
Recombination Events in Putative Tail Fibre Gene in Litunavirus Phages Infecting Pseudomonas aeruginosa and Their Phylogenetic Consequences

Abstract
:1. Introduction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phage Name | Phage Origin | Phage Species | GenBank Number | Genome Size (bp) | BLASTn Results (KX171209.1 as Query) | Capsid Size (nm) | Tail Length (nm) | Tail Structures | Reference | |
|---|---|---|---|---|---|---|---|---|---|---|
| Query Coverage (%) | Sequence Identity (%) | |||||||||
| vB_Pae575P-3 | Gdańsk (Poland) | UC | KX171209.1 | 72,728 | 100 | 100.00 | 60 | 30 | nd * | [9] |
| vB_Pae1396P-5 | Gdańsk (Poland) | UC | KX171210.1 | 72,508 | 100 | 99.96 | 60 | 30 | nd * | [9] |
| PA26 | Naju City (South Korea) | Litunavirus PA26 | NC_041907.1 | 72,321 | 98 | 95.86 | - | - | - ** | [10] |
| PAP02 | Seoul (South Korea) | UC | MT080102.1 | 73,345 | 99 | 95.63 | - | - | - | [11] |
| YH30 | Beijing (China) | Litunavirus PA26 | KP994390.1 | 72,192 | 99 | 96.06 | 65 | 40 | tail fibre-like structures *** | [4] |
| vB_PaeP_PYO2 | Milan (Italy) | UC | MF490236.1 | 72,697 | 97 | 96.17 | 72 | 18 | nd | [7] |
| vB_PaeP_DEV | Milan (Italy) | Litunavirus Ab09 | MF490238.1 | 72,697 | 97 | 96.10 | 72 | 18 | nd | [7] |
| RWG | Lubbock, Texas (U.S.) | Litunavirus Ab09 | KM411958.1 | 72,646 | 94 | 97.36 | - | - | - | [8] |
| PEV2 | Olympia, Washington (U.S.) | Litunavirus Ab09 | KU948710.1 | 72,697 | 94 | 97.34 | 70 | 30 | nd | [12] |
| vB_PaeP_C2-10_Ab09 | Indénié-Djuablin (Ivory Coast) | Litunavirus Ab09 | HG962375.1 | 72,028 | 94 | 97.05 | 70 | - | nd | [13] |
| ph_P3P1 | Paris (France) | UC | LT594787.1 | 72,778 | 95 | 96.92 | 74 | - | nd | [14] |
| vB_PaeP_TUMS_P121 | Tehran (Iran) | UC | MZ955867.1 | 73,001 | 97 | 96.93 | - | - | - | [15] |
| vB_PaeS_TUMS_P81 | Tehran (Iran) | UC | OL519844.1 | 73,167 | 97 | 96.94 | - | - | - | [16] |
| DL64 | Bath (UK) | Litunavirus LIT1 | KR054032.1 | 72,378 | 94 | 96.69 | 65 | 12–13 | fibres protruding from capsid | [17] |
| YH6 | Changchun (China) | UC | KM974184.1 | 73,050 | 95 | 96.55 | 65 | 25 | nd | [5] |
| LY218 | Alexandria Creek, Alabama (U.S.) | UC | MN906996.1 | 73,083 | 95 | 96.18 | 84–85 | 10–12 | neck with collar | [18] |
| phi176 | Lubbock, Texas (U.S.) | Litunavirus Ab09 | KM411960.1 | 73,048 | 95 | 96.03 | - | - | - | [8] |
| Pa2 | Lubbock, Texas (U.S.) | Litunavirus Ab09 | NC_027345.1 | 73,008 | 96 | 96.00 | - | - | - | [8] |
| VB_PaeS_VL1 | Nakhon Pathom (Thailand) | UC | OK665488.1 | 73,308 | 97 | 95.74 | - | - | - | [19] |
| LP14 | Qingdao (China) | UC | MH356729.1 | 73,080 | 97 | 95.63 | 60 | 30 | tail fibre-like structures *** | [20] |
| vB_PaeP_MAG4 | Puławy (Poland) | UC | KR052142.1 | 72,979 | 94 | 97.59 | 63 | 36 | collar and ring-like structure with appendages | [21] |
| LIT1 | Leuven (Belgium) | Litunavirus LIT1 | FN422399.1 | 72,544 | 95 | 96.82 | 70 | 30 | nd | [12] |
| CMS1 | Stanford, California (U.S.) | UC | OM937766.1 | 72,673 | 97 | 96.11 | - | - | - | [22] |
| vB_PaeS_TUMS_P6 | Tehran (Iran) | Luzseptimavirus KPP21 | OL519842.1 | 73,885 | 17 | 85.47 | - | - | - | [23] |
| vB_PaeS_TUMS_P10 | Tehran (Iran) | Luzseptimavirus KPP21 | OM782452.1 | 74,200 | 17 | 85.60 | - | - | - | [24] |
| vB_Pae_AM.P2 | Kollam, Kerala (India) | Luzseptimavirus KPP21 | MT416090.1 | 73,308 | 17 | 85.37 | - | - | tail fibre-like structures *** | [25] |
| KPP21 | Kochi City (Japan) | Luzseptimavirus KPP21 | LC064302.1 | 73,420 | 18 | 84.91 | - | - | tail fibre-like structures *** | [26] |
| LUZ7 | Leuven (Belgium) | Luzseptimavirus LUZ7 | FN422398.1 | 74,901 | 12 | 78.82 | 70 | 30 | nd | [12] |
2. Materials and Methods
3. Results
3.1. Recombination Detection
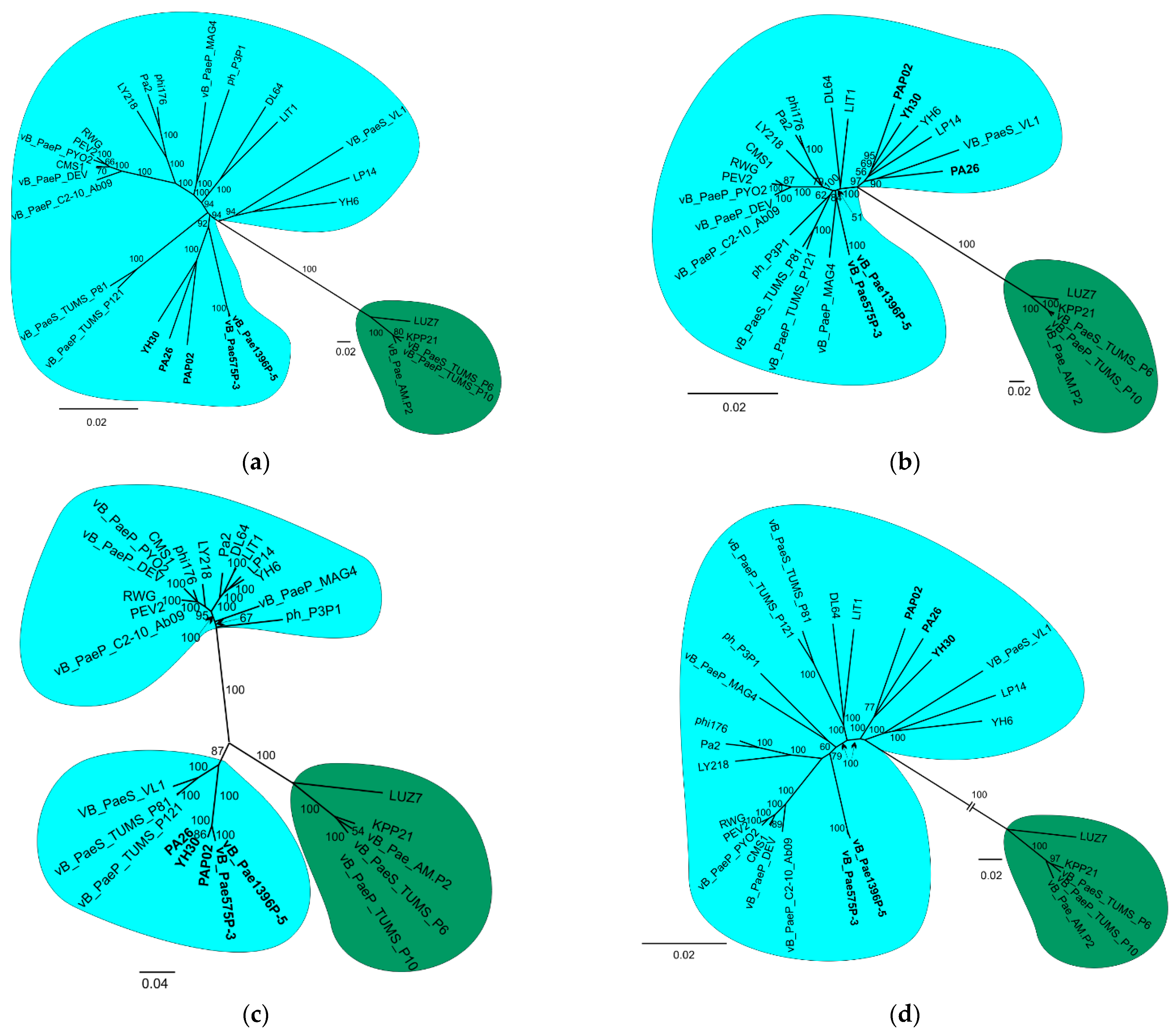
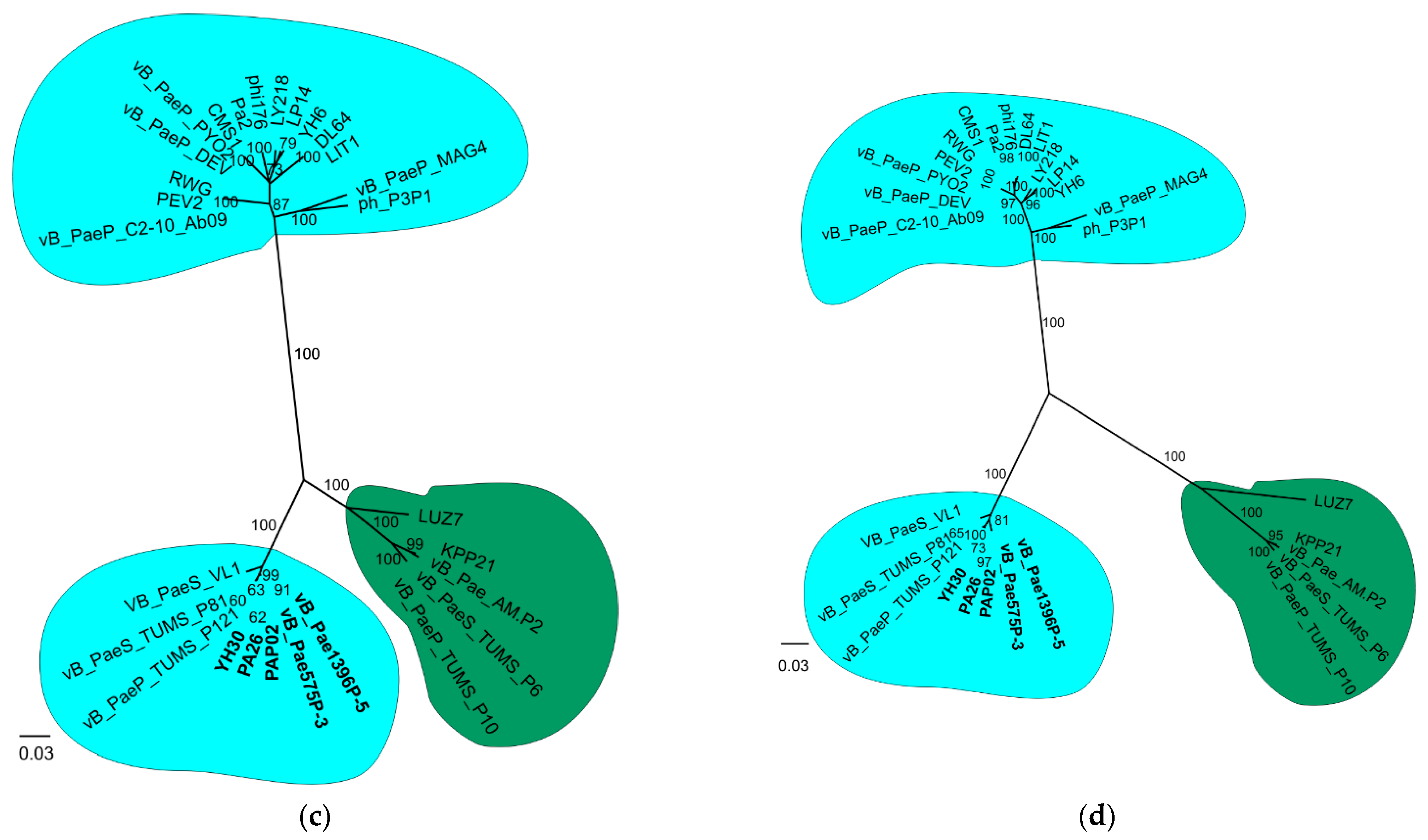
3.2. Phylogenetic Analysis
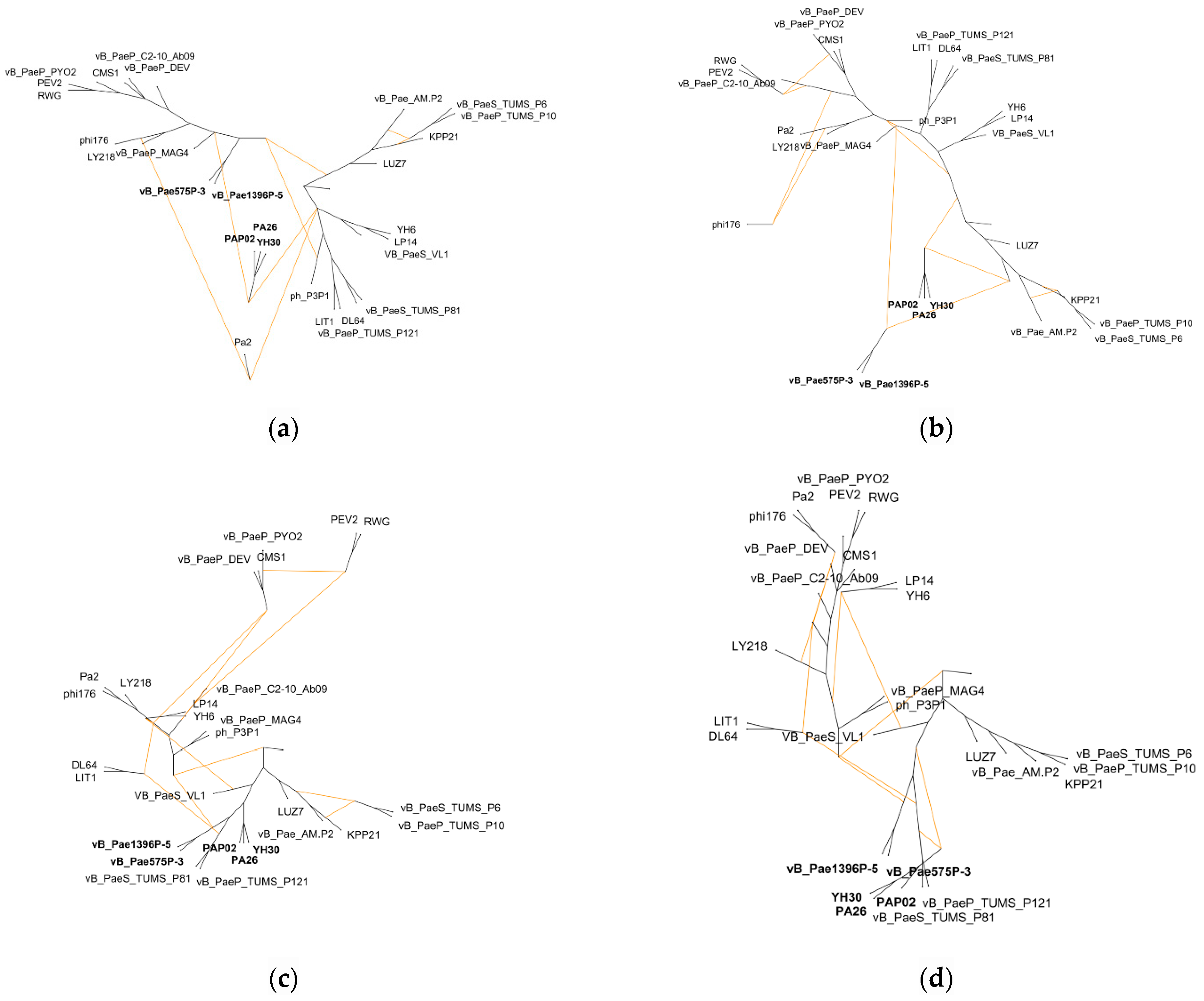
3.3. Hybridization Network
4. Discussion
4.1. Phylogenetic Position of Litunavirus Phages
4.2. Bacteriophage Species Concept
4.3. Speculations on RBS
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wittmann, J.; Turner, D.; Millard, A.D.; Mahadevan, P.; Kropinski, A.M.; Adriaenssens, E.M. From orphan phage to a proposed new family—The diversity of N4-like viruses. Antibiotics 2020, 9, 663. [Google Scholar] [CrossRef] [PubMed]
- Thi, M.T.T.; Wibowo, D.; Rehm, B.H. Pseudomonas aeruginosa biofilms. Int. J. Mol. Sci. 2020, 21, 8671. [Google Scholar] [CrossRef] [PubMed]
- Chegini, Z.; Khoshbayan, A.; Taati Moghadam, M.; Farahani, I.; Jazireian, P.; Shariati, A. Bacteriophage therapy against Pseudomonas aeruginosa biofilms: A review. Ann. Clin. Microb. Anti. 2020, 19, 1–17. [Google Scholar] [CrossRef]
- Gu, J.; Li, X.; Yang, M.; Du, C.; Cui, Z.; Gong, P.; Xia, F.; Song, J.; Zhang, L.; Li, J.; et al. Therapeutic effect of Pseudomonas aeruginosa phage YH30 on mink hemorrhagic pneumonia. Vet. Microbiol. 2016, 190, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Du, C.; Gong, P.; Xia, F.; Sun, C.; Feng, X.; Lei, L.; Song, J.; Zhang, L.; Wang, B.; et al. Therapeutic effect of the YH6 phage in a murine hemorrhagic pneumonia model. Res. Microbiol. 2015, 166, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Antoine, C.; Laforêt, F.; Blasdel, B.; Glonti, T.; Kutter, E.; Pirnay, J.P.; Mainil, J.; Delcenserie, V.; Thiry, D. Efficacy assessment of PEV2 phage on Galleria mellonella larvae infected with a Pseudomonas aeruginosa dog otitis isolate. Res. Vet. Sci. 2021, 136, 598–601. [Google Scholar] [CrossRef]
- Forti, F.; Roach, D.R.; Cafora, M.; Pasini, M.E.; Horner, D.S.; Fiscarelli, E.V.; Rossitto, M.; Cariani, L.; Briani, F.; Debarbieux, L.; et al. Design of a broad-range bacteriophage cocktail that reduces Pseudomonas aeruginosa biofilms and treats acute infections in two animal models. Antimicrob. Agents Chemother. 2018, 62, e02573-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burrowes, B.H.; Molineux, I.J.; Fralick, J.A. Directed in vitro evolution of therapeutic bacteriophages: The Appelmans protocol. Viruses 2019, 11, 241. [Google Scholar] [CrossRef] [Green Version]
- Jurczak-Kurek, A.; Gąsior, T.; Nejman-Faleńczyk, B.; Bloch, S.; Dydecka, A.; Topka, G.; Necel, A.; Jakubowska-Deredas, M.; Narajczyk, M.; Richert, M.; et al. Biodiversity of bacteriophages: Morphological and biological properties of a large group of phages isolated from urban sewage. Sci. Rep. 2016, 6, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.S.; Cha, K.E.; Myung, H. Complete genome of Pseudomonas aeruginosa phage PA26. Virol. J. 2012, 86, 10244. [Google Scholar] [CrossRef]
- Lee, J.-H.; Jeong, H.-J. Pseudomonas Phage PAP02, Complete Genome. 2020; direct submission. Available online: https://www.ncbi.nlm.nih.gov/nuccore/1846450820 (accessed on 1 July 2022).
- Ceyssens, P.J.; Brabban, A.; Rogge, L.; Lewis, M.S.; Pickard, D.; Goulding, D.; Dougan, G.; Noben, J.-P.; Kropinski, A.; Kutter, E.; et al. Molecular and physiological analysis of three Pseudomonas aeruginosa phages belonging to the “N4-like viruses”. Virology 2010, 405, 26–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Essoh, C.; Latino, L.; Midoux, C.; Blouin, Y.; Loukou, G.; Nguetta, S.P.A.; Lathro, S.; Cablanmian, A.; Kouassi, A.K.; Vergnaud, G.; et al. Investigation of a Large Collection of Pseudomonas aeruginosa Bacteriophages Collected from a Single Environmental Source in Abidjan, Côte d’Ivoire. PLoS ONE 2015, 10, e0130548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pourcel, C.; Midoux, C.; Latino, L.; Petit, M.A.; Vergnaud, G. Complete genome sequences of Pseudomonas aeruginosa phages vB_PaeP_PcyII-10_P3P1 and vB_PaeM_PcyII-10_PII10A. Genome Announc. 2016, 4, e00916-16. [Google Scholar] [CrossRef] [Green Version]
- Kamyab, H.; Torkashvand, N.; Shahverdi, A.R.; Sepehrizadeh, Z. Pseudomonas Phage vB_PaeP_TUMS_P121, Complete Genome. 2021; direct submission. Available online: https://www.ncbi.nlm.nih.gov/nuccore/2132890761 (accessed on 1 July 2022).
- Kamyab, H.; Torkashvand, N.; Shahverdi, A.R.; Sepehrizadeh, Z. Isolation, characterization and genome analysis of Pseudomonas phage vB_PaeS_TUMS_P81, a lytic bacteriophage against Pseudomonas aeruginosa. Virus Genes 2022, 1–10. [Google Scholar] [CrossRef]
- Alves, D.R.; Perez-Esteban, P.; Kot, W.; Bean, J.E.; Arnot, T.; Hansen, L.H.; Enright, M.C.; Jenkins, A.T.A. A novel bacteriophage cocktail reduces and disperses Pseudomonas aeruginosa biofilms under static and flow conditions. Microb. Biotechnol. 2016, 9, 61–74. [Google Scholar] [CrossRef] [Green Version]
- Blair, B.; Amshaqn, A.; Murdock, C. Isolation, characterization and complete genome sequence of LY218: A bacteriophage of Pseudomonas aeruginosa ATCC 27853. J. Ala. Acad. Sci. 2020, 91, 80–88. [Google Scholar]
- Lerdsittikul, V.; Apiratwarrasakul, S.; Thongdee, M. Pseudomonas Phage VB_PaeS_VL1, Complete Genome. 2021; direct submission. Available online: https://www.ncbi.nlm.nih.gov/nuccore/2164964878 (accessed on 1 July 2022).
- Shi, X.; Zhao, F.; Sun, H.; Yu, X.; Zhang, C.; Liu, W.; Pan, Q.; Ren, H. Characterization and complete genome analysis of Pseudomonas aeruginosa bacteriophage vB_PaeP_LP14 belonging to genus Litunavirus. Curr. Microbiol. 2020, 77, 2465–2474. [Google Scholar] [CrossRef]
- Kwiatek, M.; Parasion, S.; Rutyna, P.; Mizak, L.; Gryko, R.; Niemcewicz, M.; Olender, A.; Łobocka, M. Isolation of bacteriophages and their application to control Pseudomonas aeruginosa in planktonic and biofilm models. Res. Microbiol. 2017, 168, 194–207. [Google Scholar] [CrossRef]
- Faith, D.; Kinnersley, M.; Schwartzkopf, C.M.; de Mattos, C.D.; Schmidt, A.K.; Secor, P.R. Complete Genome Sequence of the N4-like Pseudomonas aeruginosa Bacteriophage vB_PaeP_CMS1. Microbiol. Resour. Announc. 2022, 11, e00239-22. [Google Scholar] [CrossRef]
- Kamyab, H.; Torkashvand, N.; Shahverdi, A.R.; Sepehrizadeh, Z. Pseudomonas Phage vB_PaeS_TUMS_P6, Complete Genome. 2021; direct submission. Available online: https://www.ncbi.nlm.nih.gov/nuccore/2162914812 (accessed on 1 July 2022).
- Kamyab, H.; Torkashvand, N.; Shahverdi, A.R.; Sepehrizadeh, Z. Pseudomonas Phage vB_PaeP_TUMS_P10, Complete Genome. 2021; direct submission. Available online: https://www.ncbi.nlm.nih.gov/nuccore/2209084086 (accessed on 1 July 2022).
- Menon, N.D.; Kumar, M.S.; Satheesh Babu, T.G.; Bose, S.; Vijayakumar, G.; Baswe, M.; Chatterjee, M.; D’Silva, J.R.; Shetty, K.; Haripriyan, J.; et al. A Novel N4-Like Bacteriophage isolated from a wastewater source in South India with activity against several multidrug-resistant clinical Pseudomonas aeruginosa isolates. Msphere 2021, 6, e01215-20. [Google Scholar] [CrossRef]
- Shigehisa, R.; Uchiyama, J.; Kato, S.I.; Takemura-Uchiyama, I.; Yamaguchi, K.; Miyata, R.; Ujihara, T.; Sakaguchi, Y.; Okamoto, N.; Shimakura, H.; et al. Characterization of Pseudomonas aeruginosa phage KPP21 belonging to family Podoviridae genus N4-like viruses isolated in Japan. Microbiol. Immunol. 2016, 60, 64–67. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Varsani, A.; Roumagnac, P.; Botha, G.; Maslamoney, S.; Schwab, T.; Kelz, Z.; Kumar, V.; Murrell, B. RDP5: A computer program for analyzing recombination in, and removing signals of recombination from, nucleotide sequence datasets. Virus Evol. 2021, 7, veaa087. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.; Rybicki, E. RDP: Detection of recombination amongst aligned sequences. Bioinformatics 2000, 16, 562–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padidam, M.; Sawyer, S.; Fauquet, C.M. Possible emergence of new geminiviruses by frequent recombination. Virology 1999, 265, 218–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, J.M. Analyzing the mosaic structure of genes. J. Mol. Evol. 1992, 34, 126–129. [Google Scholar] [CrossRef]
- Martin, D.P.; Posada, D.; Crandall, K.A.; Williamson, C. A modified bootscan algorithm for automated identification of recombinant sequences and recombination breakpoints. J. Virol. 2005, 95, 18. [Google Scholar] [CrossRef] [Green Version]
- Gibbs, M.J.; Armstrong, J.S.; Gibbs, A.J. Sister-scanning: A Monte Carlo procedure for assessing signals in recombinant sequences. Bioinformatics 2000, 16, 573–582. [Google Scholar] [CrossRef] [Green Version]
- Posada, D.; Crandall, K.A. Evaluation of methods for detecting recombination from DNA sequences: Computer simulations. Proc. Natl. Acad. Sci. USA 2001, 98, 13757–13762. [Google Scholar] [CrossRef] [Green Version]
- Lam, H.M.; Ratmann, O.; Boni, M.F. Improved algorithmic complexity for the 3SEQ recombination detection algorithm. Mol. Biol. Evol. 2018, 35, 247–251. [Google Scholar] [CrossRef] [Green Version]
- Darling, A.C.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef] [Green Version]
- Huson, D.H.; Bryant, D. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 2006, 23, 254–267. [Google Scholar] [CrossRef] [PubMed]
- Posada, D. jModelTest: Phylogenetic model averaging. Mol. Biol. Evol. 2008, 25, 1253–1256. [Google Scholar] [CrossRef] [PubMed]
- Huson, D.H.; Linz, S. Autumn Algorithm—Computation of Hybridization Networks for Realistic Phylogenetic Trees. IEEE/ACM Trans. Comput. Biol. Bioinf. 2018, 15, 398–420. [Google Scholar] [CrossRef]
- Swofford, D.L. PAUP*. Phylogenetic Analysis Using Parsimony (*and Other Methods). Version 4; Sinauer Associates: Sunderland, MA, USA, 2003. [Google Scholar]
- Dion, M.B.; Oechslin, F.; Moineau, S. Phage diversity, genomics and phylogeny. Nat. Rev. Microbiol. 2020, 18, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Gratia, J.-P. Genetic recombinational events in prokaryotes and their viruses: Insight into the study of evolution and biodiversity. Antonie Van Leeuwenhoek 2017, 110, 1493–1514. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Hawley, A.K.; Torres Beltran, M.; Scofield, M.; Schwientek, P.; Stepanauskas, R.; Woyke, T.; Hallam, S.J.; Sullivan, M.B. Ecology and evolution of viruses infecting uncultivated SUP05 bacteria as revealed by single-cell- and meta-genomics. eLife 2014, 3, e03125. [Google Scholar] [CrossRef]
- Hunter, M.; Fusco, D. Superinfection exclusion: A viral strategy with short-term benefits and long-term drawbacks. PLoS Comput. Biol. 2022, 18, e1010125. [Google Scholar] [CrossRef]
- Bobay, L.M.; Ochman, H. Biological species in the viral world. Proc. Natl. Acad. Sci. USA 2018, 115, 6040–6045. [Google Scholar] [CrossRef] [Green Version]
- Hyman, P.; van Raaij, M. Bacteriophage T4 long tail fiber domains. Biophys. Rev. 2018, 10, 463–471. [Google Scholar] [CrossRef] [Green Version]
- Holtzman, T.; Globus, R.; Molshanski-Mor, S.; Ben-Shem, A.; Yosef, I.; Qimron, U. A continuous evolution system for contracting the host range of bacteriophage T7. Sci. Rep. 2020, 10, 307. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Tu, J.; Liu, J.; Molineux, I.J. Structural Dynamics of Bacteriophage P22 Virions during the Initiation of Infection. Nat. Microbiol. 2019, 4, 1049–1056. [Google Scholar] [CrossRef]
- McPartland, J.; Rothman-Denes, L.B. The tail sheath of bacteriophage N4 interacts with the Escherichia coli receptor. J. Bacteriol. 2009, 191, 525–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perrody, E.; Cirinesi, A.M.; Desplats, C.; Keppel, F.; Schwager, F.; Tranier, S.; Georgopoulos, C.; Genevaux, P. A bacteriophage-encoded J-domain protein interacts with the DnaK/Hsp70 chaperone and stabilizes the heat-shock factor σ32 of Escherichia coli. PLoS Genet. 2012, 8, e1003037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellas, C.M.; Schroeder, D.C.; Edwards, A.; Barker, G.; Anesio, A.M. Flexible genes establish widespread bacteriophage pan-genomes in cryoconite hole ecosystems. Nat. Commun. 2020, 11, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Buth, S.A.; Shneider, M.M.; Scholl, D.; Leiman, P.G. Structure and analysis of R1 and R2 pyocin receptor-binding fibers. Viruses 2018, 10, 427. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Górniak, M.; Zalewska, A.; Jurczak-Kurek, A. Recombination Events in Putative Tail Fibre Gene in Litunavirus Phages Infecting Pseudomonas aeruginosa and Their Phylogenetic Consequences. Viruses 2022, 14, 2669. https://doi.org/10.3390/v14122669
Górniak M, Zalewska A, Jurczak-Kurek A. Recombination Events in Putative Tail Fibre Gene in Litunavirus Phages Infecting Pseudomonas aeruginosa and Their Phylogenetic Consequences. Viruses. 2022; 14(12):2669. https://doi.org/10.3390/v14122669
Chicago/Turabian StyleGórniak, Marcin, Aleksandra Zalewska, and Agata Jurczak-Kurek. 2022. "Recombination Events in Putative Tail Fibre Gene in Litunavirus Phages Infecting Pseudomonas aeruginosa and Their Phylogenetic Consequences" Viruses 14, no. 12: 2669. https://doi.org/10.3390/v14122669
APA StyleGórniak, M., Zalewska, A., & Jurczak-Kurek, A. (2022). Recombination Events in Putative Tail Fibre Gene in Litunavirus Phages Infecting Pseudomonas aeruginosa and Their Phylogenetic Consequences. Viruses, 14(12), 2669. https://doi.org/10.3390/v14122669