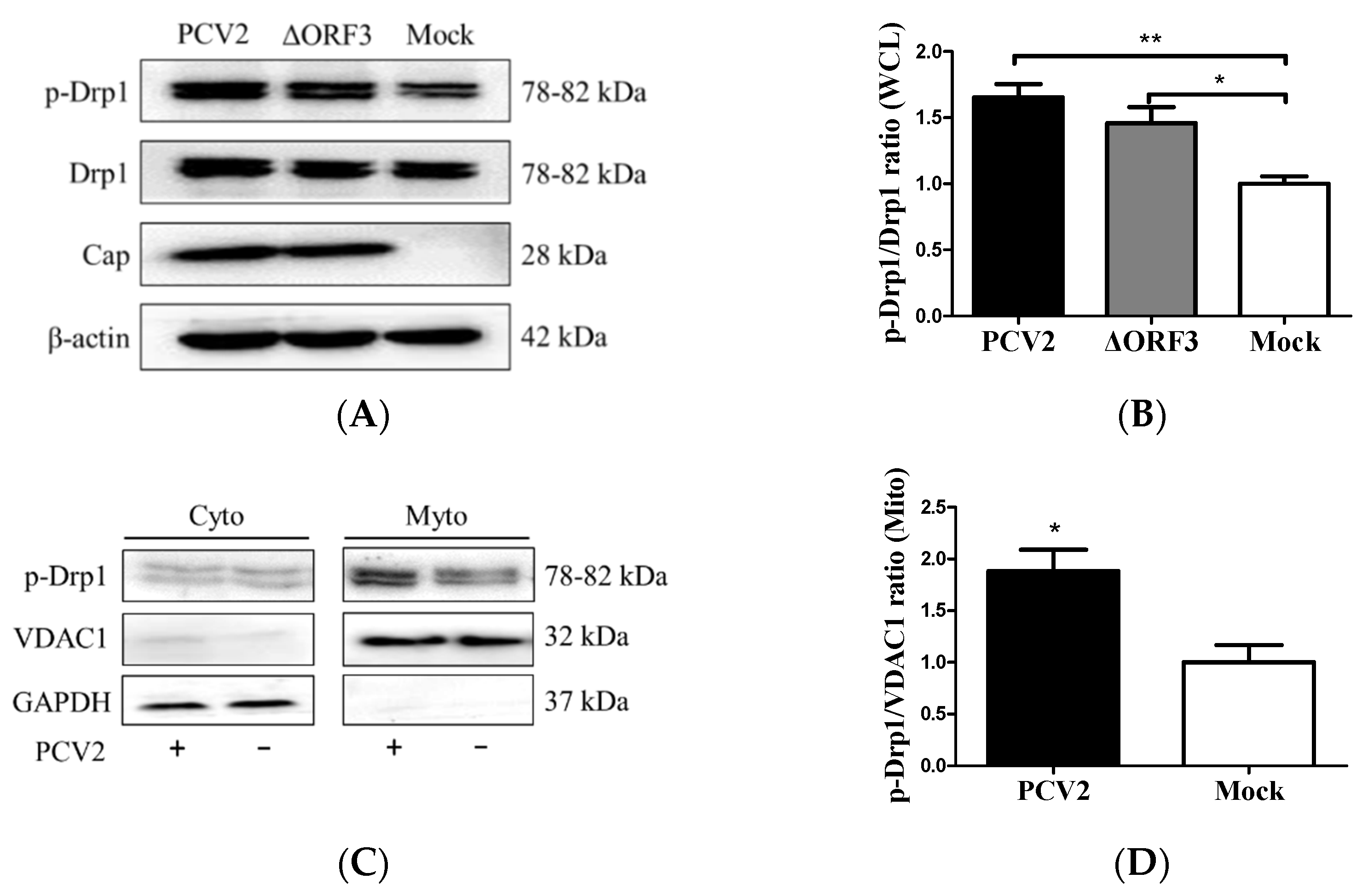
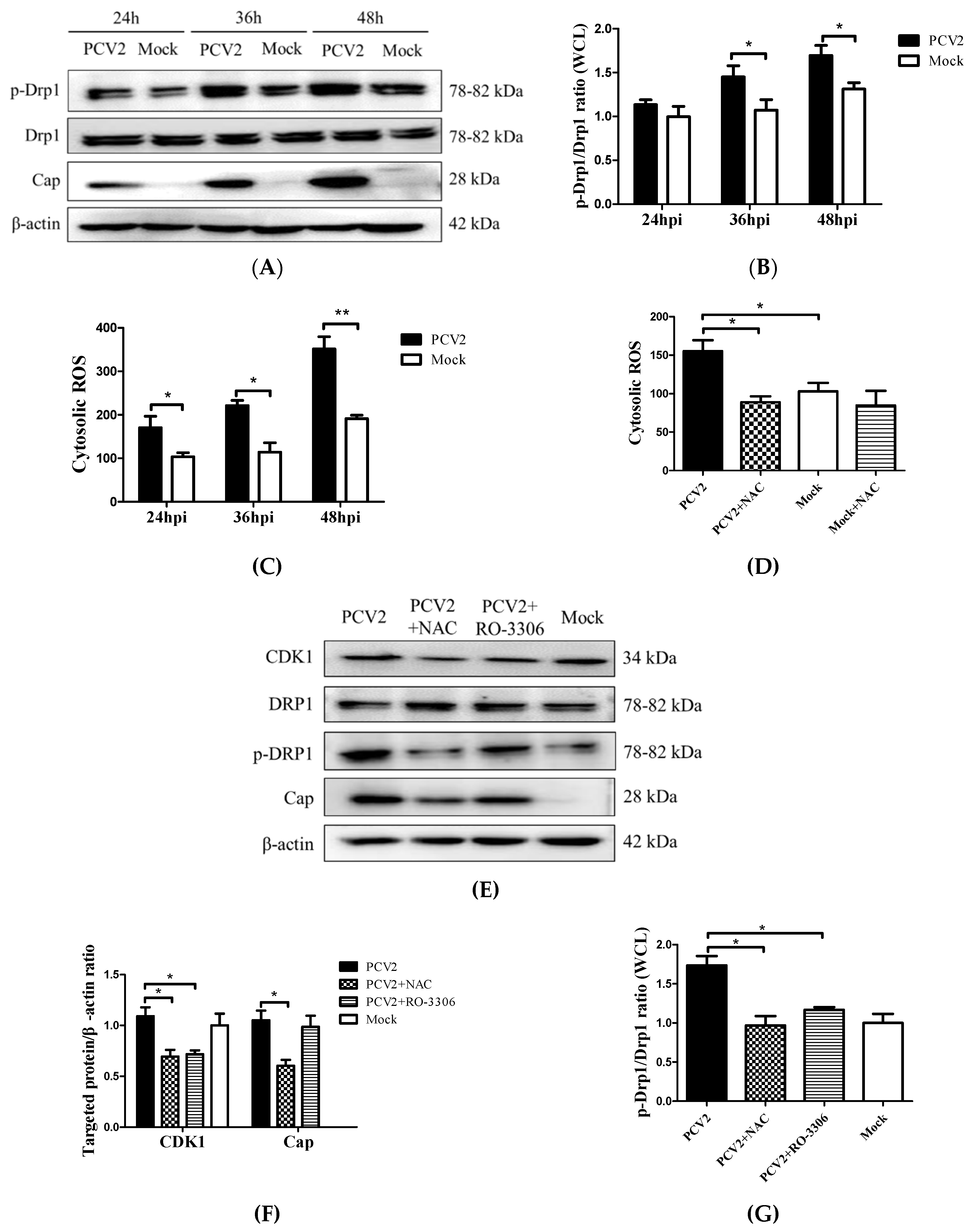
3.2. PCV2 Enhanced Drp1 Ser616 Phosphorylation and Its Mitochondrial Translocation
3.3. PCV2-Induced ROS was Involved in Drp1 Phosphorylation
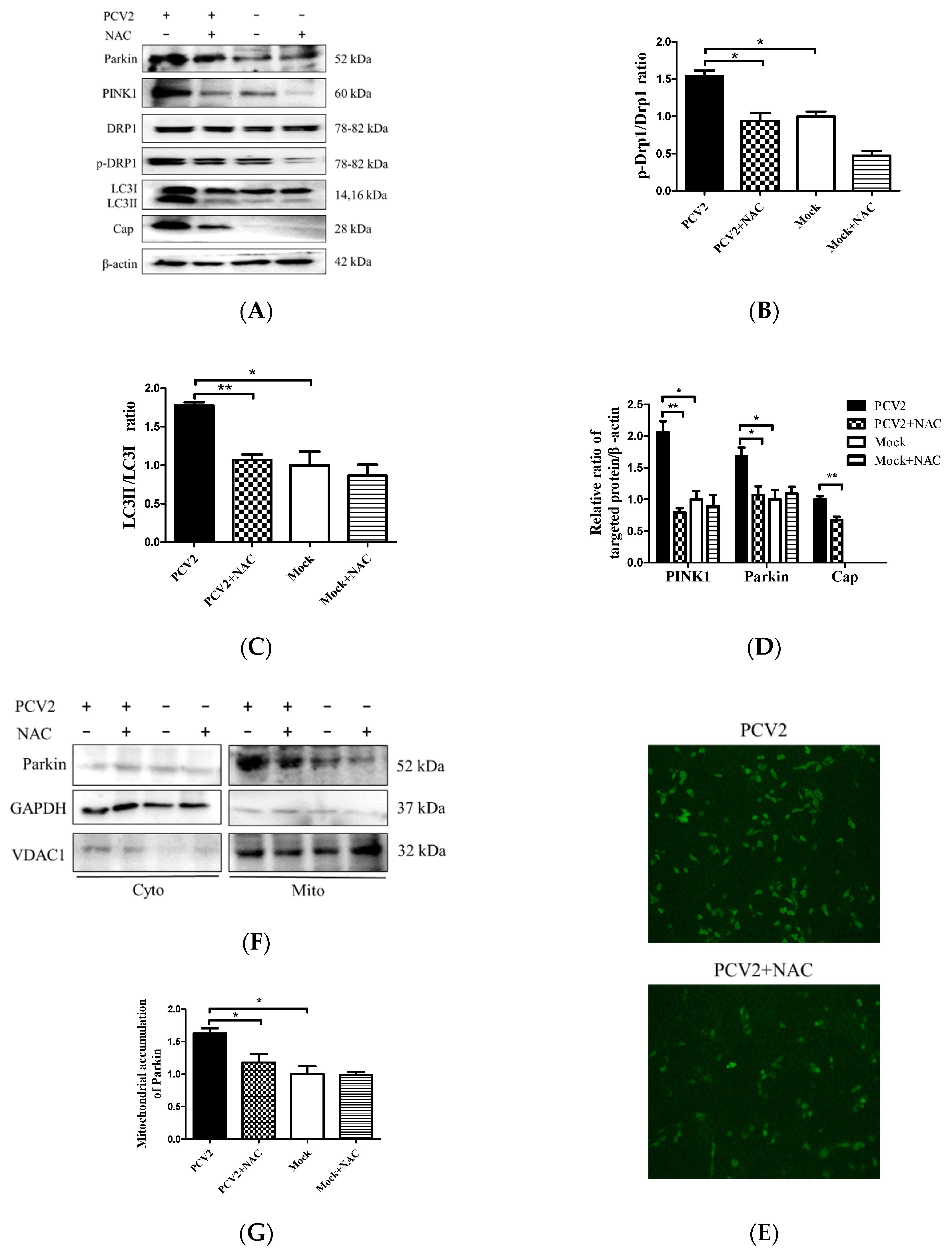
3.4. PCV2-Induced Drp1 Phosphorylation and Mitophagy Could be Reversed Using ROS Scavenging N-acetyl-L-cysteine
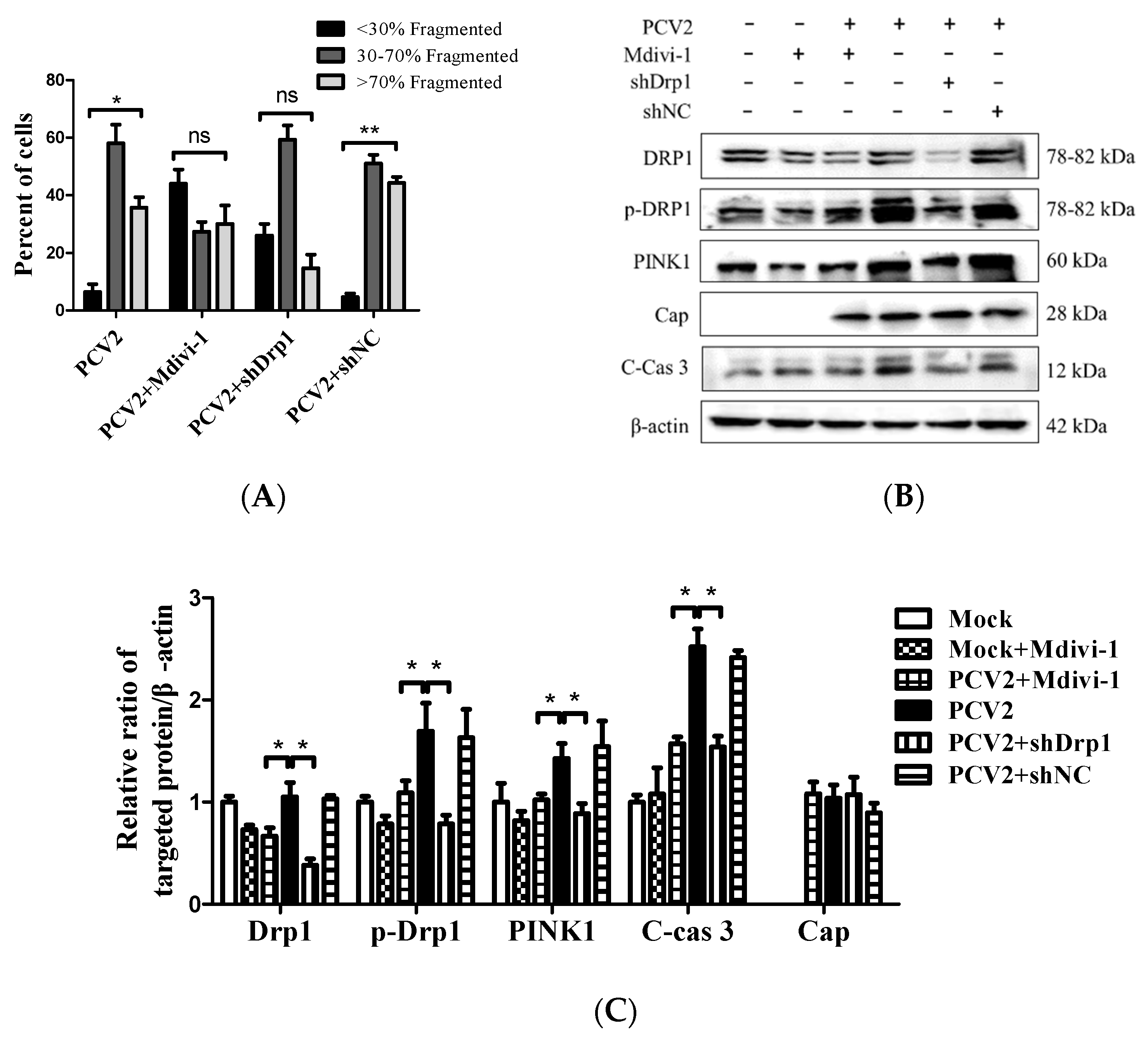
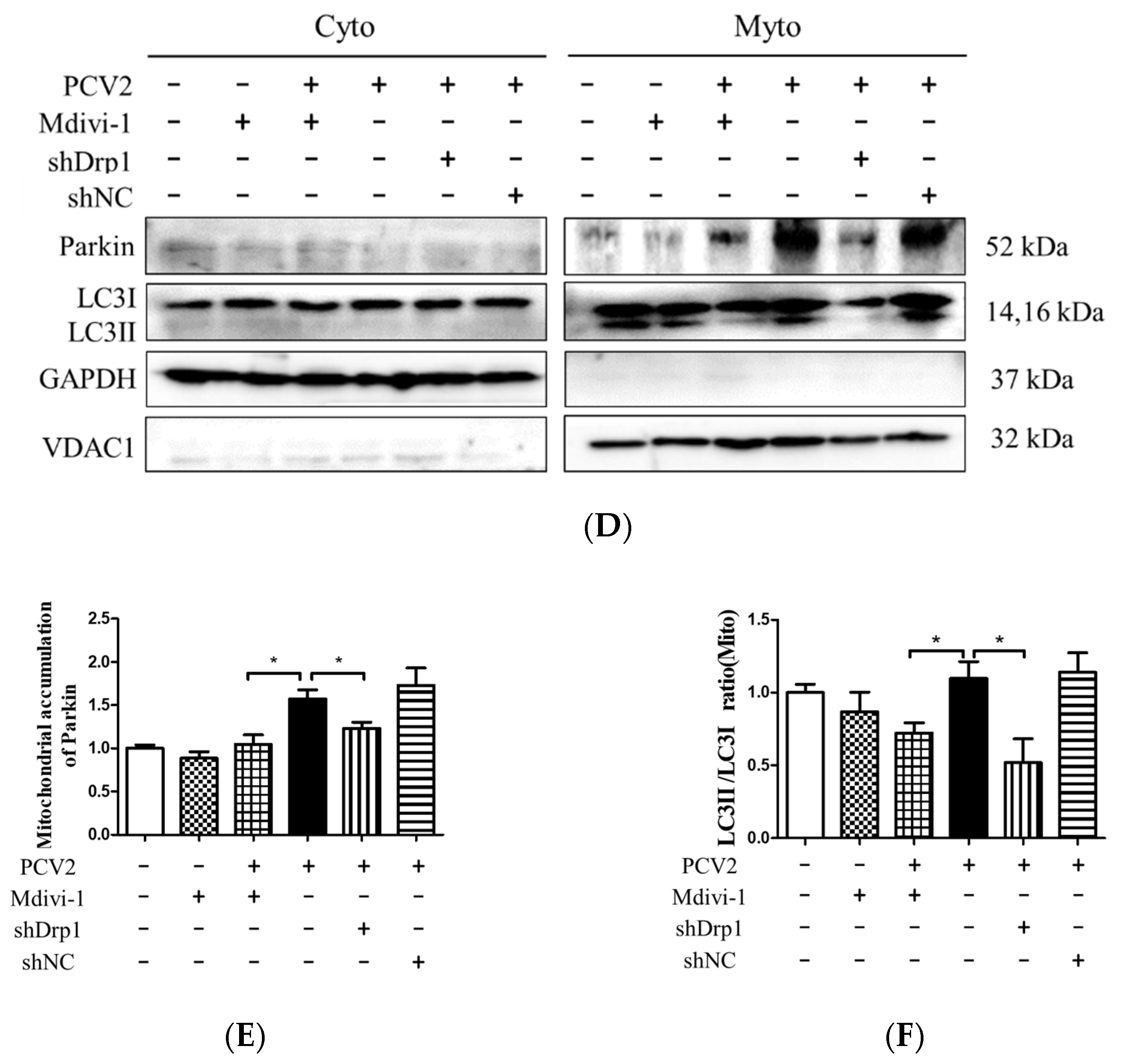
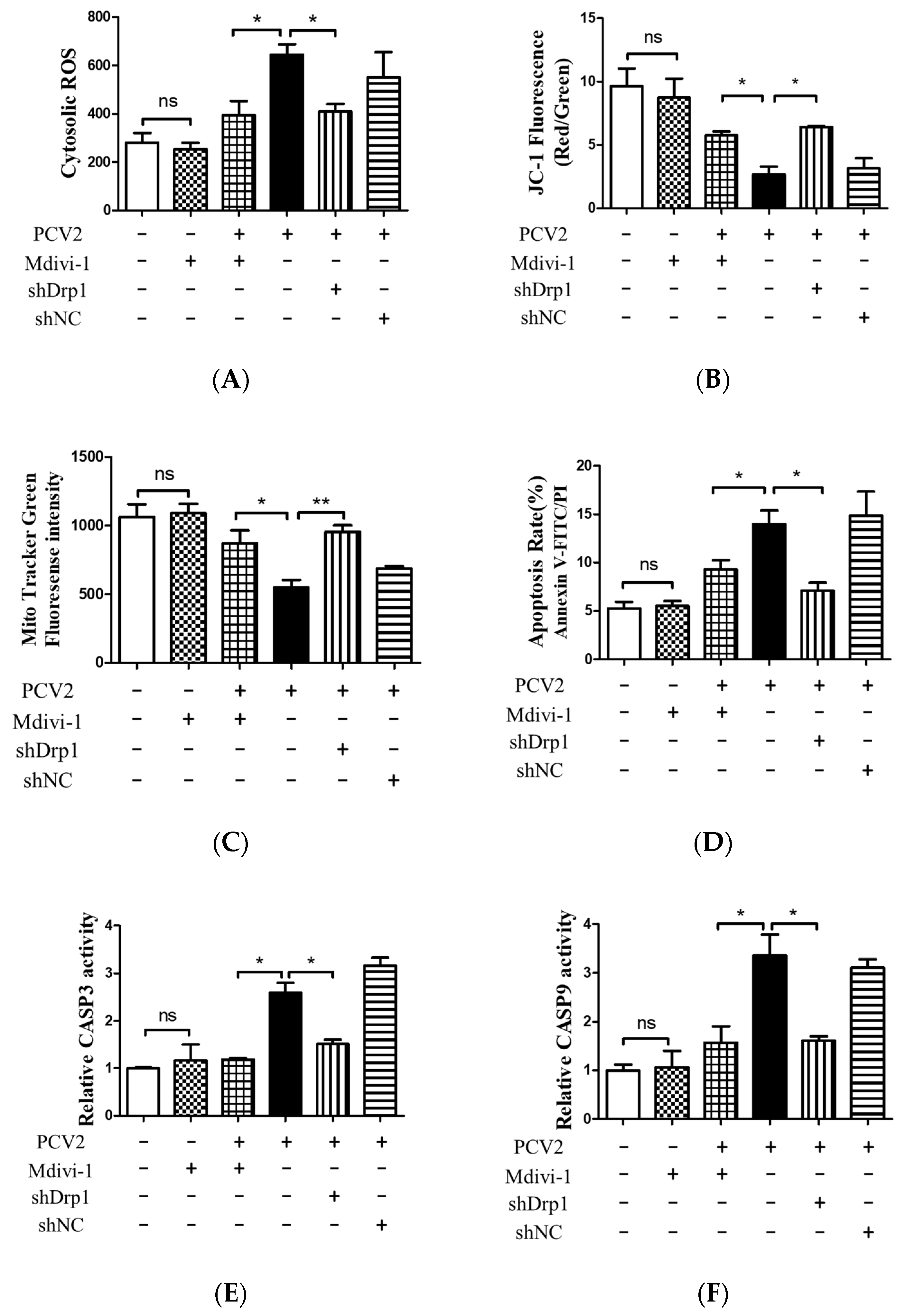
3.5. Suppression of the Mitochondrial Fission Protein Drp1 Inhibited PCV2-Induced Mitophagy and Mitochondrial Apoptosis
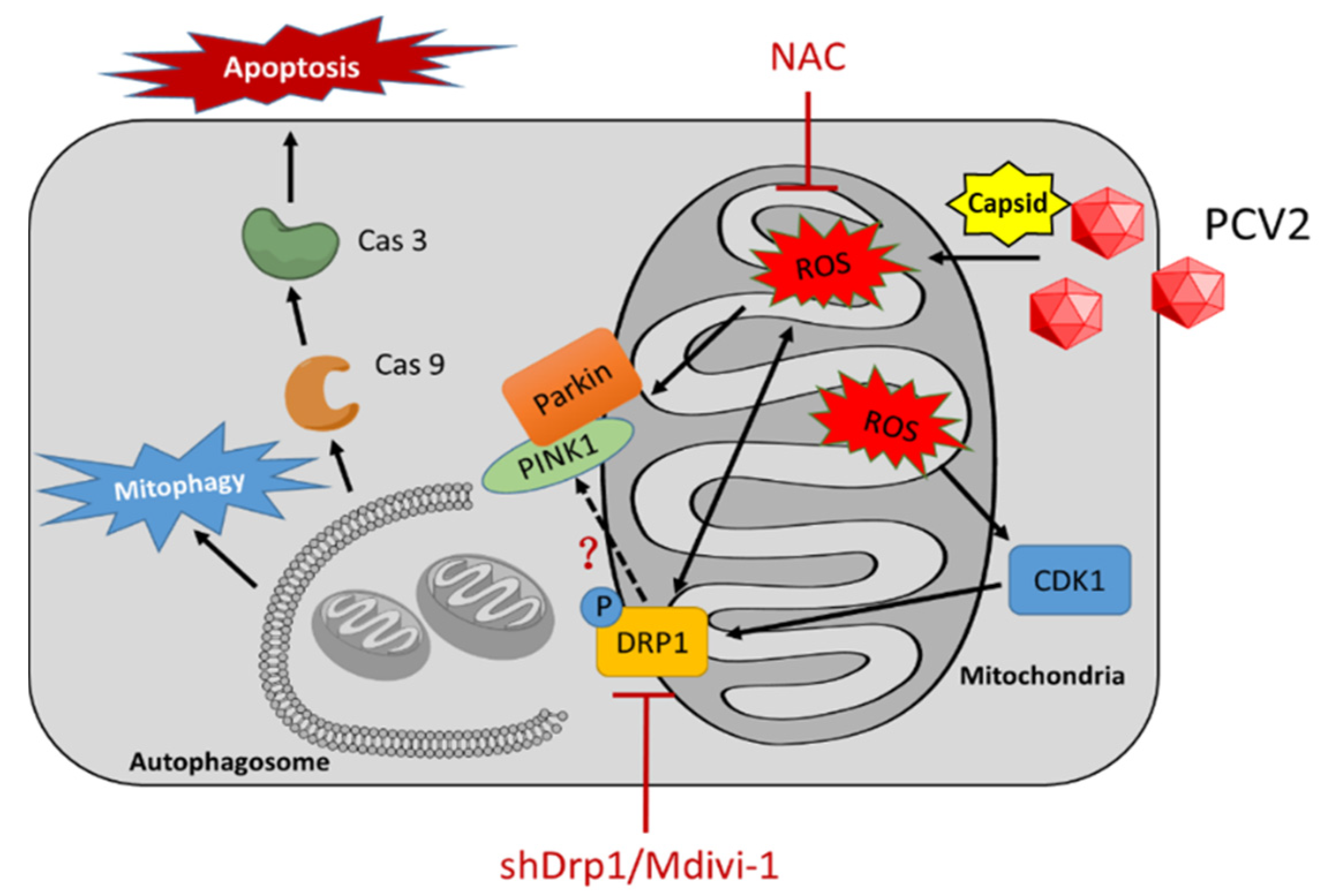
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Meng, X.J. Porcine circovirus type 2 (PCV2): Pathogenesis and interaction with the immune system. Annu. Rev. Anim. Biosci. 2013, 1, 43–64. [Google Scholar] [CrossRef]
- Liu, J.; Chen, I.; Du, Q.; Chua, H.; Kwang, J. The ORF3 protein of porcine circovirus type 2 is involved in viral pathogenesis in vivo. J. Virol. 2006, 80, 5065–5073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Chen, I.; Kwang, J. Characterization of a previously unidentified viral protein in porcine circovirus type 2-infected cells and its role in virus-induced apoptosis. J. Virol. 2005, 79, 8262–8274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Sun, R.; Geng, S.; Shan, Y.; Li, X.; Fang, W. Porcine circovirus type 2 induces ORF3-independent mitochondrial apoptosis via PERK activation and elevation of cytosolic calcium. J. Virol. 2019, 93, e01784-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, B.; Zhou, Y.; Xu, F.; Shuai, J.; Li, X.; Fang, W. Porcine circovirus type 2 induces autophagy via the AMPK/ERK/TSC2/mTOR signaling pathway in PK-15 cells. J. Virol. 2012, 86, 12003–12012. [Google Scholar] [CrossRef] [Green Version]
- Gu, Y.; Qi, B.; Zhou, Y.; Jiang, X.; Xian, Z.; Li, X.; Fang, W. Porcine Circovirus Type 2 Activates CaMMKβ to Initiate Autophagy in PK-15 Cells by Increasing Cytosolic Calcium. Viruses 2016, 8, 135. [Google Scholar] [CrossRef] [Green Version]
- Deretic, V.; Levine, B. Autophagy, Immunity, and Microbial Adaptations. Cell Host Microbe 2009, 5, 527–549. [Google Scholar] [CrossRef] [Green Version]
- Noboru, M.; Masaaki, K. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar]
- Dalibor, M.; Mark, P.; Devenish, R.J. Microautophagy in mammalian cells: Revisiting a 40-year-old conundrum. Autophagy 2011, 7, 673–682. [Google Scholar]
- Eloy, B.; Ana Maria, C. Chaperone-mediated autophagy. Autophagy 2007, 3, 295–299. [Google Scholar]
- Yu, W.H.; Cuervo, A.M.; Kumar, A.; Peterhoff, C.M.; Schmidt, S.D.; Lee, J.H.; Mohan, P.S.; Mercken, M.; Farmery, M.R.; Tjernberg, L.O.; et al. Macroautophagy--a novel Beta-amyloid peptide-generating pathway activated in Alzheimer’s disease. J. Cell Biol. 2005, 171, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Cribbs, J.; Strack, S. Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep. 2007, 8, 939–944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaja, I.; Bai, X.; Liu, Y.; Kikuchi, C.; Dosenovic, S.; Yan, Y.; Canfield, S.G.; Bosnjak, Z.J. Cdk1, PKCdelta and calcineurin-mediated Drp1 pathway contributes to mitochondrial fission-induced cardiomyocyte death. Biochem. Biophys. Res. Commun. 2014, 453, 710–721. [Google Scholar] [CrossRef] [Green Version]
- Pickrell, A.M.; Youle, R.J. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 2015, 85, 257–273. [Google Scholar] [CrossRef] [Green Version]
- Geisler, S.; Holmstrom, K.M.; Skujat, D.; Fiesel, F.C.; Rothfuss, O.C.; Kahle, P.J.; Springer, W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 2010, 12, 119–131. [Google Scholar] [CrossRef]
- Kawajiri, S.; Saiki, S.; Sato, S.; Sato, F.; Hatano, T.; Eguchi, H.; Hattori, N. PINK1 is recruited to mitochondria with parkin and associates with LC3 in mitophagy. FEBS Lett. 2010, 584, 1073–1079. [Google Scholar] [CrossRef] [Green Version]
- Vives-Bauza, C.; Zhou, C.; Huang, Y.; Cui, M.; de Vries, R.L.; Kim, J.; May, J.; Tocilescu, M.A.; Liu, W.; Ko, H.S.; et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc. Natl. Acad. Sci. USA 2010, 107, 378–383. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.J.; Khan, M.; Quan, J.; Till, A.; Subramani, S.; Siddiqui, A. Hepatitis B virus disrupts mitochondrial dynamics: Induces fission and mitophagy to attenuate apoptosis. PLoS Pathog. 2013, 9, e1003722. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.J.; Syed, G.H.; Khan, M.; Chiu, W.W.; Sohail, M.A.; Gish, R.G.; Siddiqui, A. Hepatitis C virus triggers mitochondrial fission and attenuates apoptosis to promote viral persistence. Proc. Natl. Acad. Sci. USA 2014, 111, 6413–6418. [Google Scholar] [CrossRef] [Green Version]
- Trachootham, D.; Lu, W.; Ogasawara, M.A.; Nilsa, R.D.; Huang, P. Redox regulation of cell survival. Antioxid. Redox Signal. 2008, 10, 1343–1374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Circu, M.L.; Aw, T.Y. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic. Biol. Med. 2010, 48, 749–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Ren, F.; Hesketh, J.; Shi, X.; Li, J.; Gan, F.; Hu, Z.; Huang, K. Interaction of porcine circovirus type 2 replication with intracellular redox status in vitro. Redox Rep. Commun. Free Radic. Res. 2013, 18, 186–192. [Google Scholar]
- Zhu, B.; Xu, F.; Li, J.; Shuai, J.; Li, X.; Fang, W. Porcine circovirus type 2 explores the autophagic machinery for replication in PK-15 cells. Virus Res. 2012, 163, 476–485. [Google Scholar] [CrossRef]
- Gou, H.; Zhao, M.; Xu, H.; Yuan, J.; He, W.; Zhu, M.; Ding, H.; Yi, L.; Chen, J. CSFV induced mitochondrial fission and mitophagy to inhibit apoptosis. Oncotarget 2017, 8, 39382–39400. [Google Scholar] [CrossRef] [Green Version]
- Yamano, K.; Youle, R.J. Coupling mitochondrial and cell division. Nat. Cell Biol. 2011, 13, 1026–1027. [Google Scholar] [CrossRef]
- Taguchi, N.; Ishihara, N.; Jofuku, A.; Oka, T.; Mihara, K. Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J. Biol. Chem. 2007, 282, 11521–11529. [Google Scholar] [CrossRef] [Green Version]
- Scherz-Shouval, R.; Elazar, Z. Regulation of autophagy by ROS: Physiology and pathology. Trends Biochem. Sci. 2011, 36, 30–38. [Google Scholar] [CrossRef]
- Chen, X.; Ren, F.; Hesketh, J.; Shi, X.; Li, J.; Gan, F.; Huang, K. Reactive oxygen species regulate the replication of porcine circovirus type 2 via NF-κB pathway. Virology 2012, 426, 66–72. [Google Scholar] [CrossRef] [Green Version]
- Otera, H.; Ishihara, N.; Mihara, K. New insights into the function and regulation of mitochondrial fission. BBA-Mol. Cell Res. 2013, 1833, 1256–1268. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Chan, D.C. Mitochondrial dynamics--fusion, fission, movement, and mitophagy--in neurodegenerative diseases. Hum. Mol. Genet. 2009, 18, R169–R176. [Google Scholar] [CrossRef] [PubMed]
- Blazquez, A.B.; Escribano-Romero, E.; Merino-Ramos, T.; Saiz, J.C.; Martin-Acebes, M.A. Stress responses in flavivirus-infected cells: Activation of unfolded protein response and autophagy. Front. Microbiol. 2014, 5, 266. [Google Scholar] [CrossRef] [PubMed]
- Mehrbod, P.; Ande, S.R.; Alizadeh, J. The roles of apoptosis, autophagy and unfolded protein response in arbovirus, influenza virus, and HIV infections. Virulence 2019, 10, 376–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeganeh, B.; Rezaei Moghadam, A.; Alizadeh, J.; Wiechec, E.; Alavian, S.M.; Hashemi, M.; Geramizadeh, B.; Samali, A.; Bagheri Lankarani, K.; Post, M.; et al. Hepatitis B and C virus-induced hepatitis: Apoptosis, autophagy, and unfolded protein response. World J. Gastroenterol. 2015, 21, 13225–13239. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.V.; Valuev-Elliston, V.T. Oxidative Stress during HIV Infection: Mechanisms and Consequences. Oxidative Med. Cell. Longev. 2016, 2016, 8910396. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Chen, F.; Liu, T.; Chen, F.; Liu, S.; Yang, J. The role of oxidative stress in influenza virus infection. Microbes Infect. 2017, 19, 580–586. [Google Scholar] [CrossRef]
- Zhou, Y.; Gu, Y.; Qi, B.; Zhang, Y.; Li, X.; Fang, W. Porcine circovirus type 2 capsid protein induces unfolded protein response with subsequent activation of apoptosis. J. Zhejiang Univ. Sci. B 2017, 18, 316–323. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Qi, B.; Gu, Y.; Xu, F.; Du, H.; Li, X.; Fang, W. Porcine Circovirus 2 Deploys PERK Pathway and GRP78 for Its Enhanced Replication in PK-15 Cells. Viruses 2016, 8, 56. [Google Scholar] [CrossRef]
- Estaquier, J.; Arnoult, D. Inhibiting Drp1-mediated mitochondrial fission selectively prevents the release of cytochrome c during apoptosis. Cell Death Differ. 2007, 14, 1086–1094. [Google Scholar] [CrossRef] [Green Version]
- Kageyama, Y.; Hoshijima, M.; Seo, K.; Bedja, D.; Sysa-Shah, P.; Andrabi, S.A.; Chen, W.; Hoke, A.; Dawson, V.L.; Dawson, T.M.; et al. Parkin-independent mitophagy requires Drp1 and maintains the integrity of mammalian heart and brain. EMBO J. 2014, 33, 2798–2813. [Google Scholar] [CrossRef]
- Xie, L.; Shi, F.; Tan, Z.; Li, Y.; Bode, A.M.; Cao, Y. Mitochondrial network structure homeostasis and cell death. Cancer Sci. 2018, 109, 3686–3694. [Google Scholar] [CrossRef] [PubMed]
- Buhlman, L.; Damiano, M.; Bertolin, G.; Ferrando-Miguel, R.; Lombès, A.; Brice, A.; Corti, O. Functional interplay between Parkin and Drp1 in mitochondrial fission and clearance. Biochim. Biophys. Acta 2014, 1843, 2012–2026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, Y.S.; Choi, S.E.; Koh, H.C. PGAM5 regulates PINK1/Parkin-mediated mitophagy via DRP1 in CCCP-induced mitochondrial dysfunction. Toxicol. Lett. 2018, 284, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Xiao, B.; Goh, J.Y.; Xiao, L.; Xian, H.; Lim, K.L.; Liou, Y.C. Reactive oxygen species trigger Parkin/PINK1 pathway-dependent mitophagy by inducing mitochondrial recruitment of Parkin. J. Biol. Chem. 2017, 292, 16697–16708. [Google Scholar] [CrossRef] [Green Version]
- Xie, B.; Wang, S.; Jiang, N.; Li, J.J. Cyclin B1/CDK1-regulated mitochondrial bioenergetics in cell cycle progression and tumor resistance. Cancer Lett. 2019, 443, 56–66. [Google Scholar] [CrossRef]
- Kashatus, D.F.; Lim, K.H.; Brady, D.C.; Pershing, N.L.; Cox, A.D.; Counter, C.M. RALA and RALBP1 regulate mitochondrial fission at mitosis. Nat. Cell Biol. 2011, 13, 1108–1115. [Google Scholar] [CrossRef] [Green Version]
- Sharp, W.W.; Fang, Y.H.; Han, M.; Zhang, H.J.; Hong, Z.; Banathy, A.; Morrow, E.; Ryan, J.J.; Archer, S.L. Dynamin-related protein 1 (Drp1)-mediated diastolic dysfunction in myocardial ischemia-reperfusion injury: Therapeutic benefits of Drp1 inhibition to reduce mitochondrial fission. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2014, 28, 316–326. [Google Scholar] [CrossRef] [Green Version]
- Yu, T.; Sheu, S.S.; Robotham, J.L.; Yoon, Y. Mitochondrial fission mediates high glucose-induced cell death through elevated production of reactive oxygen species. Cardiovasc. Res. 2008, 79, 341–351. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Wang, J.; Zhou, A.; Khan, F.A.; Hu, L.; Zhang, S. Porcine reproductive and respiratory syndrome virus triggers mitochondrial fission and mitophagy to attenuate apoptosis. Oncotarget 2016, 7, 56002–56012. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Qin, Y.; Chen, M. Viral Strategies for Triggering and Manipulating Mitophagy. Autophagy 2018, 14, 1665–1673. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Mou, C.; Yang, X.; Lin, J.; Yang, Q. Mitophagy in TGEV infection counteracts oxidative stress and apoptosis. Oncotarget 2016, 7, 27122–27141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, J.; Ying, M.; Xie, N.; Lin, G.; Dong, R.; Zhang, J.; Yan, H.; Yang, X.; He, Q.; Yang, B. The oxidation states of DJ-1 dictate the cell fate in response to oxidative stress triggered by 4-hpr: Autophagy or apoptosis? Antioxid. Redox Signal. 2014, 21, 1443–1459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolgacheva, L.P.; Berezhnov, A.V.; Fedotova, E.I.; Zinchenko, V.P.; Abramov, A.Y. Role of DJ-1 in the mechanism of pathogenesis of Parkinson’s disease. J. Bioenerg. Biomembr. 2019, 51, 175–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]











© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Sun, R.; Li, X.; Fang, W. Porcine Circovirus 2 Induction of ROS Is Responsible for Mitophagy in PK-15 Cells via Activation of Drp1 Phosphorylation. Viruses 2020, 12, 289. https://doi.org/10.3390/v12030289
Zhang Y, Sun R, Li X, Fang W. Porcine Circovirus 2 Induction of ROS Is Responsible for Mitophagy in PK-15 Cells via Activation of Drp1 Phosphorylation. Viruses. 2020; 12(3):289. https://doi.org/10.3390/v12030289
Chicago/Turabian StyleZhang, Yikai, Renjie Sun, Xiaoliang Li, and Weihuan Fang. 2020. "Porcine Circovirus 2 Induction of ROS Is Responsible for Mitophagy in PK-15 Cells via Activation of Drp1 Phosphorylation" Viruses 12, no. 3: 289. https://doi.org/10.3390/v12030289
APA StyleZhang, Y., Sun, R., Li, X., & Fang, W. (2020). Porcine Circovirus 2 Induction of ROS Is Responsible for Mitophagy in PK-15 Cells via Activation of Drp1 Phosphorylation. Viruses, 12(3), 289. https://doi.org/10.3390/v12030289