
Novel Ampeloviruses Infecting Cassava in Central Africa and the South-West Indian Ocean Islands

 , , , , , ,
, , , , , , 
Abstract
:1. Introduction
2. Materials and Methods
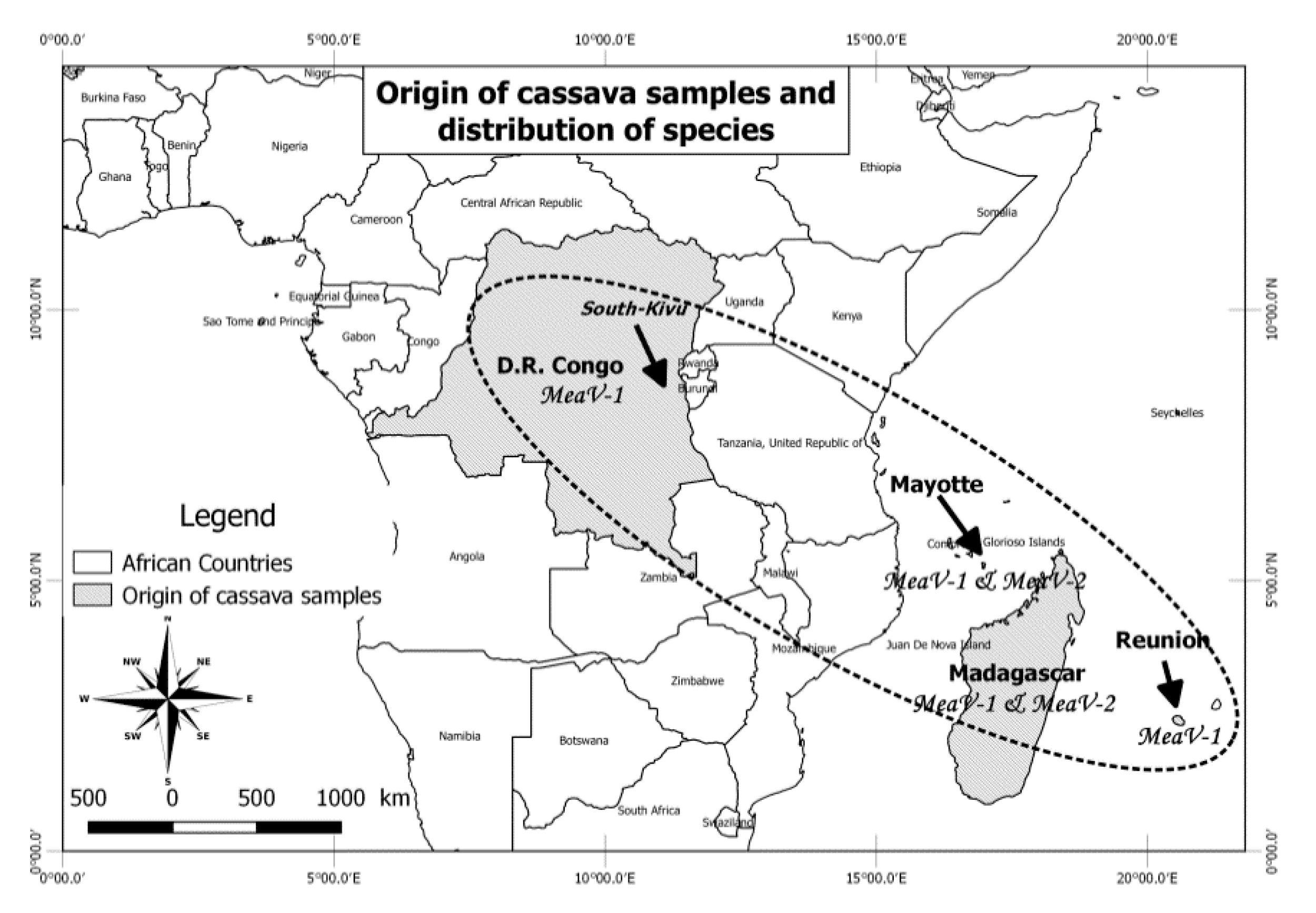
2.1. Origin of the Analyzed Cassava Samples and High-Throughput Sequencing
2.2. Bioinformatic Analyses
2.3. Confirmatory RT-PCR and Sequencing
2.4. Characterization of Defective RNA (D-RNA Molecules)
2.4.1. RNA Isolation, cDNA Synthesis and Amplification of D-RNAs
2.4.2. Sequence Analysis and Alignments
3. Results
3.1. Identification of Closteroviridae Members in Cassava
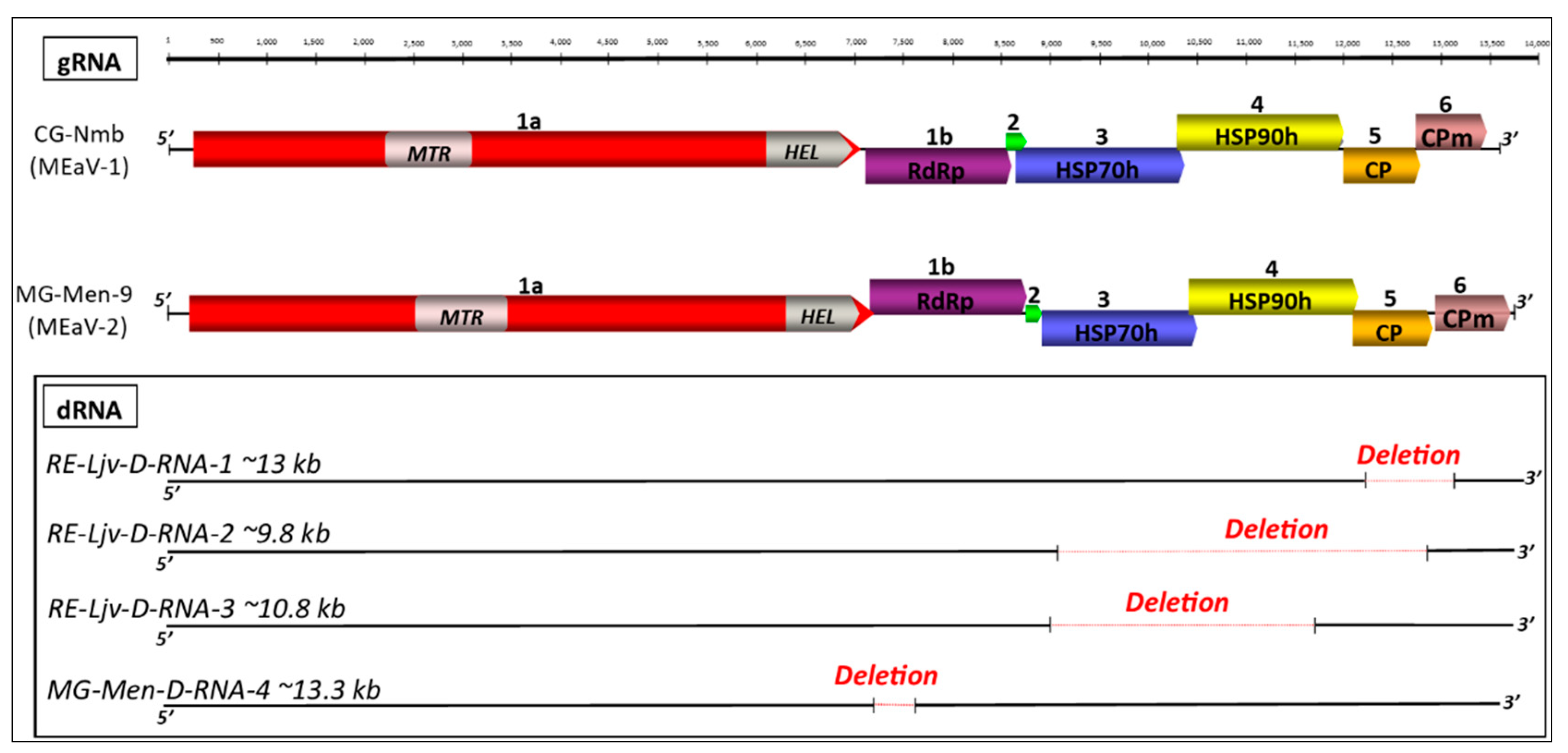
3.2. Genome Annotation
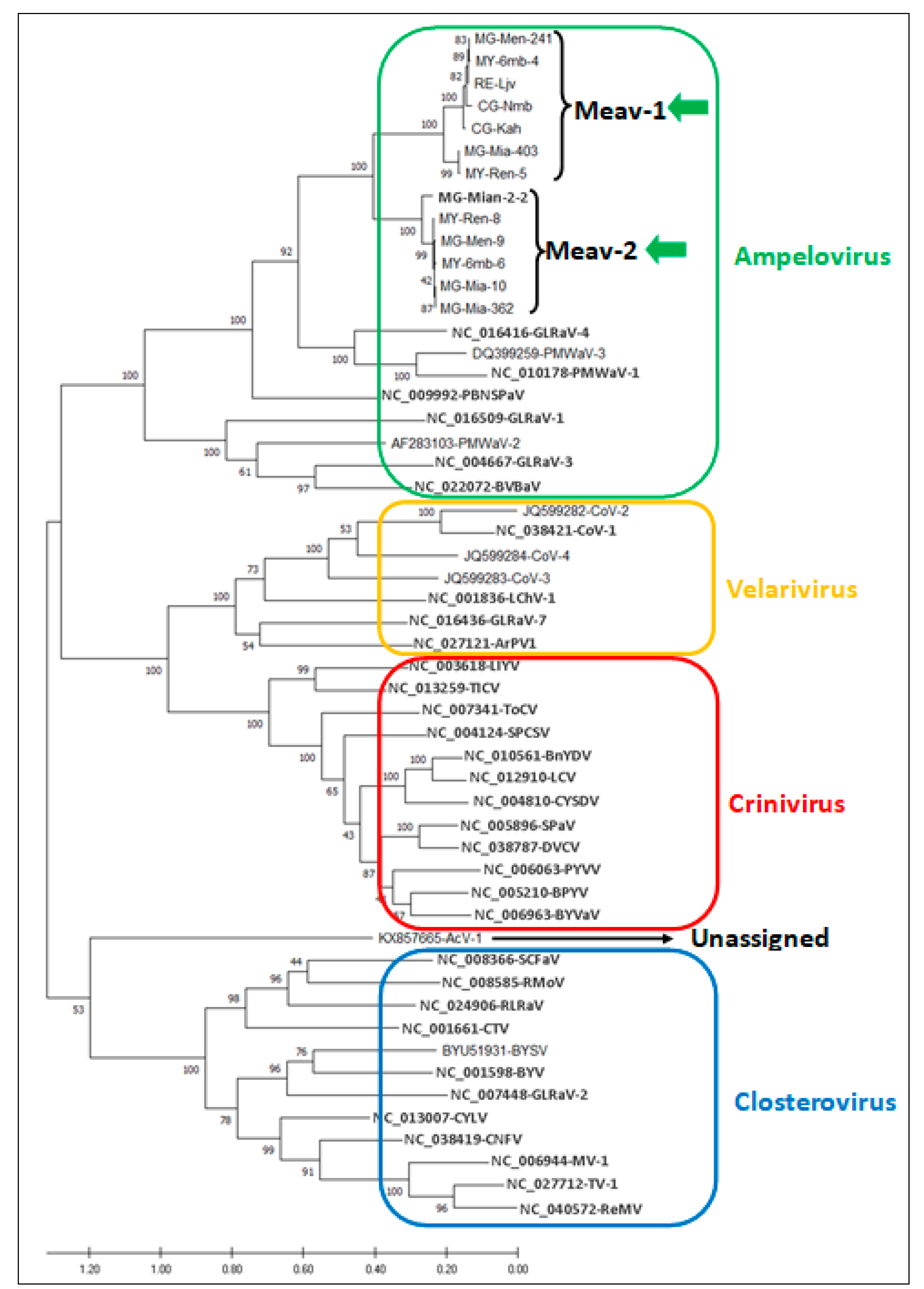
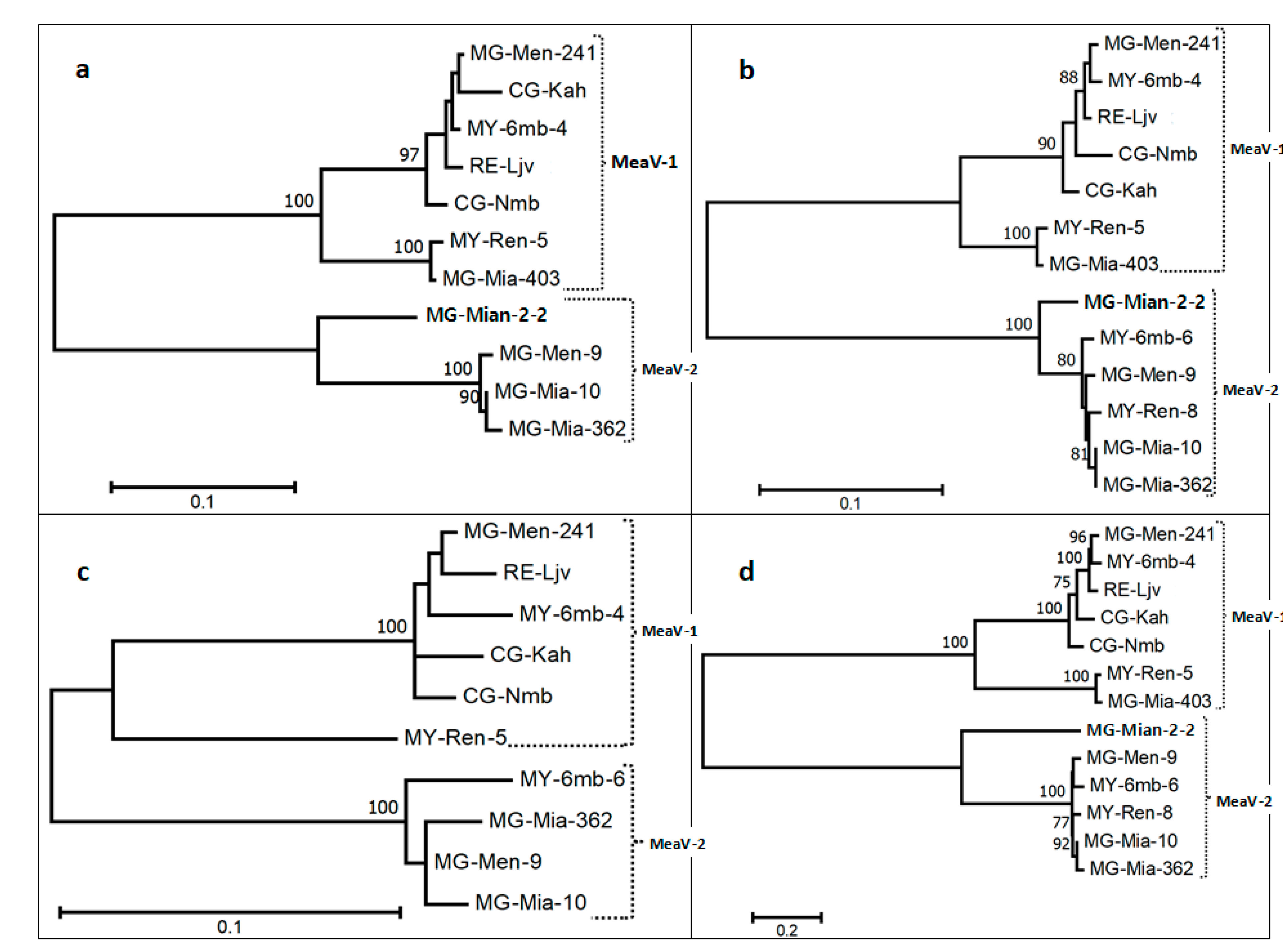
3.3. Phylogenetic Analysis of Cassava Isolates with Members of the Family Closteroviridae
3.4. Diversity of the New Isolates
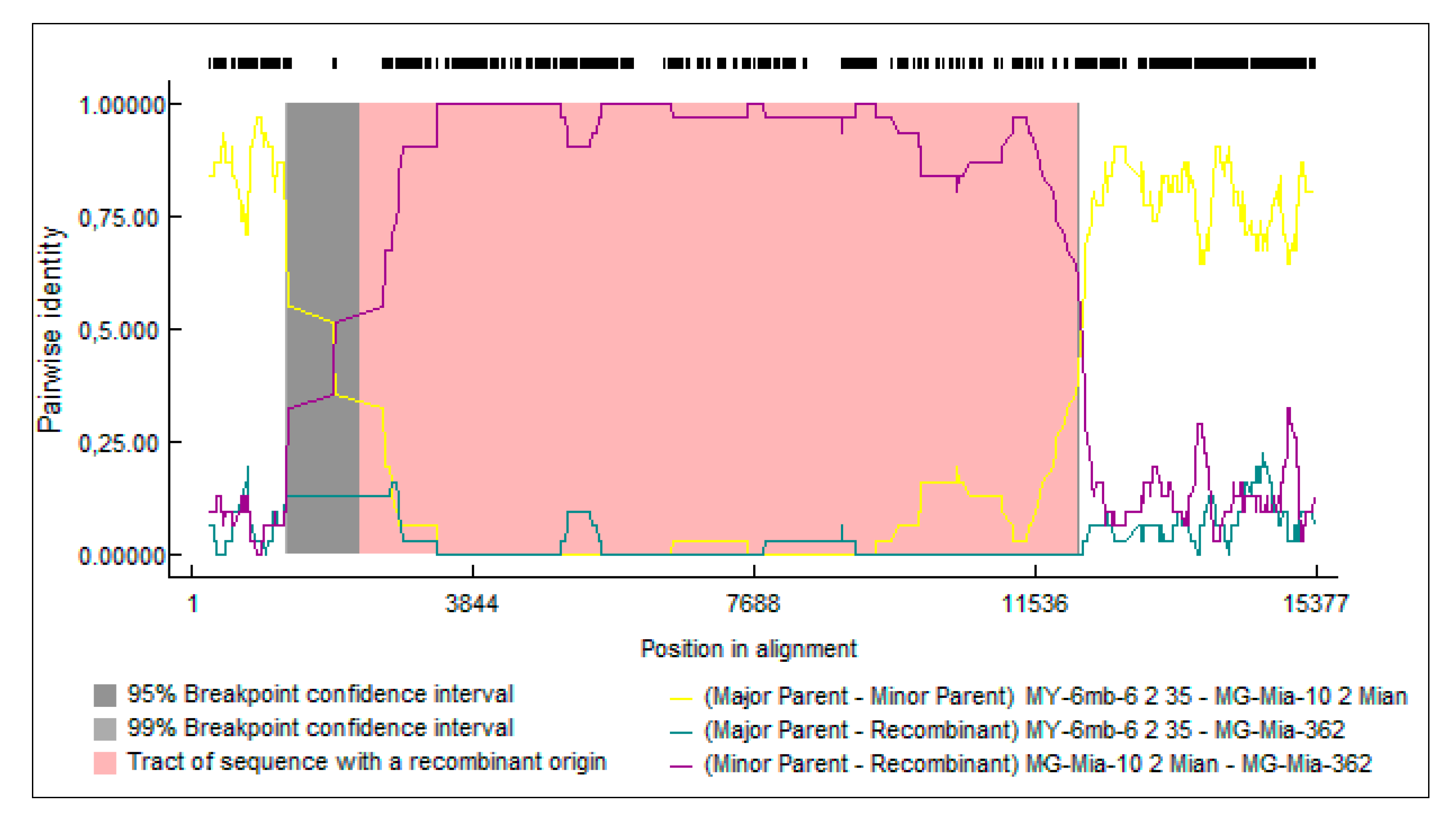
3.5. Recombination Analysis
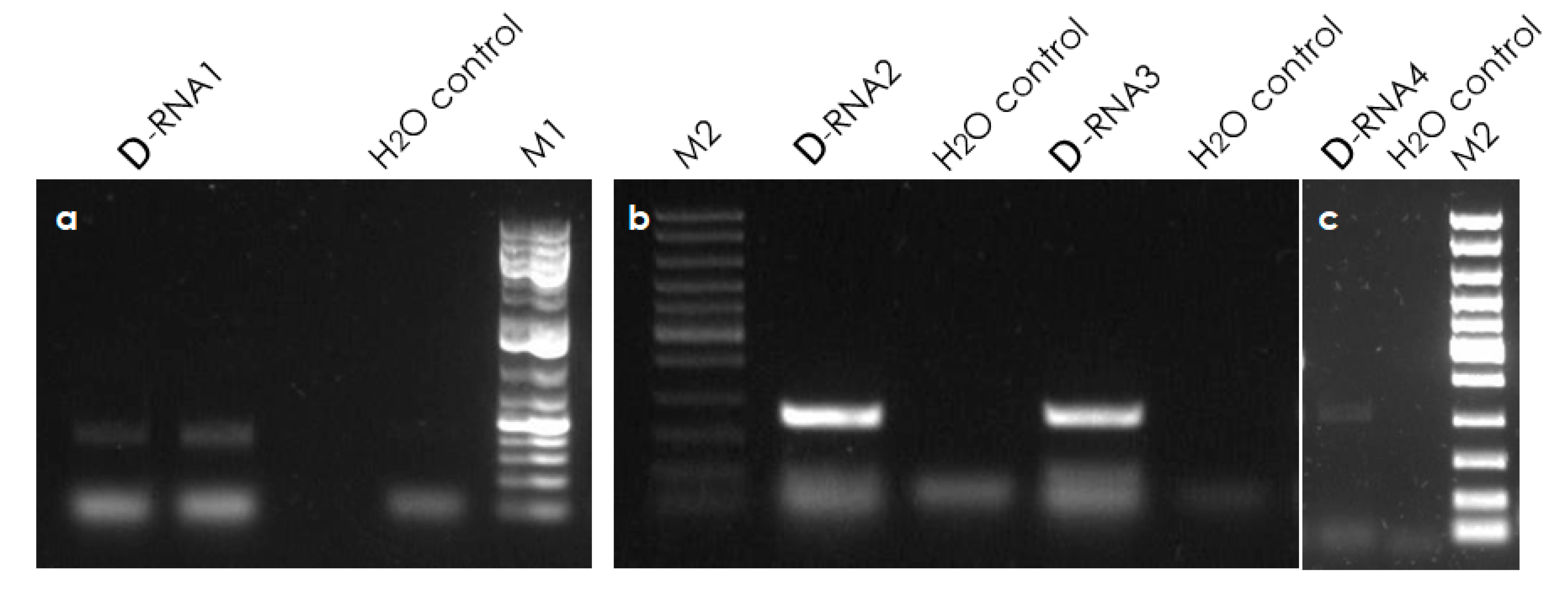
3.6. Validation of Defective RNAs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Food and Agriculture Organization of the United Nations. FAOSTAT Statistical Database; FAO: Rome, Italy, 2017. [Google Scholar]
- Burns, A.; Gleadow, R.; Cliff, J.; Zacarias, A.; Cavagnaro, T. Cassava: The Drought, War and Famine Crop in a Changing World. Sustainability 2010, 2, 3572–3607. [Google Scholar] [CrossRef] [Green Version]
- Lobell, D.B.; Burke, M.B.; Tebaldi, C.; Mastrandrea, M.D.; Falcon, W.P.; Naylor, R.L. Prioritizing Climate Change Adaptation Needs for Food Security in 2030. Science 2008, 319, 607–610. [Google Scholar] [CrossRef] [PubMed]
- Jarvis, A.; Ramirez-Villegas, J.; Campo, B.V.H.; Navarro-Racines, C. Is Cassava the Answer to African Climate Change Adaptation? Trop. Plant. Biol. 2012, 5, 9–29. [Google Scholar] [CrossRef]
- Fermont, A.; Van Asten, P.; Tittonell, P.; Van Wijk, M.; Giller, K. Closing the cassava yield gap: An analysis from smallholder farms in East Africa. Field Crop. Res. 2009, 112, 24–36. [Google Scholar] [CrossRef]
- Rey, C.; Vanderschuren, H.V. Cassava Mosaic and Brown Streak Diseases: Current Perspectives and Beyond. Annu. Rev. Virol. 2017, 4, 429–452. [Google Scholar] [CrossRef] [PubMed]
- Owor, B.; Legg, J.P.; Okao-Okuja, G.; Obonyo, R.; Ogenga-Latigo, M.W. The effect of cassava mosaic geminiviruses on symptom severity, growth and root yield of a cassava mosaic virus disease-susceptible cultivar in Uganda. Ann. Appl. Biol. 2004, 145, 331–337. [Google Scholar] [CrossRef]
- Dolja, V.V.; Karasev, A.V.; Koonin, E.V. Molecular biology and evolution of Closterovirus: Sophisticated Build-up of Large RNA Genomes. Annu. Rev. Phytopathol. 1994, 32, 261–285. [Google Scholar] [CrossRef]
- Martelli, G.P.; Agranovsky, A.A.; Bar-joseph, M. Family Closteroviridae. In Ninth Report of the International Committee on Taxonomy of Viruses; King, A.M.Q., Adams, M.J., Carstens, E.B., Lefkowitz, E.J., Eds.; Elsevier: New York, NY, USA, 2012; pp. 987–1001. [Google Scholar]
- Karasev, A.V. Genetic diversity and evolution of Closteroviruses. Annu. Rev. Phytopathol. 2000, 38, 293–324. [Google Scholar] [CrossRef]
- Blouin, A.G.; Pearson, M.N.; Chavan, R.R.; Woo, E.N.Y.; Lebas, B.S.M.; Veerakone, S.; Ratti, C.; Biccheri, R.; MacDiarmid, R.M.; Cohen, D. Viruses of Kiwifruit (Actinidia species). J. Plant. Pathol. 2013, 95, 221–235. [Google Scholar]
- Dey, K.K.; Sugikawa, J.; Kerr, C.; Melzer, M.J. Air potato (Dioscorea bulbifera) plants displaying virus-like symptoms are co-infected with a novel potyvirus and a novel ampelovirus. Virus Genes 2018, 55, 117–121. [Google Scholar] [CrossRef]
- Verdin, E.; Wipf-Scheibel, C.; Gognalons, P.; Aller, F.; Jacquemond, M.; Tepfer, M. Sequencing viral siRNAs to identify previously undescribed viruses and viroids in a panel of ornamental plant samples structured as a matrix of pools. Virus Res. 2017, 241, 19–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuchs, M.; Bar-Joseph, M.; Candresse, T.; Maree, H.J.; Martelli, G.P.; Melzer, M.J.; Menzel, W.; Minafra, A.; Sabanadzovic, S. ICTV Report Consortium ICTV Virus Taxonomy Profile: Closteroviridae. J. Gen. Virol. 2020, 101, 364–365. [Google Scholar] [CrossRef] [PubMed]
- Candresse, T.; Fuchs, M. Closteroviridae. eLS 2020, 1–10. [Google Scholar] [CrossRef]
- Fuchs, M.; Bar-Joseph, M.; Candresse, T. ICTV Virus Taxonomy Profile: Closteroviridae. J. Gen. Virol. 2020, 2. [Google Scholar] [CrossRef]
- Marais, A.; Faure, C.; Bergey, B.; Candresse, T. Viral Double-Stranded RNAs (dsRNAs) from Plants: Alternative Nucleic Acid Substrates for High-Throughput Sequencing. In Methods in Molecular Biology; Springer Science and Business Media LLC: Clifton, NJ, USA, 2018; Volume 1746, pp. 45–53. [Google Scholar]
- Palanga, E.; Filloux, D.; Martin, D.P.; Fernandez, E.; Gargani, D.; Ferdinand, R.; Zabré, J.; Bouda, Z.; Neya, J.B.; Sawadogo, M.; et al. Metagenomic-Based Screening and Molecular Characterization of Cowpea-Infecting Viruses in Burkina Faso. PLoS ONE 2016, 11, e0165188. [Google Scholar] [CrossRef] [Green Version]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef] [Green Version]
- Scussel, S.; Candresse, T.; Marais, A.; Claverie, S.; Hoareau, M.; Azali, H.A.; Verdin, E.; Tepfer, M.; Filloux, D.; Fernandez, E.; et al. High-throughput sequencing of complete genomes of ipomoviruses associated with an epidemic of cassava brown streak disease in the Comoros Archipelago. Arch. Virol. 2019, 164, 2193–2196. [Google Scholar] [CrossRef]
- Koonin, E.V.; Dolja, V.V.; Morris, T.J. Evolution and Taxonomy of Positive-Strand RNA Viruses: Implications of Comparative Analysis of Amino Acid Sequences. Crit. Rev. Biochem. Mol. Biol. 1993, 28, 375–430. [Google Scholar] [CrossRef]
- Blouin, A.G.; Biccheri, R.; Khalifa, M.E.; Pearson, M.N.; Pollini, C.P.; Hamiaux, C.; Cohen, D.; Ratti, C. Characterization of Actinidia virus 1, a new member of the family Closteroviridae encoding a thaumatin-like protein. Arch. Virol. 2017, 163, 229–234. [Google Scholar] [CrossRef]
- Peremyslov, V.V.; Pan, Y.-W.; Dolja, V.V. Movement Protein of a Closterovirus Is a Type III Integral Transmembrane Protein Localized to the Endoplasmic Reticulum. J. Virol. 2004, 78, 3704–3709. [Google Scholar] [CrossRef] [Green Version]
- Koloniuk, I.; Thekke-Veetil, T.; Reynard, J.-S.; Pleško, I.M.; Přibylová, J.; Brodard, J.; Kellenberger, I.; Sarkisova, T.; Špak, J.; Lamovšek, J.; et al. Molecular Characterization of Divergent Closterovirus Isolates Infecting Ribes Species. Viruses 2018, 10, 369. [Google Scholar] [CrossRef] [Green Version]
- Adiputra, J.; Jarugula, S.; Naidu, R.A. Intra-species recombination among strains of the ampelovirus Grapevine leafroll-associated virus 4. Virol. J. 2019, 16, 1–13. [Google Scholar] [CrossRef]
- Dey, K.K.; Green, J.C.; Melzer, M.; Borth, W.; Hu, J.S. Mealybug Wilt of Pineapple and Associated Viruses. Horticulturae 2018, 4, 52. [Google Scholar] [CrossRef] [Green Version]
- Yu, N.; Luo, Z.; Fan, H.; Zhang, Z.; Li, X.; Wang, J.; Liu, Z.; He, F. Complete genomic sequence of a Pineapple mealybug wilt-associated virus-1 from Hainan Island, China. Eur. J. Plant. Pathol. 2015, 141, 611–615. [Google Scholar] [CrossRef]
- Melzer, M.J.; Sether, D.M.; Karasev, A.V.; Borth, W.; Hu, J.S. Complete nucleotide sequence and genome organization of pineapple mealybug wilt-associated virus-1. Arch. Virol. 2008, 153, 707–714. [Google Scholar] [CrossRef] [PubMed]
- Green, J.C.; Rwahnih, M.A.; Olmedo-Velarde, A.; Melzer, M.J.; Hamim, I.; Borth, W.B.; Brower, T.M.; Wall, M.; Hu, J.S. Further genomic characterization of pineapple mealybug wilt-associated viruses using high-throughput sequencing. Trop. Plant. Pathol. 2020, 45, 64–72. [Google Scholar] [CrossRef]
- Sether, D.M.; Melzer, M.J.; Borth, W.B.; Hu, J.S. Genome organization and phylogenetic relationship of Pineapple mealybug wilt associated virus-3 with family Closteroviridae members. Virus Genes 2009, 38, 414–420. [Google Scholar] [CrossRef] [PubMed]
- Kwon, S.-J.; Jin, M.; Cho, I.-S.; Yoon, J.-Y.; Choi, G.-S. Identification of rehmannia virus 1, a novel putative member of the genus Closterovirus, from Rehmannia glutinosa. Arch. Virol. 2018, 163, 3383–3388. [Google Scholar] [CrossRef]
- Martín, S.; Sambade, A.; Rubio, L.; Vives, M.C.; Moya, P.; Guerri, J.; Elena, S.F.; Moreno, P. Contribution of recombination and selection to molecular evolution of Citrus tristeza virus. J. Gen. Virol. 2009, 90, 1527–1538. [Google Scholar] [CrossRef] [Green Version]
- Rubio, L.; Guerri, J.; Moreno, P. Genetic variability and evolutionary dynamics of viruses of the family Closteroviridae. Front. Microbiol. 2013, 4, 151. [Google Scholar] [CrossRef] [Green Version]
- Li, X.H.; Heaton, L.A.; Morris, T.J.; Simon, A.E. Turnip crinkle virus defective interfering RNAs intensify viral symptoms and are generated de novo. Proc. Natl. Acad. Sci. USA 1989, 86, 9173–9177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bar-Joseph, M.; Mawassi, M. The defective RNAs of Closteroviridae. Front. Microbiol. 2013, 4, 132. [Google Scholar] [CrossRef] [Green Version]
- White, K.A.; Morris, T.J. RNA determinants of junction site selection in RNA virus recombinants and defective interfering RNAs. RNA 1995, 1, 1029–1040. [Google Scholar]
- Rubio, L.; Yeh, H.-H.; Tian, T.; Falk, B.W. A Heterogeneous Population of Defective RNAs Is Associated with Lettuce infectious yellows virus. Virology 2000, 271, 205–212. [Google Scholar] [CrossRef] [Green Version]
- Eliasco, E.; Livieratos, I.C.; Müller, G.; Guzman, M.; Salazar, L.F.; Coutts, R.H.A. Sequences of defective RNAs associated with potato yellow vein virus. Arch. Virol. 2005, 151, 201–204. [Google Scholar] [CrossRef]
- Menzel, W.; Goetz, R.; Lesemann, D.E.; Vetten, H.J. Molecular characterization of a closterovirus from carrot and its identification as a German isolate of Carrot yellow leaf virus. Arch. Virol. 2009, 154, 1343–1347. [Google Scholar] [CrossRef] [PubMed]
- Mongkolsiriwattana, C.; Chen, A.Y.; Ng, J.C. Replication of Lettuce chlorosis virus (LCV), a crinivirus in the family Closteroviridae, is accompanied by the production of LCV RNA 1-derived novel RNAs. Virology 2011, 420, 89–97. [Google Scholar] [CrossRef] [Green Version]
- Massart, S.; Candresse, T.; Gil, J.; Lacomme, C.; Predajna, L.; Ravnikar, M.; Reynard, J.-S.; Rumbou, A.; Saldarelli, P.; Škorić, D.; et al. A Framework for the Evaluation of Biosecurity, Commercial, Regulatory, and Scientific Impacts of Plant Viruses and Viroids Identified by NGS Technologies. Front. Microbiol. 2017, 8, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cassava Landraces | Country of Origin | Collection Date DD/MM/YY | Symptoms | Sequencing Strategy and Target | Isolates Detected | Other Virus Detected |
|---|---|---|---|---|---|---|
| Menatana | Madagascar | 18/10/2017 | Asymptomatic in greenhouse | VANA | MG-Men-241, MG-Men-9 | - |
| Miandrazaka | Madagascar | 18/10/2017 | Asymptomatic in greenhouse | VANA | MG-Mia-10, MG-Mia-362, MG-Mia-403, MG-Mian 2-2 | - |
| Long Java | Reunion | 25/02/2015 | Asymptomatic in field | dsRNAs | RE-Ljv | - |
| Reunion | Mayotte | 26/03/2015 | Symptoms of CMD and CBSD | dsRNAs | MY-Ren-5, MY-Ren-8 | EACMV+UCBSV |
| 6 Mois Blanc | Mayotte | 26/03/2015 | Symptoms of CMD | dsRNAs | MY−6mb-4, MY−6mb-6 | EACMV |
| Nambiyombiyo | DR Congo | 13/01/2016 | Asymptomatic in greenhouse | Total RNA | CG-Nmb | CBSV |
| Kahunde | DR Congo | 13/01/2016 | Asymptomatic in greenhouse | Total RNA | CG-Kah | - |
| PRIMER | TARGET ORF | SEQUENCES (5′==> 3′) | Tm |
|---|---|---|---|
| 12.495 F cp | CP ORF5 | AATTTGGGAGGAGTGCGACC | 60.3 |
| 12.715 R cp | CP ORF5 | AGACCGACTTGTGCTACTCTTG | 60.0 |
| 11.735 F hsp90 | HSP90 ORF4 | GTCCCGGCCTACATGCAATT | 60.8 |
| 11.959 R hsp90 | HSP90 ORF4 | ACCGCCAGTCAACTCTCGTA | 60.9 |
| 9.021 F hsp70 | HSP70 ORF4 | TATGGTTGTTGCTCGCGACT | 60.0 |
| 9.292 R hsp70 | HSP70 ORF4 | CTGACAAACCAGCAGCAGTTG | 60.3 |
| 7.244 F rdrp | RdRp ORF1B | GGACAACCTCCGAAACCGTAT | 60.1 |
| 7.544 R rdrp | RdRp ORF1B | CTTTCGCTGCCATTGGTGTC | 60.1 |
| Primers Name | Sequences (5’==>3’) | Expected Amplicon Size |
|---|---|---|
| MEaV-1-RE-Ljv-D-RNA1-F | ACATCTAAATGCTAACGAACGAAGAG | 380 bp |
| MEaV-1-RE-Ljv-D-RNA1-R | CAACGCCAGAATCTTCGTACA | |
| MEaV-1-RE- Ljv-D-RNA2-F | AGGCTTTCGACAGTGAAGAAGTG | 330 bp |
| MEaV-1-RE- Ljv-D-RNA2-R | CGTAGCCATACTGAAGGATAGCA | |
| MEaV-2-RE- Ljv-D-RNA3-F | TGGAAGCCGCTGGTAAACTACA | 400 bp |
| MEaV-2-RE-Ljv-D-RNA3-R | CAAGCACGTTCAATATTAGGAATAGTAC | |
| MEaV-2-MG-Mena-D-RNA4-F | AGACATATGAAAGAGTTGCATTGGTG | 500 bp |
| MEaV-2-MG-Mena-D-RNA4-R | ACCTACAAATAATTTCGCTCGTCTG |
| Cassava Landraces | Isolate Name 1 | Accession Number (Genbank) | Virus Name | Contig Length [Nt] | Number of Reads Mapped | Percent Of Total Reads | Average Genome Coverage | Internal Undetermined Nucleotides | Internal Gaps |
|---|---|---|---|---|---|---|---|---|---|
| Menatana | MG-Men-241 | MT773584 | MEaV-1 | 13,528 | 38,475 | 12.1 | 506.8X | 0 | 0 |
| MG-Men-9 | MT773591 | MEaV-2 | 13,752 | 10,945 | 4.10 | 153.6X | 0 | 0 | |
| Miandrazaka | MG-Mia-10 | MT773592 | MEaV-2 | 13,770 | 26,987 | 4.8 | 368X | 377 | 1 |
| MG-Mia-362 | MT773594 | MEaV-2 | 12,007 | 19,421 | 8.24 | 322X | 0 | 0 | |
| MG-Mia-403 | MT773590 | MEaV-1 | 10,518 | 26,917 | 11.4 | 480.9X | 0 | 0 | |
| MG-Mian 2-2 | MT773596 | MEaV-2 | 10,417 | 7676 | 1.4 | 136.4X | 843 | 1 | |
| Long java | RE-Ljv | MT773586 | MEaV-1 | 13,640 | 21,294 | 18.5 | 312.5X | 0 | 0 |
| Reunion | MY-Ren-5 | MT773589 | MEaV-1 | 13,172 | 1976 | 3.4 | 29.6X | 72 | 1 |
| MY-Ren-8 | MT773595 | MEaV-2 | 13,762 | 377 | 0.7 | 5.4X | 2889 | 14 | |
| 6 Mois blanc | MY-6mb-4 | MT773585 | MEaV-1 | 13,615 | 4992 | 12.7 | 73.3X | 288 | 4 |
| MY-6mb-6 | MT773593 | MEaV-2 | 13,764 | 2631 | 6.7 | 39.2X | 990 | 9 | |
| Nambiyombiyo | CG-Nmb | MT773587 | MEaV-1 | 13,612 | 7502 | 0.12 | 42X | 0 | 0 |
| Kahunde | CG-Kah | MT773588 | MEaV-1 | 13,616 | 15,351 | 0.23 | 75X | 11 | 1 |
| Isolates | Genome Length (Nt) | Length of Open Reading Frames (Orfs) (Nt) and Molecular Mass of Encoded Proteins (Kda) | ||||||
|---|---|---|---|---|---|---|---|---|
| 1a | 1b | P5-P7 | HSP70h | HSP90h | CP | ORF6 | ||
| CG-Nmb (MEaV-1) | 13,616 nt | 6789 nt | 1503 nt | 204 nt | 1707 nt | 1686 nt | 774 nt | 717 nt |
| 254.6 kDa | 56.6 kDa | 7.6 kDa | 62.2 kDa | 63.4 kDa | 27.7 kDa | 27.1 kDa | ||
| MG-Men-9 (MEaV-2) | 13,752 nt | 6975 nt | 1593 nt | 147 nt | 1608 nt | 1698 nt | 816 nt | 639 nt |
| 258.8 kDa | 60.4 kDa | 5.3 kDa | 58.4 kDa | 64.3 kDa | 29.3 kDa | 24 kDa | ||
| Genus | Representative Members | Proteins | ||
|---|---|---|---|---|
| RdRp | HSP70H | CP | ||
| Ampelovirus | MEaV-1 (isolate CG-Nmb) | 66.5% | 69.9% | 80.1% |
| Ampelovirus | Grapevine leafroll-associated virus 4 (NC_016416) | 41.7% | 51.6% | 47.6% |
| Closterovirus | Citrus tristeza virus (NC_001661) | 27.2% | 25% | 12.5% |
| Crinivirus | Potato yellow vein virus (NC_006062 and NC_006063) | 25.7% | 26.5% | 17.2% |
| Velarivirus | Grapevine leafroll-associated virus 7 (NC_016436) | 27.5% | 27.7% | 13.6% |
| Within MEaV-2 with Mian 2-2 | Within MEaV-2 without Mian2-2 | Within MEaV-1 with Divergents | Within MEaV-1 without Divergents | Within MEaV-1 Divergents | Between MEaV-1 and MEaV-2 | ||
|---|---|---|---|---|---|---|---|
| RdRp | aa divergences | 7.3 +/− 0.7% | 1.1 +/− 0.3% | 7.1 +/− 0.7% | 0.8 +/− 0.4% | 0.8% | 32.3 +/− 1.7% |
| HSP70h | aa divergences | 2.8 +/− 0.4% | 1.0 +/− 0.3% | 6.0 +/− 0.6% | 2.3 +/− 0.4% | 0.7% | 30.8 +/− 2.0% |
| CP | aa divergences | na | 3.1 +/− 0.8% | 7.4 +/− 1.0% | 3.2 +/− 0.6% | na | 19 +/− 2.2% |
| Genome | nt divergences | 12.3 +/− 0.1% | 3.8 +/− 0.1% | 17.1 +/− 0.2% | 7.9 +/− 0.2% | 2.3% | 41.4 +/− 0.3% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kwibuka, Y.; Bisimwa, E.; Blouin, A.G.; Bragard, C.; Candresse, T.; Faure, C.; Filloux, D.; Lett, J.-M.; Maclot, F.; Marais, A.; et al. Novel Ampeloviruses Infecting Cassava in Central Africa and the South-West Indian Ocean Islands. Viruses 2021, 13, 1030. https://doi.org/10.3390/v13061030
Kwibuka Y, Bisimwa E, Blouin AG, Bragard C, Candresse T, Faure C, Filloux D, Lett J-M, Maclot F, Marais A, et al. Novel Ampeloviruses Infecting Cassava in Central Africa and the South-West Indian Ocean Islands. Viruses. 2021; 13(6):1030. https://doi.org/10.3390/v13061030
Chicago/Turabian StyleKwibuka, Yves, Espoir Bisimwa, Arnaud G. Blouin, Claude Bragard, Thierry Candresse, Chantal Faure, Denis Filloux, Jean-Michel Lett, François Maclot, Armelle Marais, and et al. 2021. "Novel Ampeloviruses Infecting Cassava in Central Africa and the South-West Indian Ocean Islands" Viruses 13, no. 6: 1030. https://doi.org/10.3390/v13061030
APA StyleKwibuka, Y., Bisimwa, E., Blouin, A. G., Bragard, C., Candresse, T., Faure, C., Filloux, D., Lett, J.-M., Maclot, F., Marais, A., Ravelomanantsoa, S., Shakir, S., Vanderschuren, H., & Massart, S. (2021). Novel Ampeloviruses Infecting Cassava in Central Africa and the South-West Indian Ocean Islands. Viruses, 13(6), 1030. https://doi.org/10.3390/v13061030