
The Algerian Chapter of SARS-CoV-2 Pandemic: An Evolutionary, Genetic, and Epidemiological Prospect

 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sequence Selection and Maximum Likelihood Phylogeny
2.2. Temporal Signal Assessment, Time-Calibrated Phylogeny Reconstruction, and Phylogeographic Analysis in Discrete Space
2.3. Genome Investigations
2.4. Haplotype Network Analysis
2.5. Epidemiological Analysis and Preventive Measures Assessment
3. Results
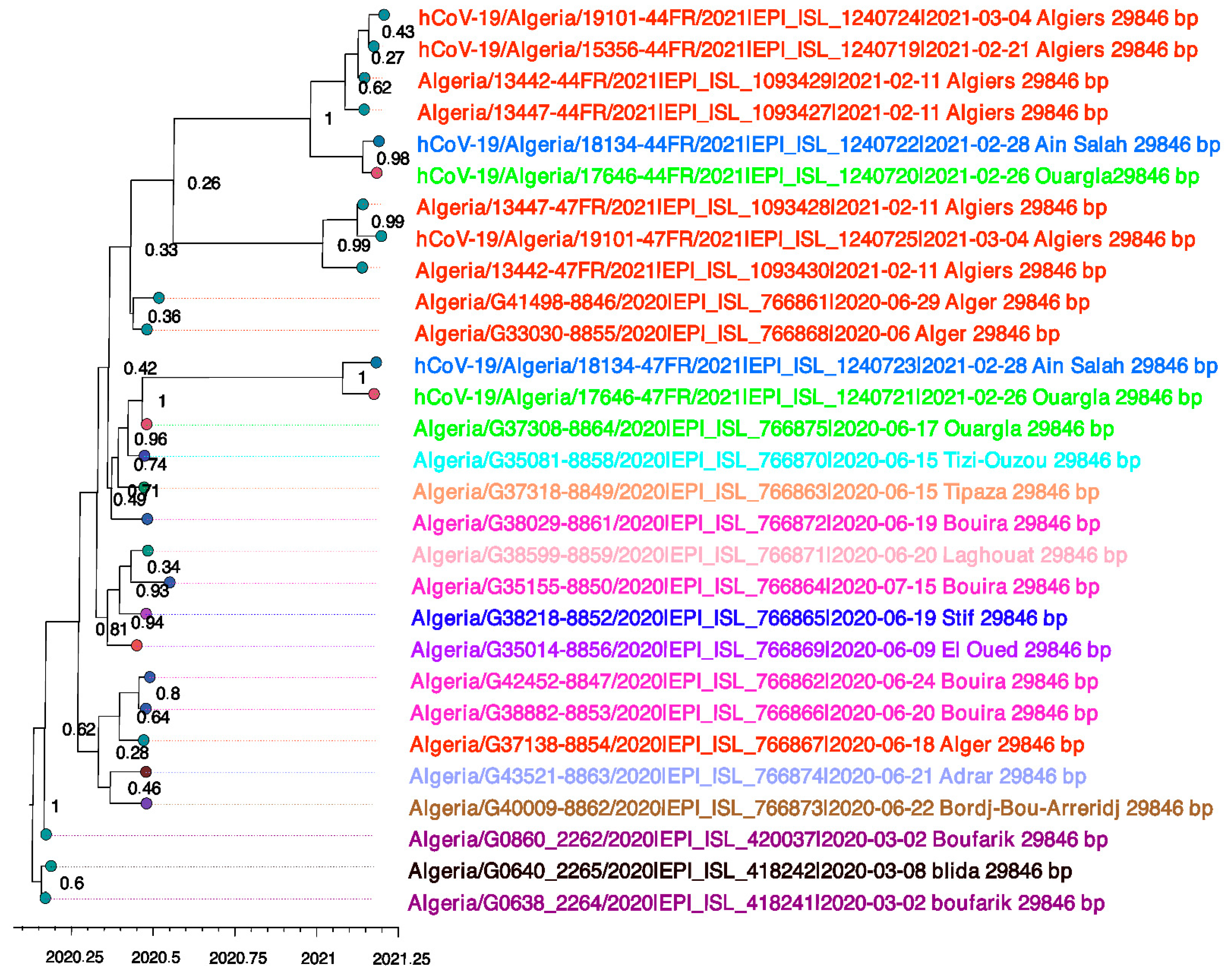
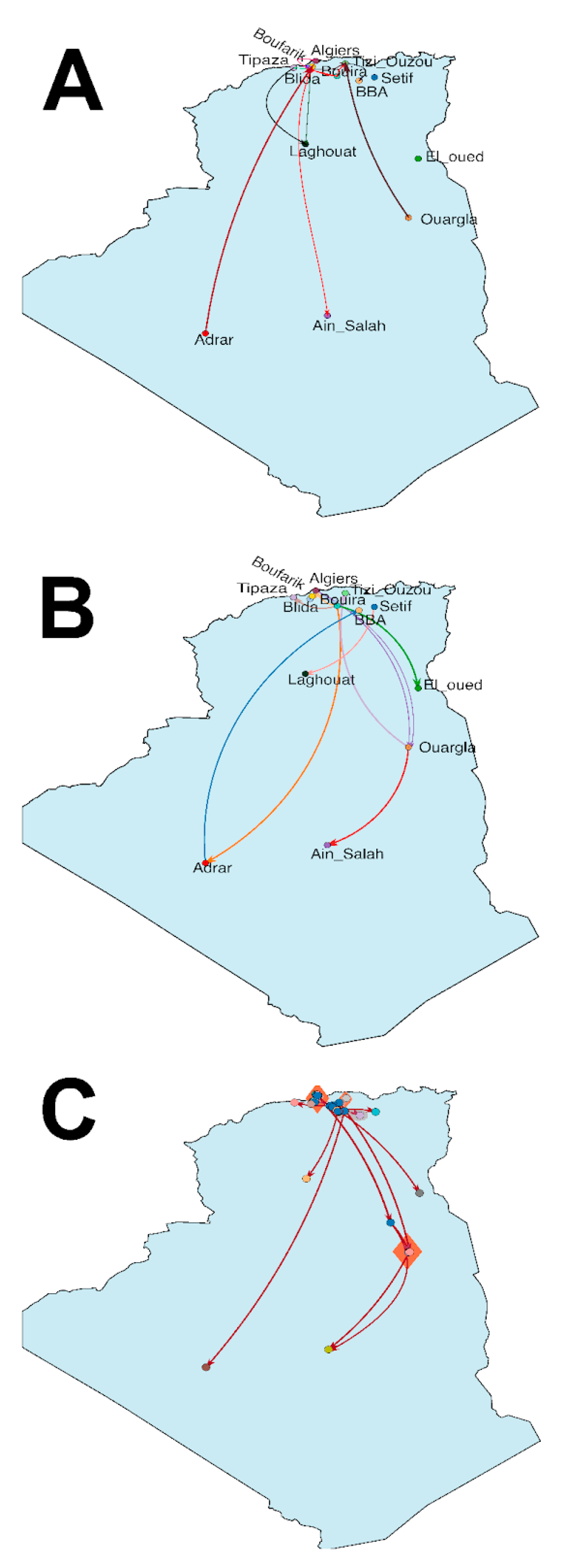
3.1. Evolutionary Phylogenetic and Phylogeographic Analyses
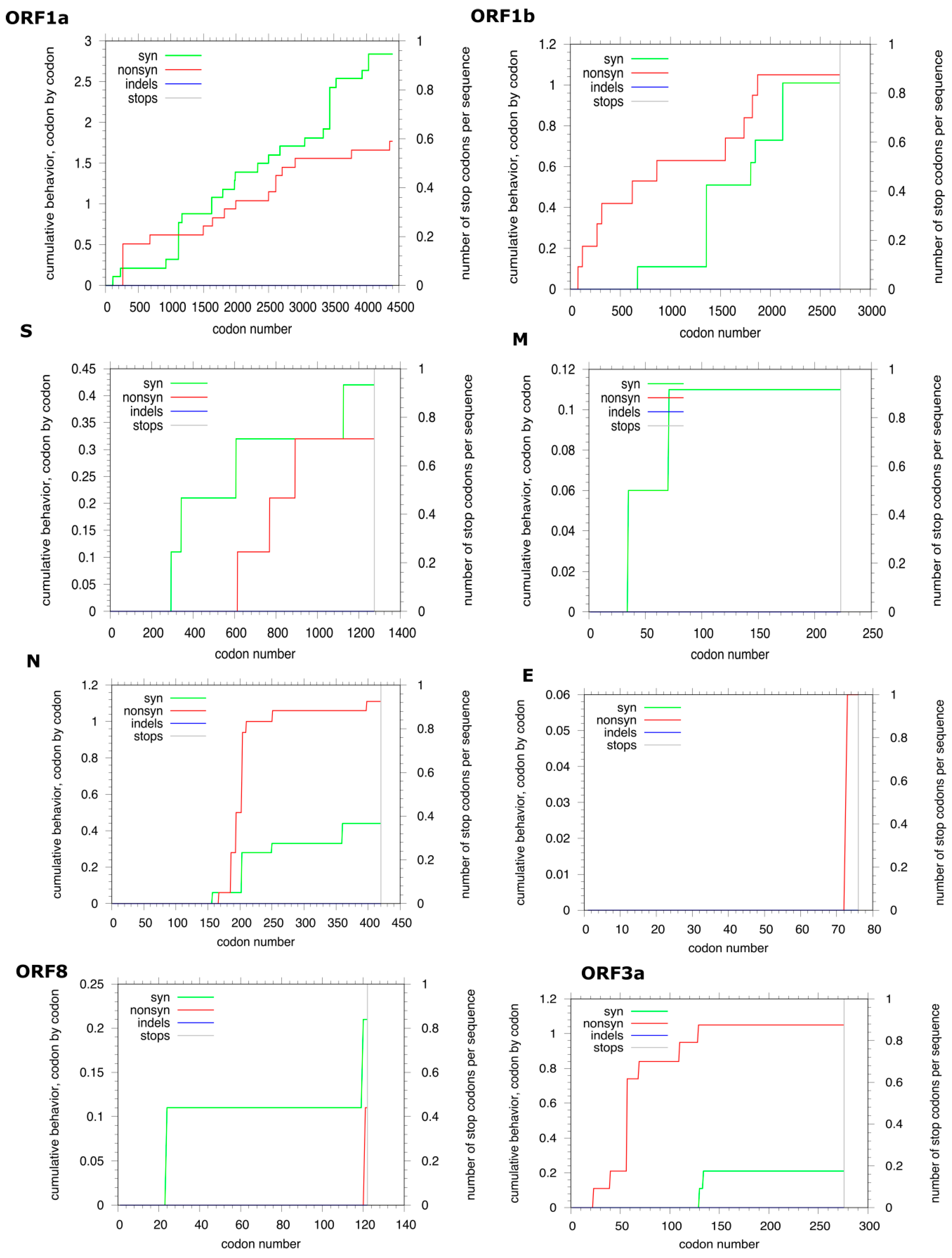
3.2. Genome Analyses
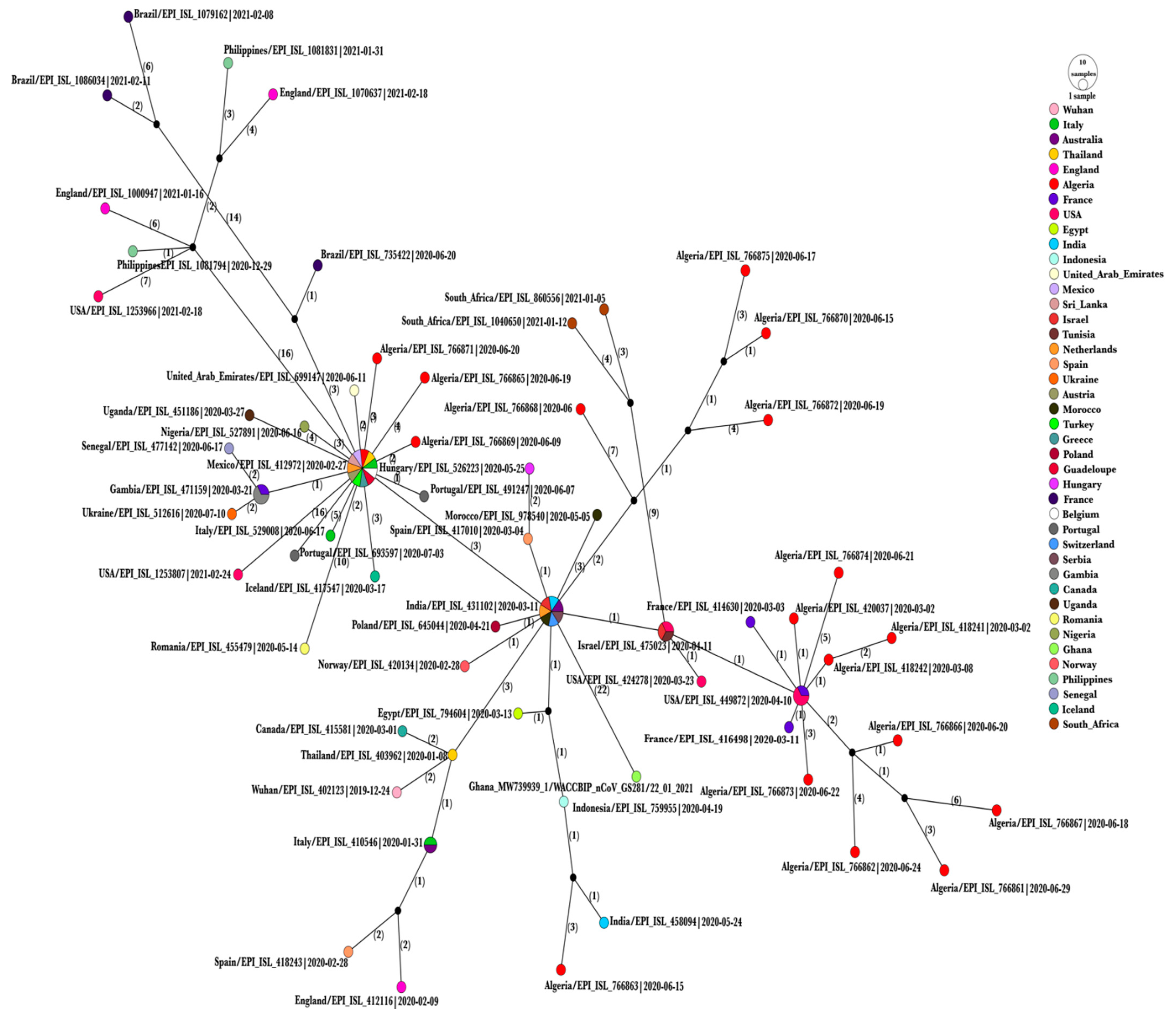
3.3. Haplotype Network Analysis
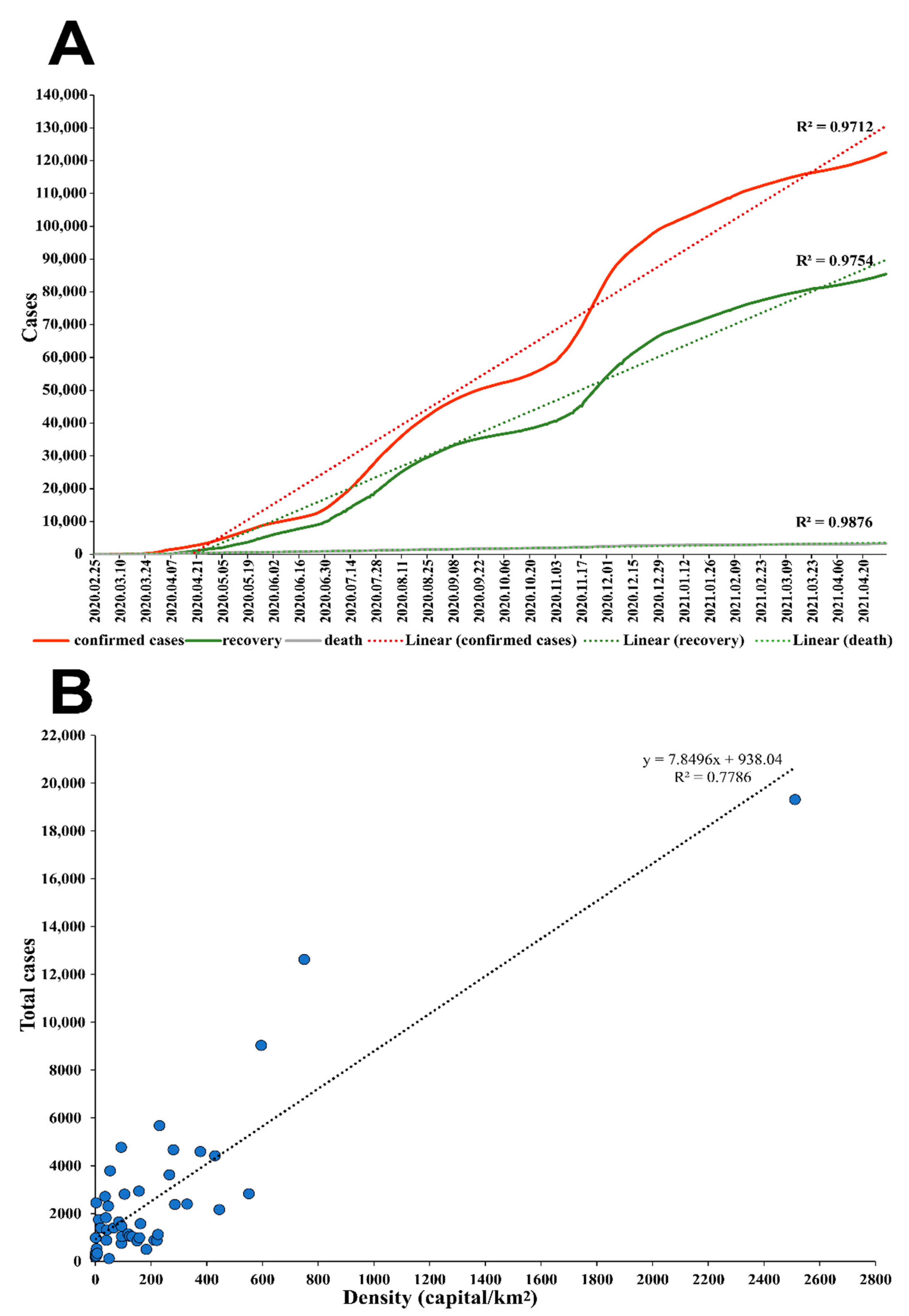
3.4. Epidemiological Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.-M.; Wang, W.; Song, Z.-G.; Hu, Y.; Tao, Z.-W.; Tian, J.-H.; Pei, Y.-Y.; et al. A New Coronavirus Associated with Human Respiratory Disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coronaviridae Study Group of the International Committee on Taxonomy of Viruses The Species Severe Acute Respiratory Syndrome-Related Coronavirus: Classifying 2019-NCoV and Naming It SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [CrossRef] [PubMed] [Green Version]
- COVID-19 Situation Reports. Available online: https://www.who.int/emergencies/diseases/novel-coronavirus-2019/situation-reports (accessed on 12 May 2020).
- Sharun, K.; Dhama, K.; Pawde, A.M.; Gortázar, C.; Tiwari, R.; Bonilla-Aldana, D.K.; Rodriguez-Morales, A.J.; de la Fuente, J.; Michalak, I.; Attia, Y.A. SARS-CoV-2 in Animals: Potential for Unknown Reservoir Hosts and Public Health Implications. Vet. Q. 2021, 41, 181–201. [Google Scholar] [CrossRef] [PubMed]
- The COVID Lab-Leak Hypothesis: What Scientists Do and Don’t Know. Available online: https://www.nature.com/articles/d41586-021-01529-3 (accessed on 29 June 2021).
- Hamidouche, M. COVID-19 Epidemic in Algeria: Assessment of the Implemented Preventive Strategy. medRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Kames, J.; Holcomb, D.D.; Kimchi, O.; DiCuccio, M.; Hamasaki-Katagiri, N.; Wang, T.; Komar, A.A.; Alexaki, A.; Kimchi-Sarfaty, C. Sequence Analysis of SARS-CoV-2 Genome Reveals Features Important for Vaccine Design. Sci. Rep. 2020, 10, 15643. [Google Scholar] [CrossRef]
- Shu, Y.; McCauley, J. GISAID: Global Initiative on Sharing All Influenza Data—From Vision to Reality. Eurosurveillance 2017, 22, 30494. [Google Scholar] [CrossRef] [Green Version]
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian Evolutionary Analysis by Sampling Trees. BMC Evol. Biol. 2007, 7, 214. [Google Scholar] [CrossRef] [Green Version]
- Leigh, J.W.; Bryant, D. Popart: Full-Feature Software for Haplotype Network Construction. Methods Ecol. Evol. 2015, 6, 1110–1116. [Google Scholar] [CrossRef]
- Elbe, S.; Buckland-Merrett, G. Data, Disease and Diplomacy: GISAID’s Innovative Contribution to Global Health. Glob. Chall. 2017, 1, 33–46. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A Novel Method for Rapid Multiple Sequence Alignment Based on Fast Fourier Transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Lam, T.T.; Max Carvalho, L.; Pybus, O.G. Exploring the Temporal Structure of Heterochronous Sequences Using TempEst (Formerly Path-O-Gen). Virus Evol. 2016, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian Phylogenetic and Phylodynamic Data Integration Using BEAST 1.10. Virus Evol. 2018, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drummond, A.J.; Ho, S.Y.W.; Phillips, M.J.; Rambaut, A. Relaxed Phylogenetics and Dating with Confidence. PLoS Biol. 2006, 4, e88. [Google Scholar] [CrossRef]
- Drummond, A.J. Bayesian Coalescent Inference of Past Population Dynamics from Molecular Sequences. Mol. Biol. Evol. 2005, 22, 1185–1192. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [Green Version]
- Lemey, P.; Rambaut, A.; Drummond, A.J.; Suchard, M.A. Bayesian Phylogeography Finds Its Roots. PLoS Comput. Biol. 2009, 5, e1000520. [Google Scholar] [CrossRef] [Green Version]
- Bielejec, F.; Rambaut, A.; Suchard, M.A.; Lemey, P. SPREAD: Spatial Phylogenetic Reconstruction of Evolutionary Dynamics. Bioinformatics 2011, 27, 2910–2912. [Google Scholar] [CrossRef] [Green Version]
- Simple Methods for Estimating the Numbers of Synonymous and Nonsynonymous Nucleotide Substitutions. Mol. Biol. Evol. 1986. [CrossRef]
- Yang, Z.; Bielawski, J.P. Statistical Methods for Detecting Molecular Adaptation. Trends Ecol. Evol. 2000, 15, 496–503. [Google Scholar] [CrossRef]
- Bromberg, Y.; Rost, B. SNAP: Predict Effect of Non-Synonymous Polymorphisms on Function. Nucleic Acids Res. 2007, 35, 3823–3835. [Google Scholar] [CrossRef] [Green Version]
- Cleemput, S.; Dumon, W.; Fonseca, V.; Abdool Karim, W.; Giovanetti, M.; Alcantara, L.C.; Deforche, K.; de Oliveira, T. Genome Detective Coronavirus Typing Tool for Rapid Identification and Characterization of Novel Coronavirus Genomes. Bioinformatics 2020, 36, 3552–3555. [Google Scholar] [CrossRef] [PubMed]
- Singer, J.; Gifford, R.; Cotten, M.; Robertson, D. CoV-GLUE: A Web Application for Tracking SARS-CoV-2 Genomic Variation. Preprints 2020. [Google Scholar] [CrossRef]
- Hanada, K.; Gojobori, T.; Li, W.-H. Radical Amino Acid Change versus Positive Selection in the Evolution of Viral Envelope Proteins. Gene 2006, 385, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Grantham, R. Amino Acid Difference Formula to Help Explain Protein Evolution. Science 1974, 185, 862–864. [Google Scholar] [CrossRef] [PubMed]
- Miyata, T.; Miyazawa, S.; Yasunaga, T. Two Types of Amino Acid Substitutions in Protein Evolution. J. Mol. Evol. 1979, 12, 219–236. [Google Scholar] [CrossRef]
- Capriotti, E.; Fariselli, P. PhD-SNPg: A Webserver and Lightweight Tool for Scoring Single Nucleotide Variants. Nucleic Acids Res. 2017, 45, W247–W252. [Google Scholar] [CrossRef] [Green Version]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting Functional Effect of Human Missense Mutations Using PolyPhen-2. Curr. Protoc. Hum. Genet 2013, 76, 7–20. [Google Scholar] [CrossRef] [Green Version]
- Ng, P.C.; Henikoff, S. SIFT: Predicting Amino Acid Changes That Affect Protein Function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef] [Green Version]
- PredictSNP: Robust and Accurate Consensus Classifier for Prediction of Disease-Related Mutations. Available online: https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1003440 (accessed on 21 June 2021).
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA Sequence Polymorphism Analysis of Large Data Sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and Analysis of Recombination Patterns in Virus Genomes. Virus Evol. 2015, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bandelt, H.J.; Forster, P.; Rohl, A. Median-Joining Networks for Inferring Intraspecific Phylogenies. Mol. Biol. Evol. 1999, 16, 37–48. [Google Scholar] [CrossRef] [PubMed]
- An Interactive Web-Based Dashboard to Track COVID-19 in Real Time|Elsevier Enhanced Reader. Available online: https://reader.elsevier.com/reader/sd/pii/S1473309920301201?token=376C61D71B1CC278C07B9C6C5D58FE8583E9CD7429F5971C8D3EA12FB3903A4D7DA908608F00013BBB0DEF3202E2EC6F (accessed on 17 August 2020).
- Algerian Ministry of Health. Available online: http://covid19.sante.gov.dz/carte/ (accessed on 15 May 2021).
- Démographie en Algérie. Available online: https://fr.wikipedia.org/wiki/Démographie_de_l’Algérie (accessed on 3 May 2021).
- Rambaut, A.; Holmes, E.C.; Hill, V.; O’Toole, Á.; McCrone, J.; Ruis, C.; du Plessis, L.; Pybus, O.G. A Dynamic Nomenclature Proposal for SARS-CoV-2 to Assist Genomic Epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, W.; Zhao, X.; Zai, J.; Zhao, Q.; Li, Y.; Chaillon, A. Transmission Dynamics and Evolutionary History of 2019-NCoV. J. Med Virol. 2020, 92, 501–511. [Google Scholar] [CrossRef] [PubMed]
- Time Dependence of SARS-CoV-2 Substitution Rates—SARS-CoV-2 Coronavirus/NCoV-2019 Evolutionary History. Available online: https://virological.org/t/time-dependence-of-sars-cov-2-substitution-rates/542 (accessed on 17 June 2021).
- Ghafari, M.; Simmonds, P.; Pybus, O.G.; Katzourakis, A. Prisoner of War Dynamics Explains the Time-Dependent Pattern of Substitution Rates in Viruses. bioRxiv 2021. [Google Scholar] [CrossRef]
- Gámbaro, F.; Behillil, S.; Baidaliuk, A.; Donati, F.; Albert, M.; Alexandru, A.; Vanpeene, M.; Bizard, M.; Brisebarre, A.; Barbet, M.; et al. Introductions and Early Spread of SARS-CoV-2 in France. Eurosurveillance 2020, 25, 2001200. [Google Scholar]
- Pandémie de Covid-19 en Algérie. Available online: https://fr.wikipedia.org/wiki/Pandémie_de_Covid-19_en_Algérie (accessed on 26 June 2021).
- Zhang, M.; Li, L.; Luo, M.; Liang, B. Genomic Characterization and Evolution of SARS-CoV-2 of a Canadian Population. PLoS ONE 2021, 16, e0247799. [Google Scholar] [CrossRef]
- Miao, M.; Clercq, E.D.; Li, G. Genetic Diversity of SARS-CoV-2 over a One-Year Period of the COVID-19 Pandemic: A Global Perspective. Biomedicines 2021, 9, 412. [Google Scholar] [CrossRef]
- Kaushal, N.; Gupta, Y.; Goyal, M.; Khaiboullina, S.F.; Baranwal, M.; Verma, S.C. Mutational Frequencies of SARS-CoV-2 Genome during the Beginning Months of the Outbreak in USA. Pathogens 2020, 9, 565. [Google Scholar] [CrossRef] [PubMed]
- Issa, E.; Merhi, G.; Panossian, B.; Salloum, T.; Tokajian, S. SARS-CoV-2 and ORF3a: Nonsynonymous Mutations, Functional Domains, and Viral Pathogenesis. mSystems 2020, 5. [Google Scholar] [CrossRef]
- Fonville, J.M. Expected Effect of Deleterious Mutations on Within-Host Adaptation of Pathogens. J. Virol. 2015, 89, 9242–9251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koelle, K.; Rasmussen, D.A. The Effects of a Deleterious Mutation Load on Patterns of Influenza A/H3N2′s Antigenic Evolution in Humans. eLife 2015, 4, e07361. [Google Scholar] [CrossRef]
- Jensen, J.D.; Lynch, M. Considering Mutational Meltdown as a Potential SARS-CoV-2 Treatment Strategy. Heredity 2020, 124, 619–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, J.K.; Sengupta, A.; Choudhury, P.P.; Roy, S. Characterizing Genomic Variants and Mutations in SARS-CoV-2 Proteins from Indian Isolates. Gene Rep. 2021, 101044. [Google Scholar] [CrossRef]
- CDC Coronavirus Disease 2019 (COVID-19). Available online: https://www.cdc.gov/coronavirus/2019-ncov/variants/variant-info.html (accessed on 22 June 2021).
- Lau, B.T.; Pavlichin, D.; Hooker, A.C.; Almeda, A.; Shin, G.; Chen, J.; Sahoo, M.K.; Huang, C.H.; Pinsky, B.A.; Lee, H.J.; et al. Profiling SARS-CoV-2 Mutation Fingerprints That Range from the Viral Pangenome to Individual Infection Quasispecies. Genome Med. 2021, 13, 62. [Google Scholar] [CrossRef]
- Megna, R. First Month of the Epidemic Caused by COVID-19 in Italy: Current Status and Real-Time Outbreak Development Forecast. Glob. Health Res. Policy 2020, 5, 43. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Query | Gisaid Clade | Pangolin Lineage | SNPs | Query | Gisaid Clade | Pangolin Lineage | SNPs |
|---|---|---|---|---|---|---|---|
| Algeria/EPI_ISL_766874 | GH | B.1 | C601T, C1059T, C3037T, C6255T, C8290T, C10582T, A13693G, C13829T, C14408T, A23403G, C25511T, G25563T, C29025T | Algeria/EPI_ISL_766861 | G | B.1.597 | C3037T, C3619T, C5144T, C10582T, C12367T, C14408T, C17550T, A23403G, C27804T, C28830A |
| Algeria/EPI_ISL_766862 | GH | B.1.597 | C1059T, C3037T, A4762C, C7765T, C10582T, C14408T, G15327T, A23403G, G25459A, G25563T, C27804T, C28830A | Algeria/EPI_ISL_420037 | GH | B.1 | C1059T, C3037T, C5730T, C10582T, C14408T, A23403G, G25563T |
| Algeria/EPI_ISL_766866 | GH | B.1.597 | C1059T, C3037T, T6199C, C10582T, C14408T, A23403G, G25563T, C25782T, C27804T, C28830A | Algeria/EPI_ISL_418242 | GH | B.1 | C1059T, C3037T, C10582T, C14408T, A23403G, G25563T, C29353T |
| Algeria/EPI_ISL_766873 | GH | B.1 | C1059T, C3037T, C10582T, C13335T, C14408T, A23403G, C24937T, G25563T, G25599T, A27965G | Algeria/EPI_ISL_418241 | GH | B.1 | C1059T, C3037T, C10582T, C14408T, C18115T, A23403G, G25563T, C25777T, C26461T, C29353T |
| Algeria/EPI_ISL_766867 | GH | B.1.597 | C1059T, C3037T, C5144T, T7264C, C7764T, C10279T, C10582T, G10870T, C12367T, C14408T, A23403G, G24236T, G25563T, C27804T, C28830A, C29466T | Algeria/EPI_ISL_1240721 | G | A | G23012A, A23403G |
| Algeria/EPI_ISL_766871 | GR | B.1.1 | A949G, C3037T, C14408T, G18677T, T19839C, A23403G, G28881A, G28882A, G28883C, G28903T | Algeria/EPI_ISL_1240723 | G | A | G23012A, A23403G |
| Algeria/EPI_ISL_766864 | GR | B.1.1 | C3037T, C14408T, T19839C, A23403G, G28881A, G28882A, G28883C | Algeria/EPI_ISL_1240725 | G | A | A23063T, C23271A, A23403G |
| Algeria/EPI_ISL_766869 | GR | B.1.1 | G2305T, C3037T, C14408T, C18928T, A23403G, G28881A, G28882A, G28883C | Algeria/EPI_ISL_1093430 | G | A | A23063T, C23277T, A23403G |
| Algeria/EPI_ISL_766865 | GR | B.1.1 | C3037T, C5654T, G12070T, C14267T, C14408T, T19839C, A23403G, G26501C, G28881A, G28882A, G28883C, T29023G | Algeria/EPI_ISL_1093428 | G | A | C23271A, A23403G |
| Algeria/EPI_ISL_766872 | G | B.1 | C3037T, C3619T, T6232C, C10582T, C14408T, C17550T, A23403G, T25794C, A26627G, G28774T, C28854T | Algeria/EPI_ISL_1093427 | Other | A | Del |
| Algeria/EPI_ISL_766875 | G | B.1 | C3037T, C3619T, C8097T, C14408T, C15480T, A16060C, C17550T, C19017T, A23403G, C28854T | Algeria/EPI_ISL_1240719 | Other | A | Del |
| Algeria/EPI_ISL_766870 | G | B.1 | C3037T, C3619T, C8097T, C14408T, C17550T, G19086T, A23403G, C25721T, C28854T | Algeria/EPI_ISL_1240720 | Other | A | A21717G, C21762T |
| Algeria/EPI_ISL_766863 | GH | B.1.36 | C3037T, C3619T, C11580T, C14408T, C18877T, C22444T, C22591T, A23403G, G25563T, C26735T, C28854T | Algeria/EPI_ISL_1240722 | Other | A | A21717G, C21762T |
| Algeria/EPI_ISL_766868 | G | B.1 | C3037T, C3619T, C5183T, C9430T, C10582T, C14408T, C17550T, G23383A, A23403G, G23868T, C28253T, A28254C, C28744T | Algeria/EPI_ISL_1240724 | Other | A | Del |
| Query | Gene/Amino Acid Replacement * |
|---|---|
| Algeria/EPI_ISL_766874 | NSP2_T85I, NSP3_A1179V, NSP12_P323L, NSP12_A130V, NSP12_T85A, Spike_D614G, NS3_Q57H, NS3_S40L, N_A251V |
| Algeria/EPI_ISL_766862 | NSP2_T85I, NSP3_E681D, NSP12_P323L, NSP12_M629I, Spike_D614G, NS3_Q57H, NS3_A23T, N_S186Y |
| Algeria/EPI_ISL_766866 | NSP2_T85I, NSP12_P323L, Spike_D614G, NS3_Q57H, N_S186Y |
| Algeria/EPI_ISL_766873 | NSP2_T85I, NSP10_A104V, NSP12_P323L, Spike_D614G, NS3_W69C, NS3_Q57H |
| Algeria/EPI_ISL_766867 | NSP2_T85I, NSP3_S1682F, NSP12_P323L, Spike_D614G, Spike_A892S, NS3_Q57H, N_S186Y, N_A398V |
| Algeria/EPI_ISL_766871 | NSP12_P323L, NSP14_R213L, Spike_D614G, N_M210I, N_G204R, N_R203K |
| Algeria/EPI_ISL_766864 | NSP12_P323L, Spike_D614G, N_G204R, N_R203K |
| Algeria/EPI_ISL_766869 | NSP2_K500N, NSP12_P323L, NSP14_P297S, Spike_D614G, N_G204R, N_R203K |
| Algeria/EPI_ISL_766865 | NSP12_P323L, NSP12_T276M, Spike_D614G, N_G204R, N_R203K |
| Algeria/EPI_ISL_766872 | NSP12_P323L, Spike_D614G, N_L167F, N_S194L |
| Algeria/EPI_ISL_766875 | NSP3_T1793I, NSP12_N874H, NSP12_P323L, Spike_D614G, N_S194L |
| Algeria/EPI_ISL_766870 | NSP3_T1793I, NSP12_P323L, NSP14_K349N, Spike_D614G, NS3_A110V, N_S194L |
| Algeria/EPI_ISL_766863 | NSP6_T203I, NSP12_P323L, Spike_D614G, NS3_Q57H, N_S194L |
| Algeria/EPI_ISL_766868 | NSP3_P822S, NSP12_P323L, Spike_G769V, Spike_D614G, NS8_I121L |
| Algeria/EPI_ISL_766861 | NSP12_P323L, Spike_D614G, N_S186Y |
| Algeria/EPI_ISL_420037 | NSP2_T85I, NSP3_T1004I, NSP12_P323L, Spike_D614G, NS3_Q57H |
| Algeria/EPI_ISL_418242 | NSP2_T85I, NSP12_P323L, Spike_D614G, NS3_Q57H |
| Algeria/EPI_ISL_418241 | NSP2_T85I, NSP12_P323L, NSP14_H26Y, Spike_D614G, NS3_Q57H, NS3_L129F, E_L73F |
| Algeria/EPI_ISL_1240721 | Spike_D614G, Spike_E484K |
| Algeria/EPI_ISL_1240723 | Spike_D614G, Spike_E484K |
| Algeria/EPI_ISL_1240725 | Spike_A570D, Spike_D614G, Spike_N501Y |
| Algeria/EPI_ISL_1093430 | Spike_T572I, Spike_D614G, Spike_N501Y |
| Algeria/EPI_ISL_1093428 | Spike_A570D, Spike_D614G |
| Algeria/EPI_ISL_1093427 | Spike_V70del, Spike_H69del |
| Algeria/EPI_ISL_1240719 | Spike_Y144del, Spike_V70del, Spike_H69del |
| Algeria/EPI_ISL_1240720 | Spike_Y144del, Spike_A67V, Spike_V70del, Spike_H69del, Spike_Q52R |
| Algeria/EPI_ISL_1240722 | Spike_Y144del, Spike_A67V, Spike_V70del, Spike_H69del, Spike_Q52R |
| Algeria/EPI_ISL_1240724 | Spike_Y144del, Spike_V70del, Spike_H69del |
| Mutation | PredictSNP | PhD-SNP | PolyPhen-1 | PolyPhen-2 | SIFT | SNAP |
|---|---|---|---|---|---|---|
| NSP12_A130V | 64% | 59% | 67% | 45% | 79% | 62% |
| NSP12_N874H | 72% | 58% | 74% | 60% | 79% | 62% |
| NSP3_E681D | 74% | 78% | 67% | 75% | 65% | 55% |
| NS3_A23T | 55% | 72% | 59% | 54% | 53% | 50% |
| NSP14_H26Y | 75% | 83% | 74% | 87% | 87% | 71% |
| E_L73F | 65% | 89% | 68% | 87% | 65% | 56% |
| NS3_L129F | 72% | 68% | 59% | 68% | 45% | 72% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeghbib, S.; Somogyi, B.A.; Zana, B.; Kemenesi, G.; Herczeg, R.; Derrar, F.; Jakab, F. The Algerian Chapter of SARS-CoV-2 Pandemic: An Evolutionary, Genetic, and Epidemiological Prospect. Viruses 2021, 13, 1525. https://doi.org/10.3390/v13081525
Zeghbib S, Somogyi BA, Zana B, Kemenesi G, Herczeg R, Derrar F, Jakab F. The Algerian Chapter of SARS-CoV-2 Pandemic: An Evolutionary, Genetic, and Epidemiological Prospect. Viruses. 2021; 13(8):1525. https://doi.org/10.3390/v13081525
Chicago/Turabian StyleZeghbib, Safia, Balázs A. Somogyi, Brigitta Zana, Gábor Kemenesi, Róbert Herczeg, Fawzi Derrar, and Ferenc Jakab. 2021. "The Algerian Chapter of SARS-CoV-2 Pandemic: An Evolutionary, Genetic, and Epidemiological Prospect" Viruses 13, no. 8: 1525. https://doi.org/10.3390/v13081525
APA StyleZeghbib, S., Somogyi, B. A., Zana, B., Kemenesi, G., Herczeg, R., Derrar, F., & Jakab, F. (2021). The Algerian Chapter of SARS-CoV-2 Pandemic: An Evolutionary, Genetic, and Epidemiological Prospect. Viruses, 13(8), 1525. https://doi.org/10.3390/v13081525