
Rapid and Robust Continuous Purification of High-Titer Hepatitis B Virus for In Vitro and In Vivo Applications

 , , ,
, , ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
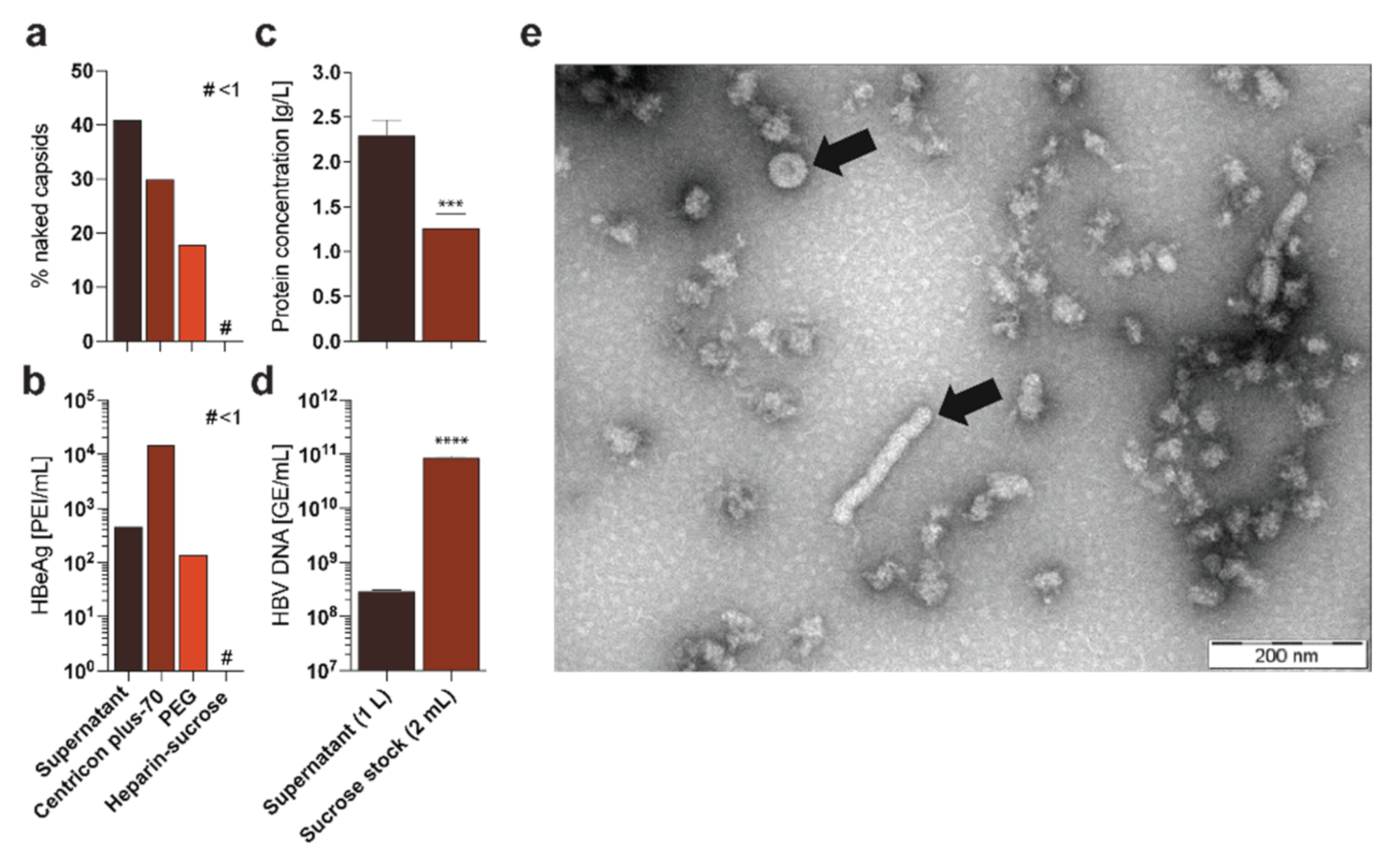
2.1. Analysis of Different HBV Purification and Concentration Methods
2.1.1. Centrifugal Filter Devices (e.g., Centricon Plus-70)
2.1.2. Precipitation Approaches via Chemical Polymers (e.g., Polyethylene Glycol 6000—PEG 6000)
2.1.3. Heparin-Affinity Chromatography
2.1.4. Sedimentation Centrifugation via Sucrose Gradients
2.1.5. Combination of Heparin-Affinity Chromatography and Sedimentation Centrifugation via Sucrose Gradient
2.2. Development of This Protocol
2.3. Overview of the Protocol and Optimization of Each Individual Part
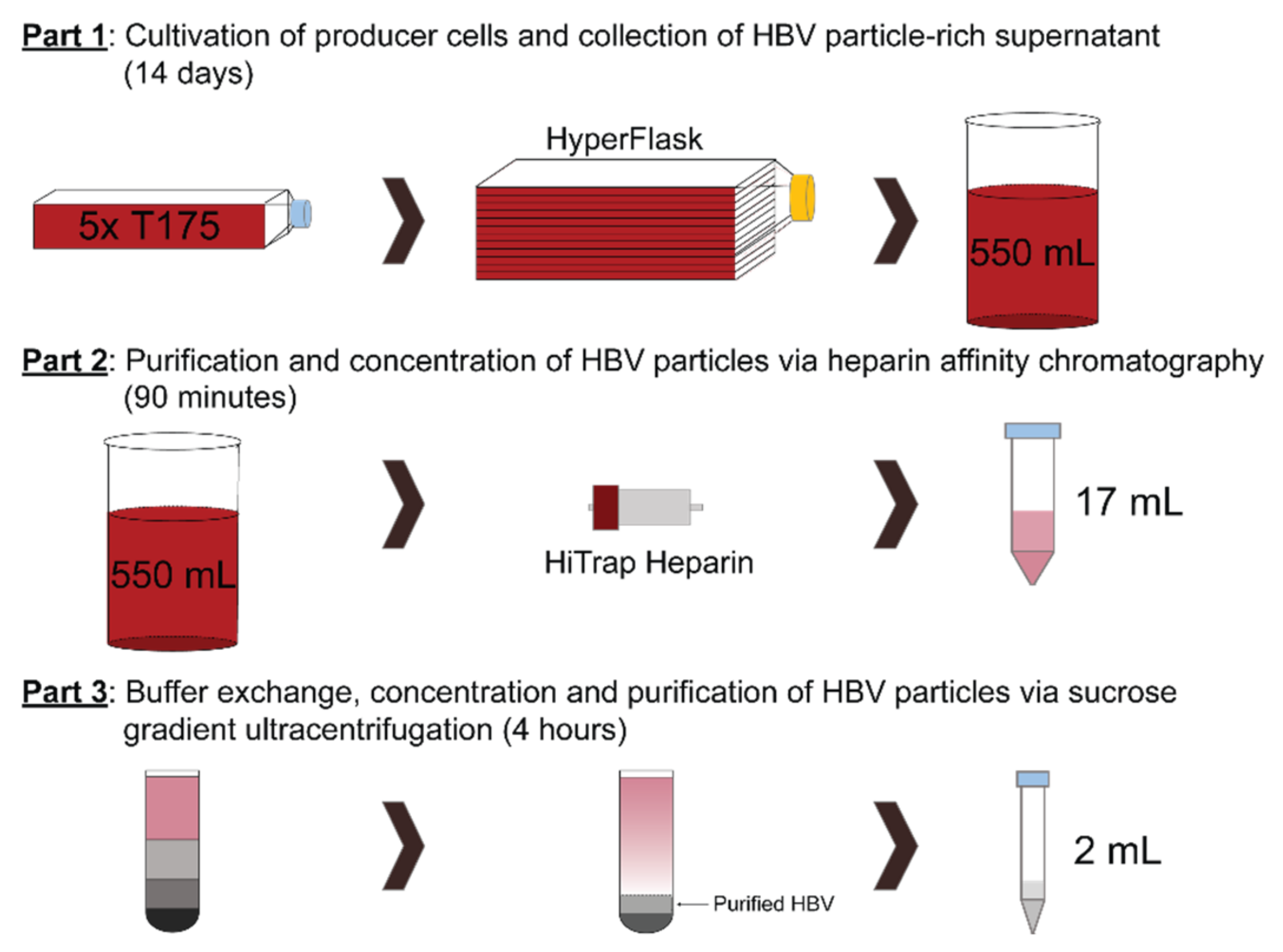
2.3.1. Cultivation of Producer Cells and Collection of HBV Particle-Rich Supernatant
2.3.2. Purification and Concentration of HBV Particles via Heparin-Affinity Chromatography
2.3.3. Purification, Buffer Exchange and Concentration of HBV Particles via Sucrose Gradient Ultracentrifugation
2.3.4. Storage of HBV Stocks
3. Materials and Methods
3.1. Reagents
3.1.1. HBV-Producer Cell Line Cultivation Medium (~550 mL)
3.1.2. Elution Buffer (100 mL)
3.1.3. Wash Solution
3.1.4. Heparin Column Storage Buffer (100 mL)
3.1.5. Sucrose
3.1.6. Cell Culture Solutions
3.2. Equipment
3.3. Virus Purification via Heparin-Affinity and Sucrose Gradient Ultracentrifugation
3.3.1. Cultivation of Producer Cells and Collection of HBV Particle-Rich Supernatant (Timing 14 Days)
- Step 1: Proliferate producer cells until they reach confluence on 5 collagen-coated T175 flasks.
- CRITICAL Cultivate all HepG2-based cell lines on collagen-coated plates and flasks. It is sufficient to collagen-coat the flasks by rinsing them with a H2O/0.02% Collagen R solution for 5 min followed by aspiration of the remaining Collagen solution.
- Step 2: Collagen coat a 10-layer Corning HYPERflask with 550 mL of H2O/0.01%-Collagen R solution and incubate overnight at room temperature. Take care that upper and lower surfaces are equally well coated. Collect H2O/0.01%-Collagen R solution and store at 4 °C for future HYPERflask collagen-coatings. Solution can be reused approx. 5 times.
- Step 3: Pre-warm 550 mL of cultivation medium to 37 °C and add 90% of the cells trypsinized off the 5 collagen-coated T175 flasks into the pre-warmed medium bottle. Cultivate the remaining 10% cells again in the 5 collagen-coated T175 flasks for step 6.
- CRITICAL Dispense the cells homogeneously by pipetting them against the wall of the T175 flask.
- Step 4: Pour 550 mL of the warm cell-suspension into the HYPERflask.
- TROUBLESHOOTING
- CRITICAL Remove all remaining air by applying pressure to the center of the HYPERflask, enabling homogenous cell spreading and guaranteeing space for medium extension during 37 °C incubation to avoid a pressure burst of the HYPERflask.
- Step 5: Culture the cells in the HYPERflask for 4 days at 37 °C in a 5% CO2 atmosphere to achieve confluence on the upper surfaces of the flask.
- Step 6: Pre-warm 550 mL of cultivation medium to 37 °C, trypsinize the cells regrown in the 5 collagen-coated T175 flasks (step 3) and suspend cells in the pre-warmed medium. Discard medium from the HYPERflask and add 550 mL of cell-suspension to the HYPERflask, flip the flask and cultivate it upside down overnight to allow attachment and growth of cells on the lower surfaces of the flask.
- CRITICAL Dispense the cells homogeneously by pipetting them against the wall of the T175 flask.
- CRITICAL Avoid drying out of cells in the HYPERflask by minimizing time between medium exchange.
- Step 7: Cultivate HYPERflask at 37 °C in a 5% CO2 atmosphere and harvest the supernatant in 4 day intervals.
- CRITICAL Discard the first medium collected after each cell plating cycle because it contains a high number of dead and non-attached cells.
3.3.2. Purification and Concentration of HBV Particles via Heparin-Affinity Chromatography (Timing 90 min)
- Step 8: Filter cold, HBV-containing supernatant through a 0.45 µm sterile filter to remove precipitated proteins and remaining cell debris.
- TROUBLESHOOTING
- CRITICAL For best filtration results and saving of filter devices, we recommend pre-filter inlays.
- Step 9: Assemble the purification apparatus without connecting the heparin columns and wash the system with 50 mL of 70% ethanol followed by 50 mL 1× PBS solution (Figure 7a,b).
- TROUBLESHOOTING
- CRITICAL Ensure that the tubing is completely filled with PBS and all remaining air bubbles are removed. Failure to do so will lead to damage of the heparin columns.
- Step 10: Connect 2 heparin columns (HiTrap heparin 5 mL) to the stopcocks in a parallel connection (Figure 7a,b) for every 550 mL of supernatant. To facilitate a weekly routine, we recommend using 4 heparin columns for the weekly volume of 1100 mL.
- TROUBLESHOOTING
- Step 11: Store filtered supernatant on ice and perfuse it through the heparin columns at a flow rate of approx. 10 mL/min per column (e.g., 40 mL/min for 4 heparin columns).
- TROUBLESHOOTING
- CRITICAL Higher loading speed may lower the lifetime of the columns.
- Step 12: After a complete perfusion of the supernatant, remove the columns and flush the system with elution buffer.
- CRITICAL It is important that the columns do not run dry. Ensure that no air is left in the system before you reattach the columns.
- Optional To increase HBV stock purity, but at costs of lower final concentration, columns can be washed with 10 mL of 1× PBS per column.
- Step 13: For elution, assemble columns using a serial connection (Figure 7c,d) and elute with 2 mL/min elution buffer. If you use more than 1 column, discard for each additional column 4 mL of the eluate due to the dead volume and collect the next 17 mL (e.g., using 4 columns, discard 12 mL and collect the following 17 mL (Figure 4e)).
- TROUBLESHOOTING
- CRITICAL Due to variations in dead volume, do not discard any eluate if you elute from a single column.
- CRITICAL Do not exceed the 2 mL/min elution speed in serial connection to avoid damaging the columns and tubing.
- CRITICAL Do not exceed more than 4 columns per elution since they can obstruct during serial elution due to high protein aggregation.
- CRITICAL Do not store the eluent on ice since HBV particles can precipitate due to high salt concentration. Keep eluent at room temperature until step 14.
- PAUSE POINT Purifying and concentrating HBV via heparin-affinity chromatography is now completed and eluent is ready to be further purified, concentrated and buffer exchanged via sucrose gradient ultracentrifugation. We recommend to not exceed storage of eluent for more than 2 h.
3.3.3. Buffer Exchange, Concentration and Purification of HBV Particles via Sucrose Gradient Ultracentrifugation (Timing 4 h)
- Step 14: Carefully overlay sucrose solutions and collected eluate in a SW32Ti ultracentrifugation tube by the following instructions: (from bottom to top) 3 mL sucrose 60%; 7 mL sucrose 25%; 9 mL sucrose 15% and 17 mL eluate (Figure 5a).
- Step 15: Centrifuge the gradient at 175,000× g (32,000 rpm) at 10 °C for 3.5 h to perform a separation (Figure 5b).
- TROUBLESHOOTING
- CRITICAL Use fast acceleration and brake.
- Step 16: Fractionate the gradient in 2 mL fractions using a fraction recovery system. The second fraction from the bottom of the gradient is the HBV-rich fraction (Figure 5b).
- TROUBLESHOOTING
- CRITICAL If you do not have access to a fraction recovery system, you may carefully insert a long blunt cannula into the tube and aspirate 2 mL from the bottom. Discard this fraction and aspirate another 2 mL from the bottom to obtain the HBV-rich fraction.
- Step 17: Clean Heparin HiTrap columns by applying 20 mL of 10× PBS solution per column at 5 mL/min in parallel connection. Flush them afterwards with heparin column storage buffer and store them air-tight at 4 °C.
- Step 18: Clean tubings, stop-cocks and fraction recovery system with 70% ethanol to inactivate potentially remaining HBV and store at room temperature.
- Optional Divide HBV stock into smaller aliquots prior to freezing. For in vitro infection experiments, we recommend diluting the sucrose stock 1:1 with fetal bovine serum to lower viscosity and to stabilize HBV particles by covering them with lipoproteins.
- PAUSE POINT Generation of HBV-rich stock is now completed. Stocks should have titers up to 100-times higher than the supernatant (typically higher than 1010 GE/mL) and can be stored for more than three years at −80 °C without a notable loss in infectivity. However, we recommend to avoid multiple freeze-thaw cycles and storage in non-frozen stages due to the loss of infectivity (Figure 6b,c).
3.3.4. Troubleshooting
- Overall → Problem: HBV is not infectious → Possible reasons: HBV has been precipitated or denatured → Solution: Check all used buffers. For correct osmolarity, it is crucial to prepare sucrose gradient solutions in 1× PBS. Due to high-salt concentration during elution, it is important to use elution buffer at room temperature and not to store eluate on ice. Thoroughly wash out ethanol-containing buffer with 1× PBS prior to usage of the purification apparatus.
- Step 6 → Problem: Supernatant is cloudy (note: supernatant can be slightly cloudy due to detached cells without affecting the purification process) → Possible reasons: Dead detached cells or bacterial contamination → Solution: Cultivate a 25 mL supernatant-sample in a collagen-coated T175 flask for 4 days and examine it under the microscope.
- → In the case of bacterial contamination (obvious cloudiness and visible bacteria at 40× magnification): start over again from Step 1 and ensure to thoroughly clean spilled medium in the screw thread of the lid after each medium exchange.
- → In the case of dead cells: Check HBV titer (HBsAg or GE/mL) of the supernatant at the next medium exchanges. If titer is further decreasing start over at Step 1. If titer is stable, continue with the protocol and try to avoid to keep too much air in the flask after each medium exchange. Carefully pat out the bubbles in front of the air trap of the HYPERflask. Consider cells are growing on both sides of the layers such that remaining bubbles will lead to punctual drying-out and death of some cells. Additionally, try to minimize time during medium exchange to avoid overall drying-out of the cells.
- → In the case of living cells: Cells probably did not attach adequately to the HYPERflask or have been detached. Check HBV titer in the supernatant of the next medium exchange. If titer has been decreased further start over at Step 1, check collagen solution and increase collagen incubation time in the HYPERflask for up to 2 days. If titer is stable or has been increased, pay attention to a more gentle medium exchange and avoid shaking or friction of the flask.
- Step 6 → Problem: HBV titer is less than 106 GE/mL → Possible reasons: Cells are not dense enough (consider that the bottom layer of the HYPERflask can be examined under the microscope) or do not produce sufficient amounts of HBV → Solution: Keep cells in culture and determine HBV titer at each medium exchange. If titer is not increasing over time until reaching a plateau due to producer cell confluence, use a lower passage of the cell line, escalate antibiotic selection or perform a single-cell selection of a high-producer cell clone or replace the producer cell line (note: a high-producing cell line (HepG2-pB-HBV1.3) is available on request). Check for contamination with other cells, bacteria or mycoplasma.
- Step 10 → Problem: Supernatant is running through parallel heparin columns at different flow-rates (note: slight differences in flow-rate are due to manufacturing and are unproblematic since it will balance during the purification process)→ Possible reasons: One or more columns are blocked or have been used too many times → Solution: Wash blocked column with 50 mL of 10× PBS. If problems remain, exchange the blocked column.
- Step 10 → Problem: Blocking of heparin columns during loading → Possible reasons: Remaining cell debris or proteins plugging the column → Solution: Cooling of the supernatant overnight at 4°C to allow clotting of cell debris and proteins prior to filtering (Step7). Carefully wash blocked column with 50 mL of 10× PBS. If problems remain, exchange it with a new heparin column.
- Step 12 → Problem: Blocking of heparin columns during elution → Possible reasons: HBV concentration is too high and virus is precipitating inside the columns → Solution: Reduce the volume of supernatant applied to each column during loading or elute each column individually. Carefully wash blocked column with 50 mL of 10× PBS. If problems remain, exchange with new heparin columns.
- Step 12 → Problem: HBV is not present in the eluate → Possible reasons: HBV does not bind to the columns or HBV does not elute → Solution: Check HBV titer pre- and post-column. If HBV did not bind to the columns and titers do not differ, exchange the columns. If titers differ, check elution buffer.
3.3.5. Timing
- Step 1–7: Cultivation of producer cells and collection of HBV particle-rich supernatant: 14 days → Production of supernatant every 3 to 4 days for more than 12 months.
- Step 8–13: Purification and concentration of HBV particles via heparin-affinity chromatography: 90 min
- Step 14–18: Buffer exchange, concentration and purification of HBV particles via sucrose gradient ultracentrifugation: 4 h
3.4. Virus Purification Using Other Published Protocols
3.5. Virus Infection
3.6. Quantification of HBV GE via Dot-Blot Analysis
3.7. Quantification of HBV GE via qPCR
3.8. Quantification of HBsAg and HBeAg
3.9. Protein Quantification
3.10. Electron Microscpy
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Trépo, C.; Chan, H.L.Y.; Lok, A. Hepatitis B virus infection. Lancet 2014, 384, 2053–2063. [Google Scholar] [CrossRef]
- WHO. Global Hepatitis Report 2017; WHO: Geneva, Switzerland, 2017; Available online: www.who.int:WHO (accessed on 28 July 2021).
- Ott, J.J.; Stevens, G.A.; Groeger, J.; Wiersma, S.T. Global epidemiology of hepatitis B virus infection: New estimates of age-specific HBsAg seroprevalence and endemicity. Vaccine 2012, 30, 2212–2219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, C.; Chakraborty, A.; Chou, W.M.; Hasreiter, J.; Wettengel, J.M.; Stadler, D.; Bester, R.; Asen, T.; Zhang, K.; Wisskirchen, K.; et al. Hepatitis B virus (HBV) genome recycling and de novo secondary infection events maintain stable cccDNA levels. J. Hepatol. 2018, 69, 1231–1241. [Google Scholar] [CrossRef] [PubMed]
- Bruss, V. Hepatitis B virus morphogenesis. World J. Gastroenterol. 2007, 13, 65–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganem, D.; Prince, A.M. Hepatitis B Virus Infection—Natural History and Clinical Consequences. N. Engl. J. Med. 2004, 350, 1118–1129. [Google Scholar] [CrossRef] [Green Version]
- Kräusslich, H.G. Morphogenesis and Maturation of Retroviruses; Springer: Berlin/Heidelberg, Germany, 2012. [Google Scholar]
- Rydell, G.E.; Prakash, K.; Norder, H.; Lindh, M. Hepatitis B surface antigen on subviral particles reduces the neutralizing effect of anti-HBs antibodies on hepatitis B viral particles in vitro. Virology 2017, 509, 67–70. [Google Scholar] [CrossRef]
- Bruns, M.; Miska, S.; Chassot, S.; Will, H. Enhancement of hepatitis B virus infection by noninfectious subviral particles. J. Virol. 1998, 72, 1462–1468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hossain, M.G.; Ueda, K. Investigation of a Novel Hepatitis B Virus Surface Antigen (HBsAg) Escape Mutant Affecting Immunogenicity. PLoS ONE 2017, 12, e0167871. [Google Scholar] [CrossRef]
- Michler, T.; Kosinska, A.D.; Festag, J.; Bunse, T.; Su, J.; Ringelhan, M.; Imhof, H.; Grimm, D.; Steiger, K.; Mogler, C.; et al. Knockdown of Virus Antigen Expression Increases Therapeutic Vaccine Efficacy in High-Titer Hepatitis B Virus Carrier Mice. Gastroenterology 2020, 158, 1762–1775.e9. [Google Scholar] [CrossRef] [PubMed]
- Schieck, A.; Schulze, A.; Gähler, C.; Müller, T.; Haberkorn, U.; Alexandrov, A.; Urban, S.; Mier, W. Hepatitis B virus hepatotropism is mediated by specific receptor recognition in the liver and not restricted to susceptible hosts. Hepatology 2013, 58, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Appelman, M.D.; Wettengel, J.M.; Protzer, U.; Elferink, R.P.O.; van de Graaf, S.F. Molecular regulation of the hepatic bile acid uptake transporter and HBV entry receptor NTCP. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2021, 1866, 158960. [Google Scholar]
- Hu, J.; Protzer, U.; Siddiqui, A. Revisiting Hepatitis B Virus: Challenges of Curative Therapies. J. Virol. 2019, 93, e01032-19. [Google Scholar] [CrossRef] [Green Version]
- Wing, P.A.; Liu, P.J.; Harris, J.M.; Magri, A.; Michler, T.; Zhuang, X.; Borrmann, H.; Minisini, R.; Frampton, N.R.; Wettengel, J.M.; et al. Hypoxia inducible factors regulate hepatitis B virus replication by activating the basal core promoter. J. Hepatol. 2021, 75, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Schulze, A.; Schieck, A.; Ni, Y.; Mier, W.; Urban, S. Fine Mapping of Pre-S Sequence Requirements for Hepatitis B Virus Large Envelope Protein-Mediated Receptor Interaction. J. Virol. 2010, 84, 1989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulze, A.; Gripon, P.; Urban, S. Hepatitis B virus infection initiates with a large surface protein-dependent binding to heparan sulfate proteoglycans. Hepatology 2007, 46, 1759–1768. [Google Scholar] [CrossRef] [PubMed]
- Protzer, U. Viral hepatitis: The bumpy road to animal models for HBV infection. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 327–328. [Google Scholar] [CrossRef] [PubMed]
- Wettengel, J.M.; Burwitz, B.J. Innovative HBV Animal Models Based on the Entry Receptor NTCP. Viruses 2020, 12, 828. [Google Scholar] [CrossRef]
- Burwitz, B.J.; Wettengel, J.; Mück-Häusl, M.A.; Ringelhan, M.; Ko, C.; Festag, M.M.; Hammond, K.B.; Northrup, M.; Bimber, B.N.; Jacob, T.; et al. Hepatocytic expression of human sodium-taurocholate cotransporting polypeptide enables hepatitis B virus infection of macaques. Nat. Commun. 2017, 8, 2146. [Google Scholar] [CrossRef]
- Lempp, F.A.; Wiedtke, E.; Qu, B.; Roques, P.; Chemin, I.; Vondran, F.W.R.; Le Grand, R.; Grimm, D.; Urban, S. Sodium taurocholate cotransporting polypeptide is the limiting host factor of hepatitis B virus infection in macaque and pig hepatocytes. Hepatology 2017, 66, 703–716. [Google Scholar] [CrossRef] [Green Version]
- Barker, L.F.; Maynard, J.E.; Purcell, R.H.; Hoofnagle, J.H.; Berquist, K.R.; London, W.T.; Gerety, R.J.; Krushak, D.H. Hepatitis B Virus Infection in Chimpanzees: Titration of Subtypes. J. Infect. Dis. 1975, 132, 451–458. [Google Scholar] [CrossRef]
- Dandri, M.; Burda, M.R.; Török, E.; Pollok, J.M.; Iwanska, A.; Sommer, G.; Rogiers, X.; Rogler, C.E.; Gupta, S.; Will, H.; et al. Repopulation of mouse liver with human hepatocytes and in vivo infection with hepatitis B virus. Hepatology 2001, 33, 981–988. [Google Scholar] [CrossRef] [PubMed]
- Vnek, J.; Prince, A.M. Large-scale purification of hepatitis B surface antigen. J. Clin. Microbiol. 1976, 3, 626–631. [Google Scholar] [CrossRef]
- Ladner, S.K.; Otto, M.J.; Barker, C.S.; Zaifert, K.; Wang, G.H.; Guo, J.T.; Seeger, C.; King, R.W. Inducible expression of human hepatitis B virus (HBV) in stably transfected hepatoblastoma cells: A novel system for screening potential inhibitors of HBV replication. Antimicrob. Agents Chemother. 1997, 41, 1715–1720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sells, M.A.; Chen, M.L.; Acs, G. Production of hepatitis B virus particles in Hep G2 cells transfected with cloned hepatitis B virus DNA. Proc. Natl. Acad. Sci. USA 1987, 84, 1005–1009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arzberger, S.; Hosel, M.; Protzer, U. Apoptosis of hepatitis B virus-infected hepatocytes prevents release of infectious virus. J. Virol. 2010, 84, 11994–12001. [Google Scholar] [CrossRef] [Green Version]
- Bardens, A.; Döring, T.; Stieler, J.; Prange, R. Alix regulates egress of hepatitis B virus naked capsid particles in an ESCRT-independent manner. Cell. Microbiol. 2010, 13, 602–619. [Google Scholar] [CrossRef]
- Xia, Y.; Cheng, X.; Blossey, C.K.; Wisskirchen, K.; Esser, K.; Protzer, U. Secreted Interferon-Inducible Factors Restrict Hepatitis B and C Virus Entry In Vitro. J. Immunol. Res. 2017, 2017, 4828936. [Google Scholar] [CrossRef]
- Ni, Y.; Urban, S. Hepatitis B Virus Infection of HepaRG Cells, HepaRG-hNTCP Cells, and Primary Human Hepatocytes. Methods Mol. Biol. 2017, 1540, 15–25. [Google Scholar]
- Batlle, R.; Andrés, E.; Gonzalez, L.; Llonch, E.; Igea, A.; Gutierrez-Prat, N.; Berenguer-Llergo, A.; Nebreda, A.R. Regulation of tumor angiogenesis and mesenchymal-endothelial transition by p38alpha through TGF-beta and JNK signaling. Nat. Commun. 2019, 10, 3071. [Google Scholar] [CrossRef]
- Möller, L.; Schünadel, L.; Nitsche, A.; Schwebke, I.; Hanisch, M.; Laue, M. Evaluation of virus inactivation by formaldehyde to enhance biosafety of diagnostic electron microscopy. Viruses 2015, 7, 666–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanyal, D.; Kudesia, G.; Corbitt, G. Comparison of ultracentrifugation and polyethylene glycol precipitation for concentration of hepatitis B virus (HBV) DNA for molecular hybridisation tests and the relationship of HBV-DNA to HBe antigen and anti-HBe status. J. Med. Microbiol. 1991, 35, 291–293. [Google Scholar] [CrossRef]
- Zahn, A.; Allain, J.P. Hepatitis C virus and hepatitis B virus bind to heparin: Purification of largely IgG-free virions from infected plasma by heparin chromatography. J. Gen. Virol. 2005, 86, 677–685. [Google Scholar] [CrossRef]
- Einarsson, M.; Kaplan, L.; Pertoft, H. A two-step procedure for the purificaton of hepatitis B surface antigen (HBsAg). Vox Sang. 1981, 41, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Seitz, S.; Iancu, C.; Volz, T.; Mier, W.; Dandri, M.; Urban, S.; Bartenschlager, R. A Slow Maturation Process Renders Hepatitis B Virus Infectious. Cell Host Microbe 2016, 20, 25–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winer, B.; Huang, T.S.; Pludwinski, E.; Heller, B.; Wojcik, F.; Lipkowitz, G.E.; Parekh, A.; Cho, C.; Shrirao, A.; Muir, T.W.; et al. Long-term hepatitis B infection in a scalable hepatic co-culture system. Nat. Commun. 2017, 8, 125. [Google Scholar] [CrossRef] [PubMed]
- Szabolcs, M.; Francia, I. Determination of the sedimentation coefficient and molecular weight of proteins by density gradient ultracentrifugation in fixed angle rotor. Acta Biochim. Biophys. Hung. 1989, 24, 245–258. [Google Scholar] [PubMed]
- Wing, P.A.; Davenne, T.; Wettengel, J.; Lai, A.G.; Zhuang, X.; Chakraborty, A.; D’Arienzo, V.; Kramer, C.; Ko, C.; Harris, J.; et al. A dual role for SAMHD1 in regulating HBV cccDNA and RT-dependent particle genesis. Life Sci. Alliance 2019, 2, e201900355. [Google Scholar] [CrossRef] [PubMed]
- Esser, K.; Lucifora, J.; Wettengel, J.; Singethan, K.; Glinzer, A.; Zernecke, A.; Protzer, U. Lipase inhibitor orlistat prevents hepatitis B virus infection by targeting an early step in the virus life cycle. Antivir. Res. 2018, 151, 4–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wisskirchen, K.; Kah, J.; Malo, A.; Asen, T.; Volz, T.; Allweiss, L.; Wettengel, J.M.; Lütgehetmann, M.; Urban, S.; Bauer, T.; et al. T cell receptor grafting allows virological control of hepatitis B virus infection. J. Clin. Investig. 2019, 129, 2932–2945. [Google Scholar] [CrossRef]
- Xia, Y.; Stadler, D.; Lucifora, J.; Reisinger, F.; Webb, D.; Hösel, M.; Michler, T.; Wisskirchen, K.; Cheng, X.; Zhang, K.; et al. Interferon-gamma and Tumor Necrosis Factor-alpha Produced by T Cells Reduce the HBV Persistence Form, cccDNA, Without Cytolysis. Gastroenterology 2016, 150, 194–205. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wettengel, J.M.; Linden, B.; Esser, K.; Laue, M.; Burwitz, B.J.; Protzer, U. Rapid and Robust Continuous Purification of High-Titer Hepatitis B Virus for In Vitro and In Vivo Applications. Viruses 2021, 13, 1503. https://doi.org/10.3390/v13081503
Wettengel JM, Linden B, Esser K, Laue M, Burwitz BJ, Protzer U. Rapid and Robust Continuous Purification of High-Titer Hepatitis B Virus for In Vitro and In Vivo Applications. Viruses. 2021; 13(8):1503. https://doi.org/10.3390/v13081503
Chicago/Turabian StyleWettengel, Jochen M., Bianca Linden, Knud Esser, Michael Laue, Benjamin J. Burwitz, and Ulrike Protzer. 2021. "Rapid and Robust Continuous Purification of High-Titer Hepatitis B Virus for In Vitro and In Vivo Applications" Viruses 13, no. 8: 1503. https://doi.org/10.3390/v13081503
APA StyleWettengel, J. M., Linden, B., Esser, K., Laue, M., Burwitz, B. J., & Protzer, U. (2021). Rapid and Robust Continuous Purification of High-Titer Hepatitis B Virus for In Vitro and In Vivo Applications. Viruses, 13(8), 1503. https://doi.org/10.3390/v13081503