
Development of Group B Coxsackievirus as an Oncolytic Virus: Opportunities and Challenges



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Viral Structure and Life-Cycle
3. Features That Make CVBs Attractive Oncolytic Viruses
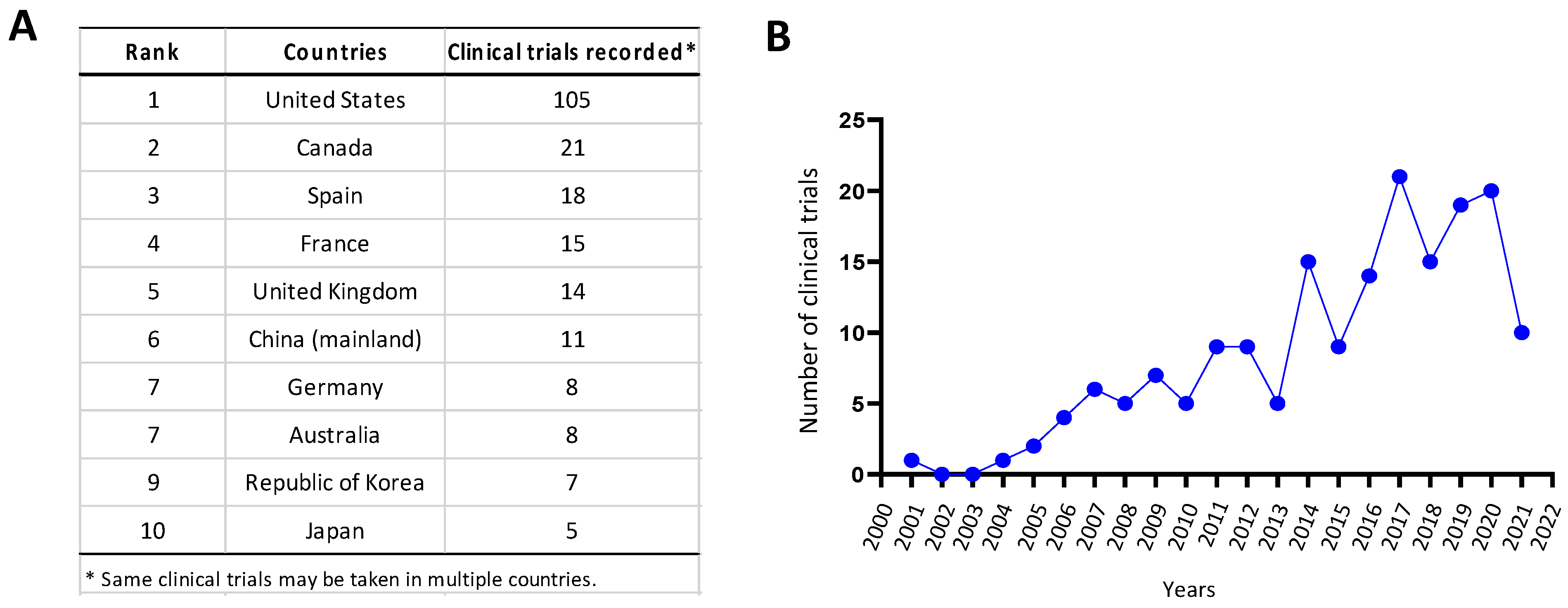
4. Opportunities for Oncolytic CVBs in Cancer Therapy
4.1. Lung Cancer
4.2. Colorectal Cancer
4.3. Breast Cancer
4.4. Other Types of Cancer
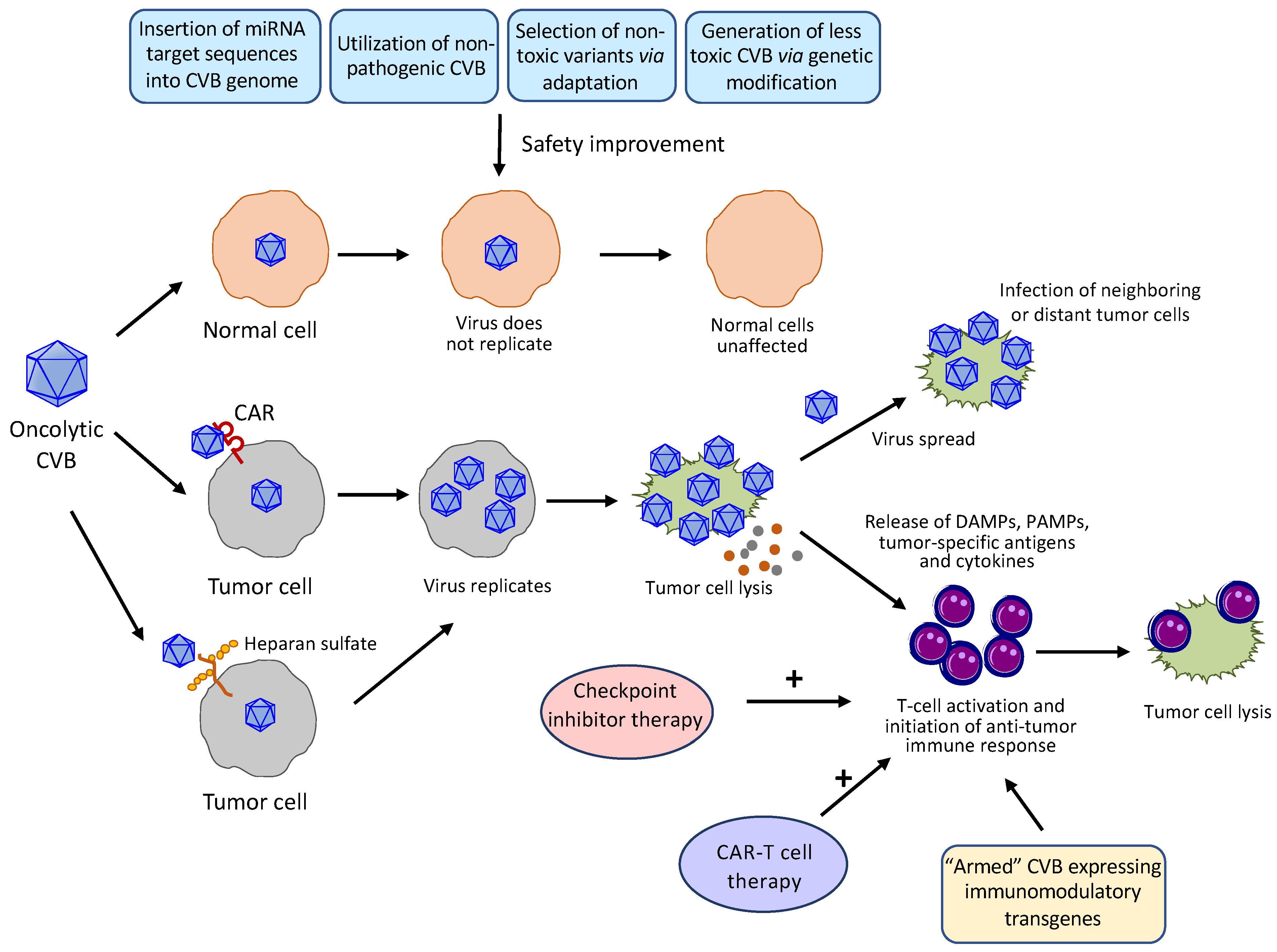
5. Challenges and Possible Solutions for the Clinical Use of Oncolytic CVBs
5.1. Improvement of Safety and Tumor Specificity
5.1.1. miRNA-Based Strategy
5.1.2. Utilization of Non-Pathogenic CVBs
5.1.3. Genetic Engineering
5.1.4. Selection of Non-Toxic Variants Via Adaptation
5.2. Expansion of Tumor Infectivity Via Adaptation
5.3. Enhancement of Oncolytic Potency
5.4. Achievement of Efficient Delivery
6. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bell, J.; McFadden, G. Viruses for tumor therapy. Cell Host Microbe 2014, 15, 260–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawler, S.E.; Speranza, M.C.; Cho, C.F.; Chiocca, E.A. Oncolytic viruses in cancer treatment: A review. JAMA Oncol. 2017, 3, 841–849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poh, A. First oncolytic viral therapy for melanoma. Cancer Discov. 2016, 6, 6. [Google Scholar] [CrossRef] [Green Version]
- Sheridan, C. First oncolytic virus edges towards approval in surprise vote. Nat. Biotechnol. 2015, 33, 569–570. [Google Scholar] [CrossRef] [PubMed]
- Schirrmacher, V.; van Gool, S.; Stuecker, W. Breaking therapy resistance: An update on oncolytic newcastle disease virus for improvements of cancer therapy. Biomedicines 2019, 7, 66. [Google Scholar] [CrossRef] [Green Version]
- Aref, S.; Bailey, K.; Fielding, A. Measles to the rescue: A review of oncolytic measles virus. Viruses 2016, 8, 294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Felt, S.A.; Grdzelishvili, V.Z. Recent advances in vesicular stomatitis virus-based oncolytic virotherapy: A 5-year update. J. Gen. Virol. 2017, 98, 2895–2911. [Google Scholar] [CrossRef] [PubMed]
- Burke, M.J. Oncolytic Seneca Valley Virus: Past perspectives and future directions. Oncolytic Virother. 2016, 5, 81–89. [Google Scholar] [CrossRef] [Green Version]
- Muller, L.; Berkeley, R.; Barr, T.; Ilett, E.; Errington-Mais, F. Past, present and future of oncolytic reovirus. Cancers 2020, 12, 3219. [Google Scholar] [CrossRef] [PubMed]
- Echchgadda, I.; Kota, S.; DeLa Cruz, I.; Sabbah, A.; Chang, T.; Harnack, R.; Mgbemena, V.; Chatterjee, B.; Bose, S. Anticancer oncolytic activity of respiratory syncytial virus. Cancer Gene Ther. 2009, 16, 923–935. [Google Scholar] [CrossRef] [Green Version]
- Brown, M.C.; Dobrikova, E.Y.; Dobrikov, M.I.; Walton, R.W.; Gemberling, S.L.; Nair, S.K.; Desjardins, A.; Sampson, J.H.; Friedman, H.S.; Friedman, A.H.; et al. Oncolytic polio virotherapy of cancer. Cancer 2014, 120, 3277–3286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradley, S.; Jakes, A.D.; Harrington, K.; Pandha, H.; Melcher, A.; Errington-Mais, F. Applications of coxsackievirus A21 in oncology. Oncolytic Virother. 2014, 3, 47–55. [Google Scholar] [CrossRef] [Green Version]
- Desjardins, A.; Gromeier, M.; Herndon, J.E., 2nd; Beaubier, N.; Bolognesi, D.P.; Friedman, A.H.; Friedman, H.S.; McSherry, F.; Muscat, A.M.; Nair, S.; et al. Recurrent glioblastoma treated with recombinant poliovirus. N. Engl. J. Med. 2018, 379, 150–161. [Google Scholar] [CrossRef]
- Holl, E.K.; Brown, M.C.; Boczkowski, D.; McNamara, M.A.; George, D.J.; Bigner, D.D.; Gromeier, M.; Nair, S.K. Recombinant oncolytic poliovirus, PVSRIPO, has potent cytotoxic and innate inflammatory effects, mediating therapy in human breast and prostate cancer xenograft models. Oncotarget 2016, 7, 79828–79841. [Google Scholar] [CrossRef] [Green Version]
- Annels, N.E.; Arif, M.; Simpson, G.R.; Denyer, M.; Moller-Levet, C.; Mansfield, D.; Butler, R.; Shafren, D.; Au, G.; Knowles, M.; et al. Oncolytic immunotherapy for bladder cancer using coxsackie A21 virus. Mol. Ther. Oncolytics 2018, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Annels, N.E.; Mansfield, D.; Arif, M.; Ballesteros-Merino, C.; Simpson, G.R.; Denyer, M.; Sandhu, S.S.; Melcher, A.A.; Harrington, K.J.; Davies, B.; et al. Phase I trial of an ICAM-1-targeted immunotherapeutic-coxsackievirus A21 (CVA21) as an oncolytic agent against non muscle-invasive bladder cancer. Clin. Cancer Res. 2019, 25, 5818–5831. [Google Scholar] [CrossRef]
- Curti, B.; Richards, J.; Hallmeyer, S.; Faries, M.; Andtbacka, R.; Daniels, G.; Grose, M.; Shafren, D.R. The MITCI (Phase 1b) study: A novel immunotherapy combination of intralesional Coxsackievirus A21 and systemic ipilimumab in advanced melanoma patients with or without previous immune checkpoint therapy treatment. Cancer Res. 2017, 77, CT114. [Google Scholar] [CrossRef]
- Elsedawy, N.B.; Nace, R.A.; Russell, S.J.; Schulze, A.J. Oncolytic activity of targeted picornaviruses formulated as synthetic infectious RNA. Mol. Ther. Oncolytics 2020, 17, 484–495. [Google Scholar] [CrossRef]
- Silk, A.W.; Kaufman, H.; Gabrail, N.; Mehnert, J.; Bryan, J.; Norrell, J.; Medina, D.; Bommareddy, P.; Shafren, D.; Grose, M.; et al. Phase 1b study of intratumoral Coxsackievirus A21 (CVA21) and systemic pembrolizumab in advanced melanoma patients: Interim results of the CAPRA clinical trial. Cancer Res. 2017, 77, CT026. [Google Scholar] [CrossRef]
- Simmonds, P.; Gorbalenya, A.E.; Harvala, H.; Hovi, T.; Knowles, N.J.; Lindberg, A.M.; Oberste, M.S.; Palmenberg, A.C.; Reuter, G.; Skern, T.; et al. Recommendations for the nomenclature of enteroviruses and rhinoviruses. Arch. Virol. 2020, 165, 793–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tariq, N.; Kyriakopoulos, C. Group B Coxsackie Virus; StatPearls: Treasure Island, FL, USA, 2021. [Google Scholar]
- Agrez, M.V.; Shafren, D.R.; Gu, X.; Cox, K.; Sheppard, D.; Barry, R.D. Integrin alpha v beta 6 enhances coxsackievirus B1 lytic infection of human colon cancer cells. Virology 1997, 239, 71–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, L.; Liu, Z. Novel recombinant coxsackievirus B3 with genetically inserted basic peptide elicits robust antitumor activity against lung cancer. Cancer Med. 2020, 9, 5210–5220. [Google Scholar] [CrossRef]
- Deng, H.; Liu, H.; de Silva, T.; Xue, Y.; Mohamud, Y.; Ng, C.S.; Qu, J.; Zhang, J.; Jia, W.W.G.; Lockwood, W.W.; et al. Coxsackievirus type B3 is a potent oncolytic virus against KRAS-mutant lung adenocarcinoma. Mol. Ther. Oncolytics 2019, 14, 266–278. [Google Scholar] [CrossRef] [Green Version]
- Hazini, A.; Dieringer, B.; Pryshliak, M.; Knoch, K.P.; Heimann, L.; Tolksdorf, B.; Pappritz, K.; El-Shafeey, M.; Solimena, M.; Beling, A.; et al. miR-375- and miR-1-regulated coxsackievirus B3 has no pancreas and heart toxicity but strong antitumor efficiency in colorectal carcinomas. Hum. Gene Ther. 2021, 32, 216–230. [Google Scholar] [CrossRef]
- Jia, Y.; Miyamoto, S.; Soda, Y.; Takishima, Y.; Sagara, M.; Liao, J.; Hirose, L.; Hijikata, Y.; Miura, Y.; Hara, K.; et al. Extremely low organ toxicity and strong antitumor activity of miR-34-regulated oncolytic Coxsackievirus B3. Mol. Ther. Oncolytics 2019, 12, 246–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.; Wang, W.; Wan, J.; Yang, Y.; Fu, W.; Pan, D.; Cai, L.; Cheng, T.; Huang, X.; Wang, Y. Oncolytic activity of a coxsackievirus B3 strain in human endometrial cancer cell lines. Virol. J. 2018, 15, 65. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Xue, Y.C.; Deng, H.; Mohamud, Y.; Ng, C.S.; Chu, A.; Lim, C.J.; Lockwood, W.W.; Jia, W.W.G.; Luo, H. MicroRNA modification of coxsackievirus B3 decreases its toxicity, while retaining oncolytic potency against lung cancer. Mol. Ther. Oncolytics 2020, 16, 207–218. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, S.; Inoue, H.; Nakamura, T.; Yamada, M.; Sakamoto, C.; Urata, Y.; Okazaki, T.; Marumoto, T.; Takahashi, A.; Takayama, K.; et al. Coxsackievirus B3 is an oncolytic virus with immunostimulatory properties that is active against lung adenocarcinoma. Cancer Res. 2012, 72, 2609–2621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sagara, M.; Miyamoto, S.; Itoh, S.; Soda, Y.; Tani, K. Development of New Oncolytic Virotherapy Targeting Breast Cancer Using Coxsackievirus B3. Anticancer Res. 2021, 41, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Fung, G.; Luo, H.; Qiu, Y.; Yang, D.; McManus, B. Myocarditis. Circ. Res. 2016, 118, 496–514. [Google Scholar] [CrossRef]
- Huang, H.I.; Shih, S.R. Neurotropic enterovirus infections in the central nervous system. Viruses 2015, 7, 6051–6066. [Google Scholar] [CrossRef] [Green Version]
- Huber, S.; Ramsingh, A.I. Coxsackievirus-induced pancreatitis. Viral Immunol. 2004, 17, 358–369. [Google Scholar] [CrossRef] [PubMed]
- Sean, P.; Semler, B.L. Coxsackievirus B RNA replication: Lessons from poliovirus. Curr. Top. Microbiol. Immunol. 2008, 323, 89–121. [Google Scholar]
- Sin, J.; Mangale, V.; Thienphrapa, W.; Gottlieb, R.A.; Feuer, R. Recent progress in understanding coxsackievirus replication, dissemination, and pathogenesis. Virology 2015, 484, 288–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altan-Bonnet, N. Lipid tales of viral replication and transmission. Trends Cell Biol. 2017, 27, 201–213. [Google Scholar] [CrossRef] [Green Version]
- Miller, S.; Krijnse-Locker, J. Modification of intracellular membrane structures for virus replication. Nat. Rev. Microbiol. 2008, 6, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Liu, Y.; Wimmer, E.; Paul, A.V. Complete protein linkage map between the P2 and P3 non-structural proteins of poliovirus. J. Gen. Virol. 2007, 88, 2259–2267. [Google Scholar] [CrossRef]
- Gruez, A.; Selisko, B.; Roberts, M.; Bricogne, G.; Bussetta, C.; Jabafi, I.; Coutard, B.; De Palma, A.M.; Neyts, J.; Canard, B. The crystal structure of coxsackievirus B3 RNA-dependent RNA polymerase in complex with its protein primer VPg confirms the existence of a second VPg binding site on Picornaviridae polymerases. J. Virol. 2008, 82, 9577–9590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pathak, H.B.; Oh, H.S.; Goodfellow, I.G.; Arnold, J.J.; Cameron, C.E. Picornavirus genome replication: Roles of precursor proteins and rate-limiting steps in oriI-dependent VPg uridylylation. J. Biol. Chem. 2008, 283, 30677–30688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossmann, M.G. Picornavirus structure overview. In Molecular Biology of Picornavirus; ASM Press: Washington, DC, USA, 2002; pp. 25–38. [Google Scholar]
- Bird, S.W.; Maynard, N.D.; Covert, M.W.; Kirkegaard, K. Nonlytic viral spread enhanced by autophagy components. Proc. Natl. Acad. Sci. USA 2014, 111, 13081–13086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.H.; Du, W.; Hagemeijer, M.C.; Takvorian, P.M.; Pau, C.; Cali, A.; Brantner, C.A.; Stempinski, E.S.; Connelly, P.S.; Ma, H.C.; et al. Phosphatidylserine vesicles enable efficient en bloc transmission of enteroviruses. Cell 2015, 160, 619–630. [Google Scholar] [CrossRef] [Green Version]
- Robinson, S.M.; Tsueng, G.; Sin, J.; Mangale, V.; Rahawi, S.; McIntyre, L.L.; Williams, W.; Kha, N.; Cruz, C.; Hancock, B.M.; et al. Coxsackievirus B exits the host cell in shed microvesicles displaying autophagosomal markers. PLoS Pathog. 2014, 10, e1004045. [Google Scholar] [CrossRef]
- Laitinen, O.H.; Svedin, E.; Kapell, S.; Nurminen, A.; Hytonen, V.P.; Flodstrom-Tullberg, M. Enteroviral proteases: Structure, host interactions and pathogenicity. Rev. Med. Virol. 2016, 26, 251–267. [Google Scholar] [CrossRef]
- Bergelson, J.M.; Cunningham, J.A.; Droguett, G.; Kurt-Jones, E.A.; Krithivas, A.; Hong, J.S.; Horwitz, M.S.; Crowell, R.L.; Finberg, R.W. Isolation of a common receptor for Coxsackie B viruses and adenoviruses 2 and 5. Science 1997, 275, 1320–1323. [Google Scholar] [CrossRef] [PubMed]
- Freimuth, P.; Philipson, L.; Carson, S.D. The coxsackievirus and adenovirus receptor. Curr. Top. Microbiol. Immunol. 2008, 323, 67–87. [Google Scholar] [CrossRef] [PubMed]
- Shafren, D.R.; Bates, R.C.; Agrez, M.V.; Herd, R.L.; Burns, G.F.; Barry, R.D. Coxsackieviruses B1, B3, and B5 use decay accelerating factor as a receptor for cell attachment. J. Virol. 1995, 69, 3873–3877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, C.J.; Shieh, J.T.; Pickles, R.J.; Okegawa, T.; Hsieh, J.T.; Bergelson, J.M. The coxsackievirus and adenovirus receptor is a transmembrane component of the tight junction. Proc. Natl. Acad. Sci. USA 2001, 98, 15191–15196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, L.; Zheng, Q.; Zhu, R.; Yin, Z.; Yu, H.; Lin, Y.; Wu, Y.; He, M.; Huang, Y.; Jiang, Y.; et al. Cryo-EM structures reveal the molecular basis of receptor-initiated coxsackievirus uncoating. Cell Host Microbe 2021, 29, 448–462.e445. [Google Scholar] [CrossRef] [PubMed]
- Muckelbauer, J.K.; Kremer, M.; Minor, I.; Tong, L.; Zlotnick, A.; Johnson, J.E.; Rossmann, M.G. Structure determination of coxsackievirus B3 to 3.5 A resolution. Acta Crystallogr. D Biol. Crystallogr. 1995, 51, 871–887. [Google Scholar] [CrossRef] [PubMed]
- Hacein-Bey-Abina, S.; Von Kalle, C.; Schmidt, M.; McCormack, M.P.; Wulffraat, N.; Leboulch, P.; Lim, A.; Osborne, C.S.; Pawliuk, R.; Morillon, E.; et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science 2003, 302, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Stephen, S.L.; Montini, E.; Sivanandam, V.G.; Al-Dhalimy, M.; Kestler, H.A.; Finegold, M.; Grompe, M.; Kochanek, S. Chromosomal integration of adenoviral vector DNA in vivo. J. Virol. 2010, 84, 9987–9994. [Google Scholar] [CrossRef] [Green Version]
- Athanasopoulos, T.; Munye, M.M.; Yanez-Munoz, R.J. Nonintegrating gene therapy vectors. Hematol. Oncol. Clin. N. Am. 2017, 31, 753–770. [Google Scholar] [CrossRef] [PubMed]
- Feuer, R.; Mena, I.; Pagarigan, R.; Slifka, M.K.; Whitton, J.L. Cell cycle status affects coxsackievirus replication, persistence, and reactivation in vitro. J. Virol. 2002, 76, 4430–4440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esfandiarei, M.; Luo, H.; Yanagawa, B.; Suarez, A.; Dabiri, D.; Zhang, J.; McManus, B.M. Protein kinase B/Akt regulates coxsackievirus B3 replication through a mechanism which is not caspase dependent. J. Virol. 2004, 78, 4289–4298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, H.; Yanagawa, B.; Zhang, J.; Luo, Z.; Zhang, M.; Esfandiarei, M.; Carthy, C.; Wilson, J.E.; Yang, D.; McManus, B.M. Coxsackievirus B3 replication is reduced by inhibition of the extracellular signal-regulated kinase (ERK) signaling pathway. J. Virol. 2002, 76, 3365–3373. [Google Scholar] [CrossRef] [Green Version]
- Opavsky, M.A.; Martino, T.; Rabinovitch, M.; Penninger, J.; Richardson, C.; Petric, M.; Trinidad, C.; Butcher, L.; Chan, J.; Liu, P.P. Enhanced ERK-1/2 activation in mice susceptible to coxsackievirus-induced myocarditis. J. Clin. Investig. 2002, 109, 1561–1569. [Google Scholar] [CrossRef] [PubMed]
- Althof, N.; Harkins, S.; Kemball, C.C.; Flynn, C.T.; Alirezaei, M.; Whitton, J.L. In vivo ablation of type I interferon receptor from cardiomyocytes delays coxsackieviral clearance and accelerates myocardial disease. J. Virol. 2014, 88, 5087–5099. [Google Scholar] [CrossRef] [Green Version]
- Deonarain, R.; Cerullo, D.; Fuse, K.; Liu, P.P.; Fish, E.N. Protective role for interferon-beta in coxsackievirus B3 infection. Circulation 2004, 110, 3540–3543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Critchley-Thorne, R.J.; Simons, D.L.; Yan, N.; Miyahira, A.K.; Dirbas, F.M.; Johnson, D.L.; Swetter, S.M.; Carlson, R.W.; Fisher, G.A.; Koong, A.; et al. Impaired interferon signaling is a common immune defect in human cancer. Proc. Natl. Acad. Sci. USA 2009, 106, 9010–9015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Parkin, D.M.; Pineros, M.; Znaor, A.; Bray, F. Cancer statistics for the year 2020: An overview. Int. J. Cancer 2021. [Google Scholar] [CrossRef] [PubMed]
- Koinis, F.; Kotsakis, A.; Georgoulias, V. Small cell lung cancer (SCLC): No treatment advances in recent years. Transl. Lung Cancer Res. 2016, 5, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Tomasini, P.; Walia, P.; Labbe, C.; Jao, K.; Leighl, N.B. Targeting the KRAS pathway in non-small cell lung cancer. Oncologist 2016, 21, 1450–1460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Colorectal cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, S.; Bao, Y.; Ni, C.; Guan, N.; Zhao, J.; Salford, L.G.; Widegren, B.; Fan, X. Coxsackievirus and adenovirus receptor expression in non-malignant lung tissues and clinical lung cancers. J. Mol. Histol. 2006, 37, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Arnold, M.; Sierra, M.S.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global patterns and trends in colorectal cancer incidence and mortality. Gut 2017, 66, 683–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hazini, A.; Pryshliak, M.; Bruckner, V.; Klingel, K.; Sauter, M.; Pinkert, S.; Kurreck, J.; Fechner, H. Heparan sulfate binding coxsackievirus B3 strain PD: A novel avirulent oncolytic agent against human colorectal carcinoma. Hum. Gene Ther. 2018, 29, 1301–1314. [Google Scholar] [CrossRef] [PubMed]
- Svyatchenko, V.A.; Ternovoy, V.A.; Kiselev, N.N.; Demina, A.V.; Loktev, V.B.; Netesov, S.V.; Chumakov, P.M. Bioselection of coxsackievirus B6 strain variants with altered tropism to human cancer cell lines. Arch. Virol. 2017, 162, 3355–3362. [Google Scholar] [CrossRef]
- Ha, M.; Kim, V.N. Regulation of microRNA biogenesis. Nat. Rev. Mol. Cell Biol. 2014, 15, 509–524. [Google Scholar] [CrossRef]
- Peng, Y.; Croce, C.M. The role of MicroRNAs in human cancer. Signal Transduct. Target. Ther. 2016, 1, 15004. [Google Scholar] [CrossRef] [Green Version]
- Reddy, K.B. MicroRNA (miRNA) in cancer. Cancer Cell Int. 2015, 15, 38. [Google Scholar] [CrossRef] [Green Version]
- Calin, G.A.; Croce, C.M. MicroRNA signatures in human cancers. Nat. Rev. Cancer 2006, 6, 857–866. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Getz, G.; Miska, E.A.; Alvarez-Saavedra, E.; Lamb, J.; Peck, D.; Sweet-Cordero, A.; Ebert, B.L.; Mak, R.H.; Ferrando, A.A.; et al. MicroRNA expression profiles classify human cancers. Nature 2005, 435, 834–838. [Google Scholar] [CrossRef] [PubMed]
- Bofill-De Ros, X.; Rovira-Rigau, M.; Fillat, C. Implications of MicroRNAs in Oncolytic Virotherapy. Front. Oncol. 2017, 7, 142. [Google Scholar] [CrossRef] [Green Version]
- Ruiz, A.J.; Russell, S.J. MicroRNAs and oncolytic viruses. Curr. Opin. Virol. 2015, 13, 40–48. [Google Scholar] [CrossRef]
- Gauntt, C.J.; Trousdale, M.D.; LaBadie, D.R.; Paque, R.E.; Nealon, T. Properties of coxsackievirus B3 variants which are amyocarditic or myocarditic for mice. J. Med. Virol. 1979, 3, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.K.; Kono, K.; Haas, E.; Kim, K.S.; Drescher, K.M.; Chapman, N.M.; Tracy, S. Characterization of an infectious cDNA copy of the genome of a naturally occurring, avirulent coxsackievirus B3 clinical isolate. J. Gen. Virol. 2005, 86, 197–210. [Google Scholar] [CrossRef]
- Cifuente, J.O.; Ferrer, M.F.; Jaquenod de Giusti, C.; Song, W.C.; Romanowski, V.; Hafenstein, S.L.; Gomez, R.M. Molecular determinants of disease in coxsackievirus B1 murine infection. J. Med. Virol. 2011, 83, 1571–1581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.S.; Nam, J.H. Characterization of attenuated coxsackievirus B3 strains and prospects of their application as live-attenuated vaccines. Expert Opin. Biol. Ther. 2010, 10, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Tu, Z.; Chapman, N.M.; Hufnagel, G.; Tracy, S.; Romero, J.R.; Barry, W.H.; Zhao, L.; Currey, K.; Shapiro, B. The cardiovirulent phenotype of coxsackievirus B3 is determined at a single site in the genomic 5’ nontranslated region. J. Virol. 1995, 69, 4607–4618. [Google Scholar] [CrossRef] [Green Version]
- Moratorio, G.; Henningsson, R.; Barbezange, C.; Carrau, L.; Borderia, A.V.; Blanc, H.; Beaucourt, S.; Poirier, E.Z.; Vallet, T.; Boussier, J.; et al. Attenuation of RNA viruses by redirecting their evolution in sequence space. Nat. Microbiol. 2017, 2, 17088. [Google Scholar] [CrossRef] [PubMed]
- Zainutdinov, S.S.; Kochneva, G.V.; Netesov, S.V.; Chumakov, P.M.; Matveeva, O.V. Directed evolution as a tool for the selection of oncolytic RNA viruses with desired phenotypes. Oncolytic Virother. 2019, 8, 9–26. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.Y.; Yousef, G.E.; Cunningham, L.; Blake, N.W.; OuYang, X.; Bayston, T.A.; Kandolf, R.; Archard, L.C. Attenuation of a reactivated cardiovirulent coxsackievirus B3: The 5’-nontranslated region does not contain major attenuation determinants. J. Med. Virol. 1993, 41, 129–137. [Google Scholar] [CrossRef]
- Zhang, H.; Blake, N.W.; Ouyang, X.; Pandolfino, Y.A.; Morgan-Capner, P.; Archard, L.C. A single amino acid substitution in the capsid protein VP1 of coxsackievirus B3 (CVB3) alters plaque phenotype in Vero cells but not cardiovirulence in a mouse model. Arch. Virol. 1995, 140, 959–966. [Google Scholar] [CrossRef]
- Zeng, J.; Chen, X.; Dai, J.; Zhao, X.; Xin, G.; Su, Y.; Wang, G.; Li, R.; Yan, Y.; Su, J.; et al. An attenuated coxsackievirus b3 vector: A potential tool for viral tracking study and gene delivery. PLoS ONE 2013, 8, e83753. [Google Scholar] [CrossRef] [Green Version]
- Polacek, C.; Ekstrom, J.O.; Lundgren, A.; Lindberg, A.M. Cytolytic replication of coxsackievirus B2 in CAR-deficient rhabdomyosarcoma cells. Virus Res. 2005, 113, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Reagan, K.J.; Goldberg, B.; Crowell, R.L. Altered receptor specificity of coxsackievirus B3 after growth in rhabdomyosarcoma cells. J. Virol. 1984, 49, 635–640. [Google Scholar] [CrossRef] [Green Version]
- Smura, T.; Natri, O.; Ylipaasto, P.; Hellman, M.; Al-Hello, H.; Piemonti, L.; Roivainen, M. Enterovirus strain and type-specific differences in growth kinetics and virus-induced cell destruction in human pancreatic duct epithelial HPDE cells. Virus Res. 2015, 210, 188–197. [Google Scholar] [CrossRef] [Green Version]
- Naidoo, J.; Page, D.B.; Wolchok, J.D. Immune modulation for cancer therapy. Br. J. Cancer 2014, 111, 2214–2219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, J.; Chehrazi-Raffle, A.; Reddi, S.; Salgia, R. Development of PD-1 and PD-L1 inhibitors as a form of cancer immunotherapy: A comprehensive review of registration trials and future considerations. J. Immunother. Cancer 2018, 6, 8. [Google Scholar] [CrossRef] [PubMed]
- Hamanishi, J.; Mandai, M.; Matsumura, N.; Abiko, K.; Baba, T.; Konishi, I. PD-1/PD-L1 blockade in cancer treatment: Perspectives and issues. Int. J. Clin. Oncol. 2016, 21, 462–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zitvogel, L.; Kroemer, G. Targeting PD-1/PD-L1 interactions for cancer immunotherapy. Oncoimmunology 2012, 1, 1223–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juneja, V.R.; McGuire, K.A.; Manguso, R.T.; LaFleur, M.W.; Collins, N.; Haining, W.N.; Freeman, G.J.; Sharpe, A.H. PD-L1 on tumor cells is sufficient for immune evasion in immunogenic tumors and inhibits CD8 T cell cytotoxicity. J. Exp. Med. 2017, 214, 895–904. [Google Scholar] [CrossRef] [PubMed]
- Filippi, C.M.; Estes, E.A.; Oldham, J.E.; von Herrath, M.G. Immunoregulatory mechanisms triggered by viral infections protect from type 1 diabetes in mice. J. Clin. Investig. 2009, 119, 1515–1523. [Google Scholar] [CrossRef] [Green Version]
- Lichtman, A.H. The heart of the matter: Protection of the myocardium from T cells. J. Autoimmun. 2013, 45, 90–96. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.Y.; Hutzen, B.; Wedekind, M.F.; Cripe, T.P. Oncolytic virus and PD-1/PD-L1 blockade combination therapy. Oncolytic Virother. 2018, 7, 65–77. [Google Scholar] [CrossRef] [Green Version]
- McDermott, D.F.; Sosman, J.A.; Sznol, M.; Massard, C.; Gordon, M.S.; Hamid, O.; Powderly, J.D.; Infante, J.R.; Fasso, M.; Wang, Y.V.; et al. Atezolizumab, an Anti-Programmed Death-Ligand 1 Antibody, in Metastatic Renal Cell Carcinoma: Long-Term Safety, Clinical Activity, and Immune Correlates From a Phase Ia Study. J. Clin. Oncol. 2016, 34, 833–842. [Google Scholar] [CrossRef]
- Moslehi, J.J.; Salem, J.E.; Sosman, J.A.; Lebrun-Vignes, B.; Johnson, D.B. Increased reporting of fatal immune checkpoint inhibitor-associated myocarditis. Lancet 2018, 391, 933. [Google Scholar] [CrossRef] [Green Version]
- Evgin, L.; Vile, R.G. Parking CAR T Cells in tumours: Oncolytic viruses as valets or vandals? Cancers 2021, 13, 1106. [Google Scholar] [CrossRef]
- Guedan, S.; Alemany, R. CAR-T cells and oncolytic viruses: Joining forces to overcome the solid tumor challenge. Front. Immunol. 2018, 9, 2460. [Google Scholar] [CrossRef] [Green Version]
- Rosewell Shaw, A.; Suzuki, M. Oncolytic viruses partner with T-Cell therapy for solid tumor treatment. Front. Immunol. 2018, 9, 2103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brudno, J.N.; Kochenderfer, J.N. Recent advances in CAR T-cell toxicity: Mechanisms, manifestations and management. Blood Rev. 2019, 34, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Ying, Z.; Huang, X.F.; Xiang, X.; Liu, Y.; Kang, X.; Song, Y.; Guo, X.; Liu, H.; Ding, N.; Zhang, T.; et al. A safe and potent anti-CD19 CAR T cell therapy. Nat. Med. 2019, 25, 947–953. [Google Scholar] [CrossRef] [PubMed]
- D’Aloia, M.M.; Zizzari, I.G.; Sacchetti, B.; Pierelli, L.; Alimandi, M. CAR-T cells: The long and winding road to solid tumors. Cell Death Dis. 2018, 9, 282. [Google Scholar] [CrossRef] [PubMed]
- Yong, C.S.M.; Dardalhon, V.; Devaud, C.; Taylor, N.; Darcy, P.K.; Kershaw, M.H. CAR T-cell therapy of solid tumors. Immunol. Cell Biol. 2017, 95, 356–363. [Google Scholar] [CrossRef]
- Evgin, L.; Huff, A.L.; Wongthida, P.; Thompson, J.; Kottke, T.; Tonne, J.; Schuelke, M.; Ayasoufi, K.; Driscoll, C.B.; Shim, K.G.; et al. Oncolytic virus-derived type I interferon restricts CAR T cell therapy. Nat. Commun. 2020, 11, 3187. [Google Scholar] [CrossRef]
- Zhang, X.; Paget, M.; Wang, C.; Zhu, Z.; Zheng, H. Innate immune evasion by picornaviruses. Eur. J. Immunol. 2020, 50, 1268–1282. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, J.; Li, Q. Immune evasion of enteroviruses under innate immune monitoring. Front. Microbiol. 2018, 9, 1866. [Google Scholar] [CrossRef]
- Boedtkjer, E.; Pedersen, S.F. The acidic tumor microenvironment as a driver of cancer. Annu. Rev. Physiol. 2020, 82, 103–126. [Google Scholar] [CrossRef] [Green Version]
- Hamada, M.; Yura, Y. Efficient delivery and replication of oncolytic virus for successful treatment of head and neck cancer. Int. J. Mol. Sci. 2020, 21, 7073. [Google Scholar] [CrossRef]
- Brouwer, L.; Moreni, G.; Wolthers, K.C.; Pajkrt, D. World-wide prevalence and genotype distribution of enteroviruses. Viruses 2021, 13, 434. [Google Scholar] [CrossRef]
- Samuelson, A.; Forsgren, M.; Johansson, B.; Wahren, B.; Sallberg, M. Molecular basis for serological cross-reactivity between enteroviruses. Clin. Diagn. Lab. Immunol. 1994, 1, 336–341. [Google Scholar] [CrossRef]
- Fu, X.; Tao, L.; Zhang, X. Genetically coating oncolytic herpes simplex virus with CD47 allows efficient systemic delivery and prolongs virus persistence at tumor site. Oncotarget 2018, 9, 34543–34553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ilett, E.J. Delivery of oncolytic reovirus by cell carriers. Methods Mol. Biol. 2020, 2058, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Power, A.T.; Wang, J.; Falls, T.J.; Paterson, J.M.; Parato, K.A.; Lichty, B.D.; Stojdl, D.F.; Forsyth, P.A.; Atkins, H.; Bell, J.C. Carrier cell-based delivery of an oncolytic virus circumvents antiviral immunity. Mol. Ther. 2007, 15, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Podshivalova, E.S.; Semkina, A.S.; Kravchenko, D.S.; Frolova, E.I.; Chumakov, S.P. Efficient delivery of oncolytic enterovirus by carrier cell line NK-92. Mol. Ther. Oncolytics 2021, 21, 110–118. [Google Scholar] [CrossRef]
- Franco-Luzon, L.; Gonzalez-Murillo, A.; Alcantara-Sanchez, C.; Garcia-Garcia, L.; Tabasi, M.; Huertas, A.L.; Chesler, L.; Ramirez, M. Systemic oncolytic adenovirus delivered in mesenchymal carrier cells modulate tumor infiltrating immune cells and tumor microenvironment in mice with neuroblastoma. Oncotarget 2020, 11, 347–361. [Google Scholar] [CrossRef] [Green Version]
- Hadrys, A.; Sochanik, A.; McFadden, G.; Jazowiecka-Rakus, J. Mesenchymal stem cells as carriers for systemic delivery of oncolytic viruses. Eur. J. Pharmacol. 2020, 874, 172991. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, H.; Luo, H. Development of Group B Coxsackievirus as an Oncolytic Virus: Opportunities and Challenges. Viruses 2021, 13, 1082. https://doi.org/10.3390/v13061082
Liu H, Luo H. Development of Group B Coxsackievirus as an Oncolytic Virus: Opportunities and Challenges. Viruses. 2021; 13(6):1082. https://doi.org/10.3390/v13061082
Chicago/Turabian StyleLiu, Huitao, and Honglin Luo. 2021. "Development of Group B Coxsackievirus as an Oncolytic Virus: Opportunities and Challenges" Viruses 13, no. 6: 1082. https://doi.org/10.3390/v13061082
APA StyleLiu, H., & Luo, H. (2021). Development of Group B Coxsackievirus as an Oncolytic Virus: Opportunities and Challenges. Viruses, 13(6), 1082. https://doi.org/10.3390/v13061082