
Grapevine Virome of the Don Ampelographic Collection in Russia Has Concealed Five Novel Viruses

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material and Preparation of mRNA Libraries
2.2. HTS Data Analysis and Assembly of Viral Genomes
2.3. Validation of Grapevine Viruses and Viroids
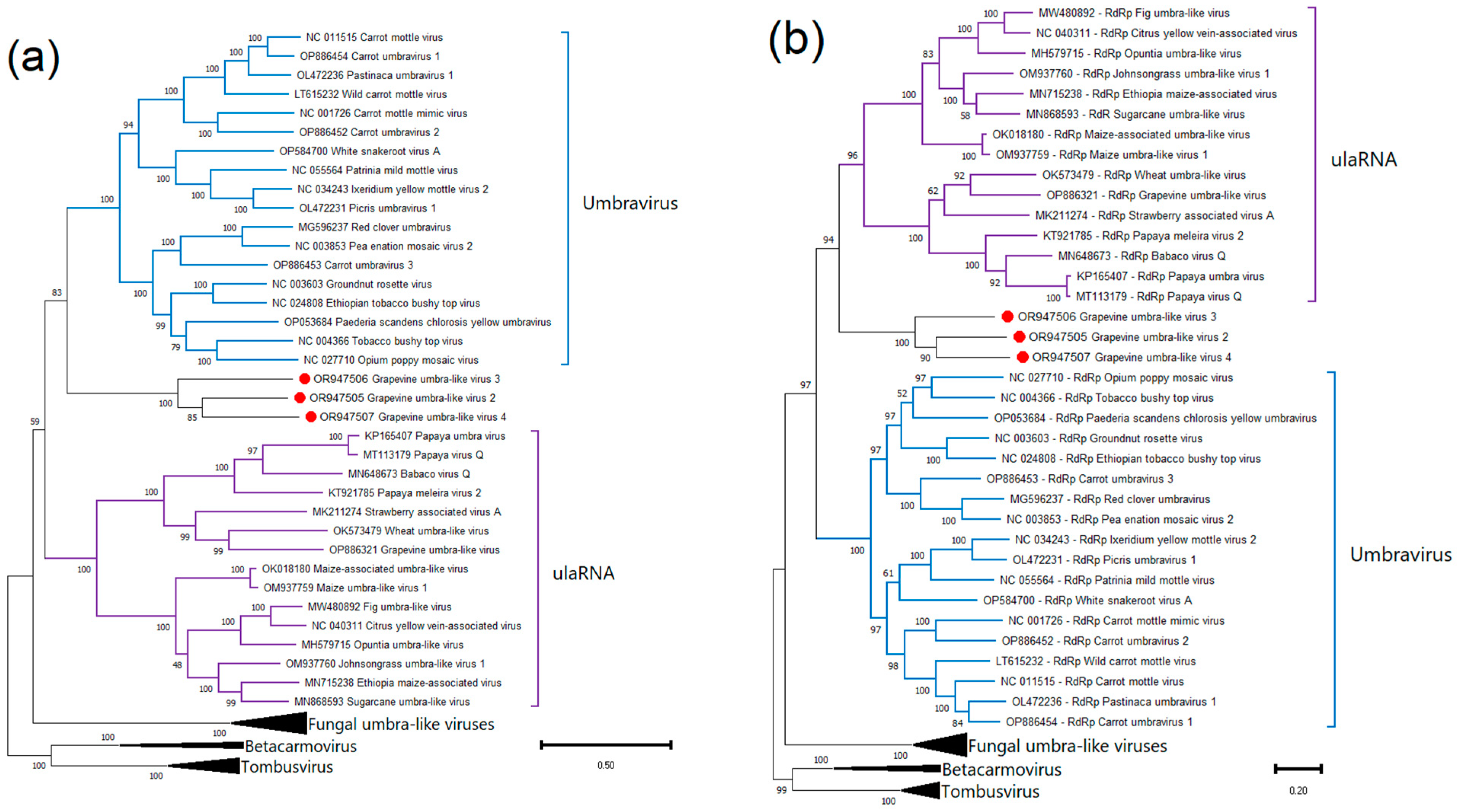
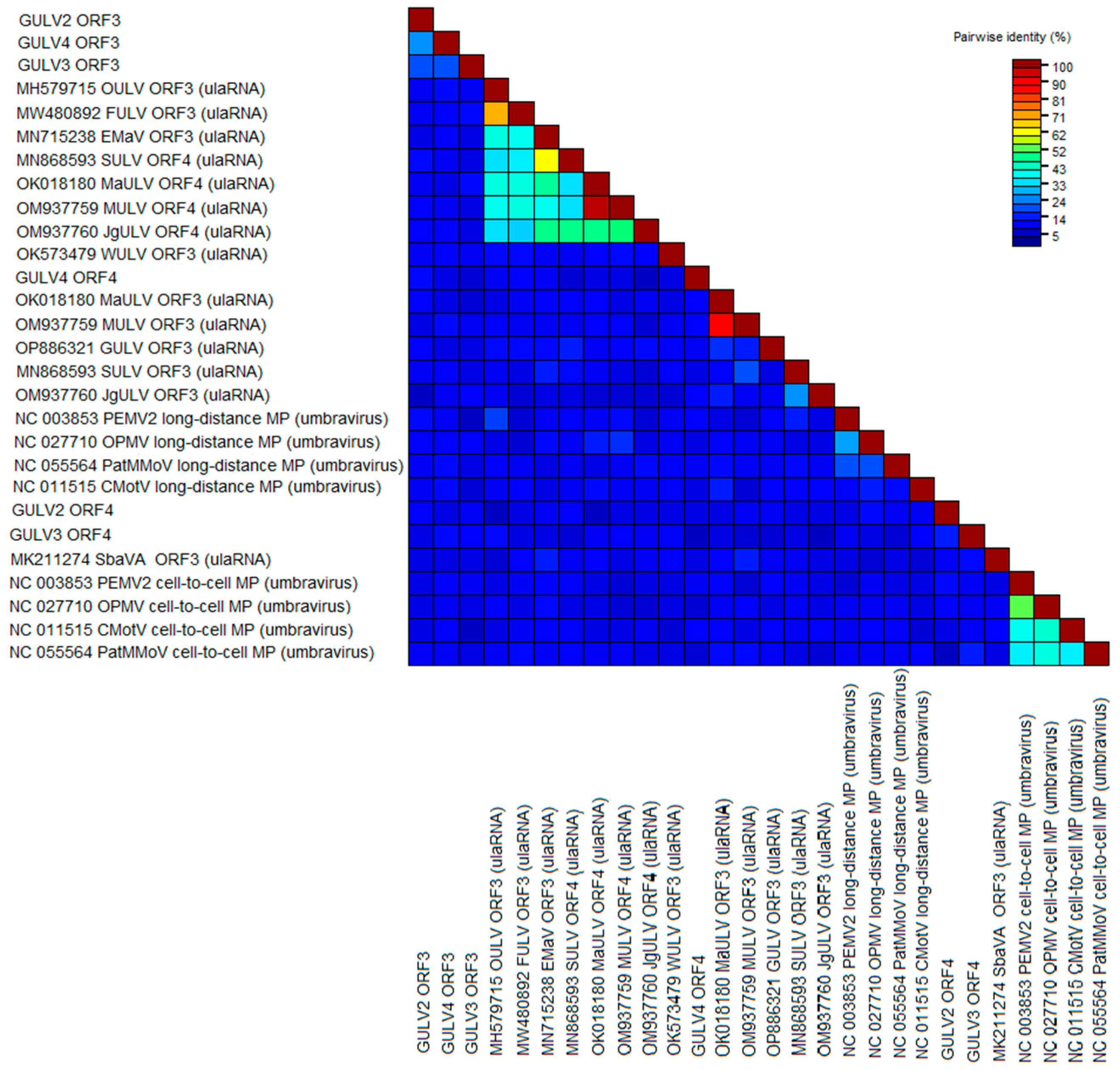
2.4. Phylogenetic Analysis and Genetic Diversity
3. Results and Discussion
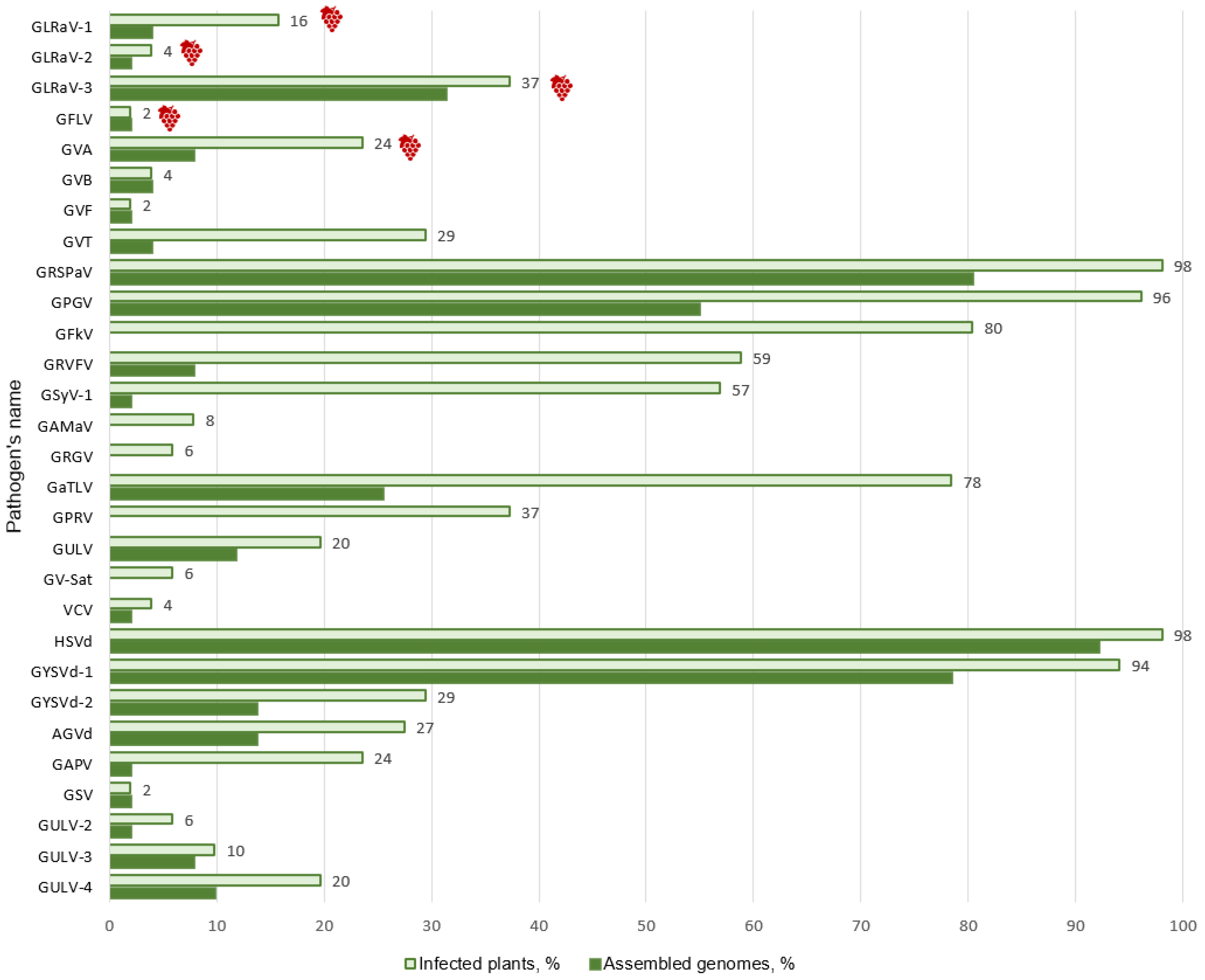
3.1. mRNA-seq Data Analysis
3.2. Identification of Known Grapevine Viruses and Viroids
3.2.1. Family: Closteroviridae
Grapevine Leafroll-Associated Virus 1
Grapevine Leafroll-Associated Virus 2
Grapevine Leafroll-Associated Virus 3
3.2.2. Family: Secoviridae
Grapevine Fanleaf Virus
3.2.3. Family: Betaflexiviridae
Grapevine Pinot Gris Virus
Grapevine Virus A
Grapevine Virus B
Grapevine Virus F
Grapevine Virus T
Grapevine Rupestris Stem Pitting-Associated Virus
3.2.4. Family: Tymoviridae
Grapevine Fleck Virus
Grapevine Red Globe Virus
Grapevine Syrah Virus 1
Grapevine Asteroid Mosaic-Associated Virus
Grapevine Rupestris Vein Feathering Virus
Grapevine-Associated Tymo-like Virus
3.2.5. Family: Partitiviridae
Vitis Cryptic Virus
3.2.6. Family: Tombusviridae
Grapevine Umbra-like Virus
3.2.7. Family: Caulimoviridae
Grapevine Pararetrovirus
3.2.8. Family: Unassigned
Grapevine Satellite Virus
3.2.9. Family: Pospiviroidae
Hop Stunt Viroid
Grapevine Yellow Speckle Viroid 1
Grapevine Yellow Speckle Viroid 2
Australian Grapevine Viroid
3.3. Detection of Novel Grapevine Viruses
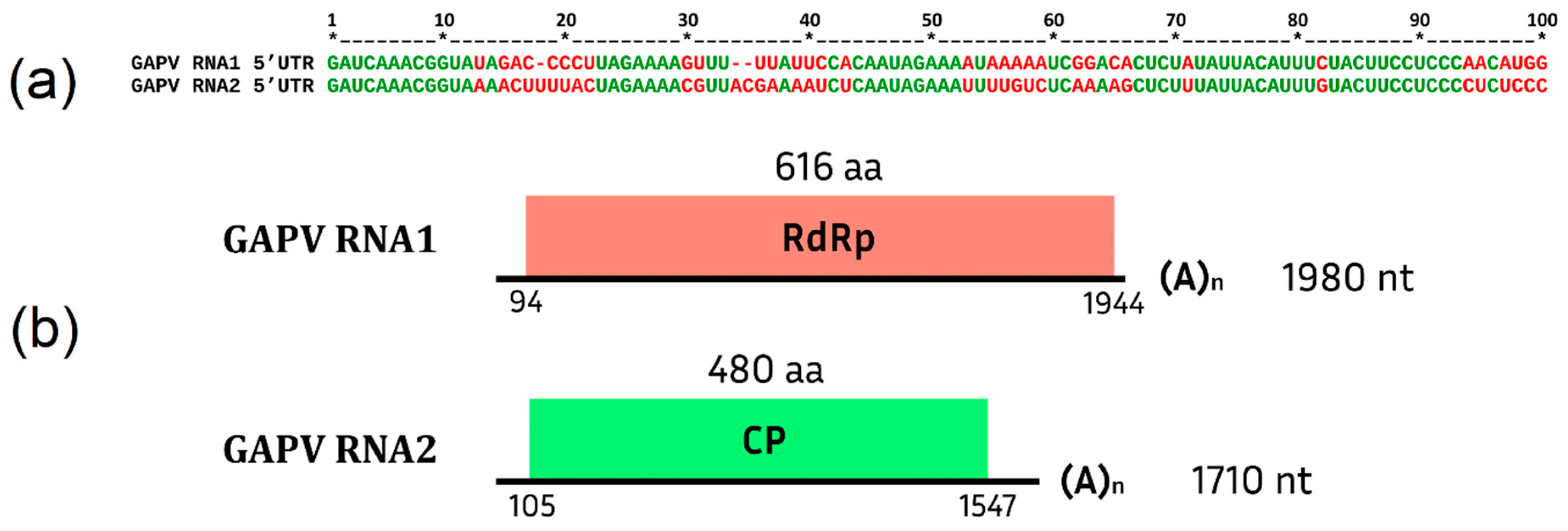
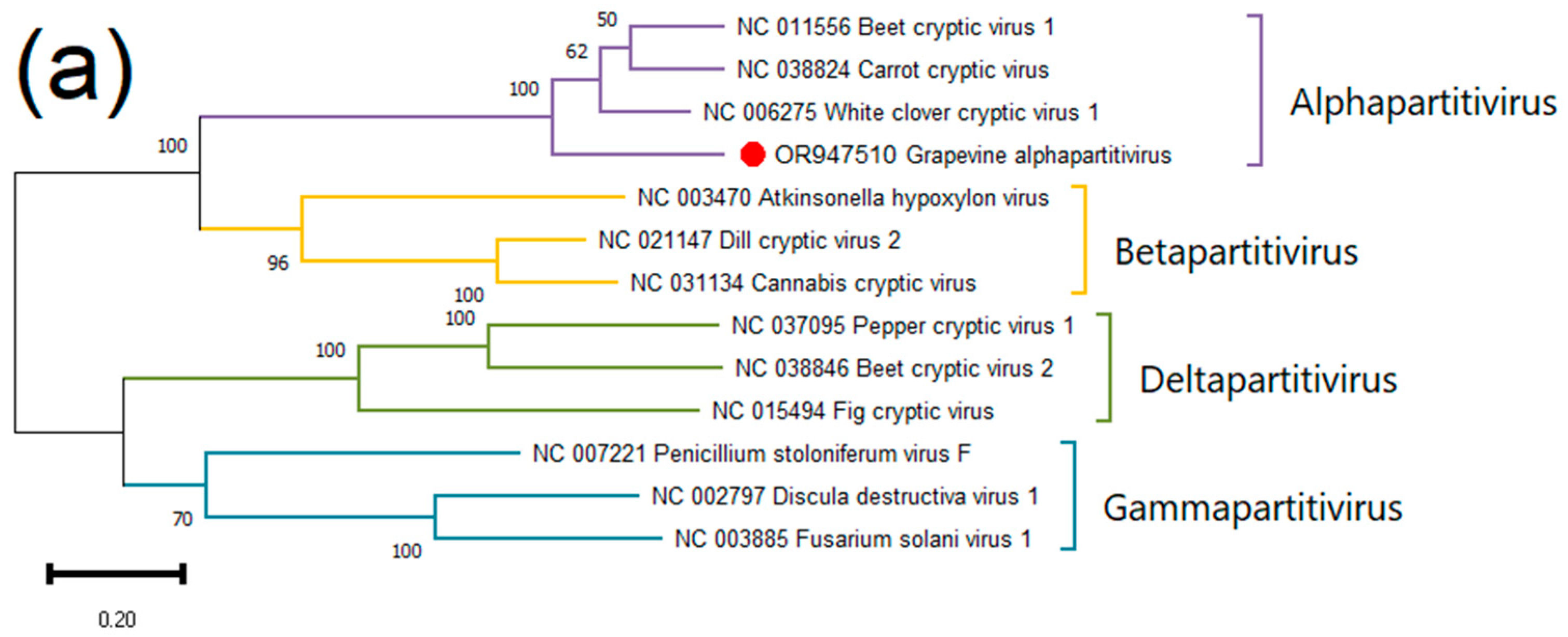
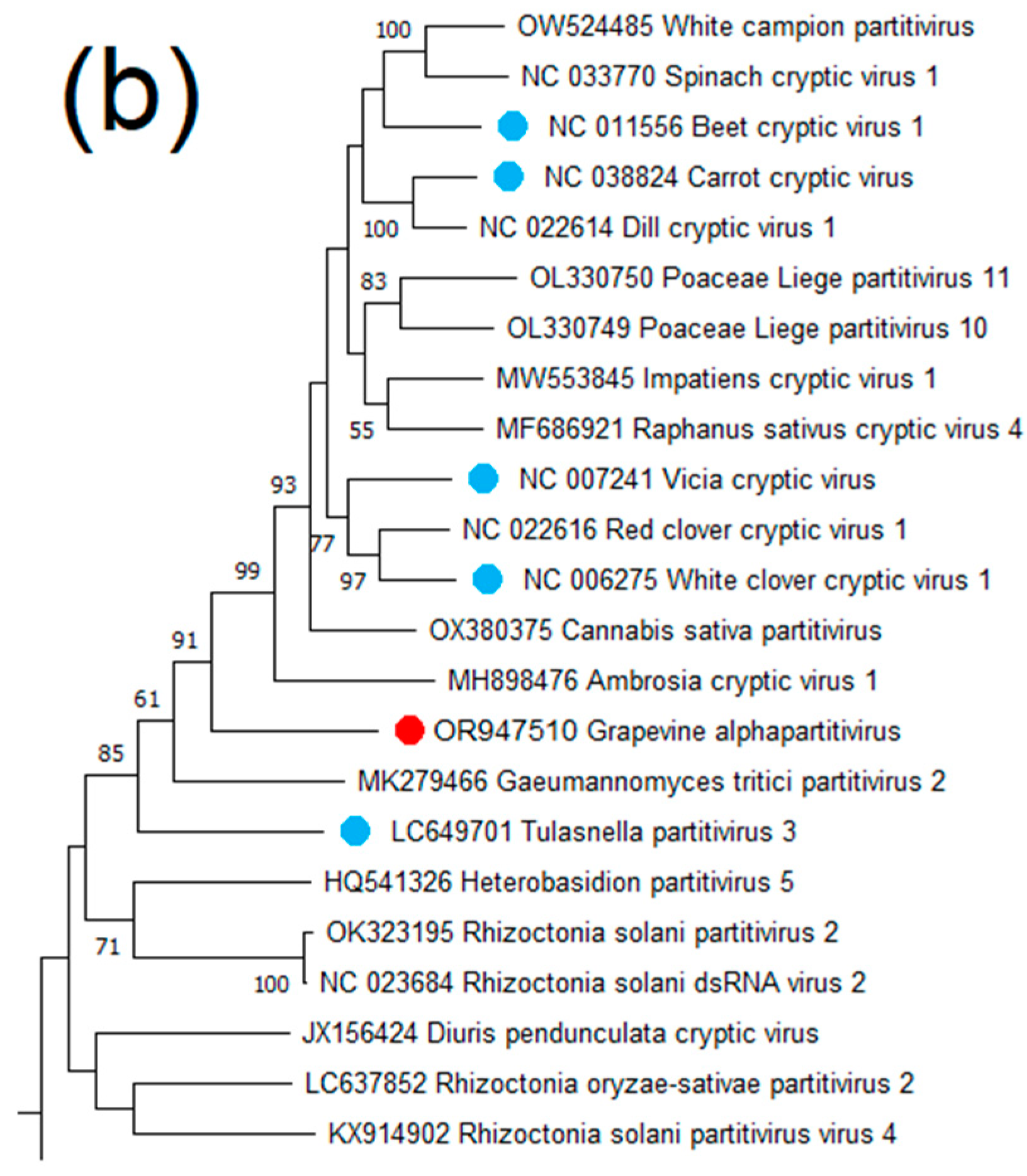
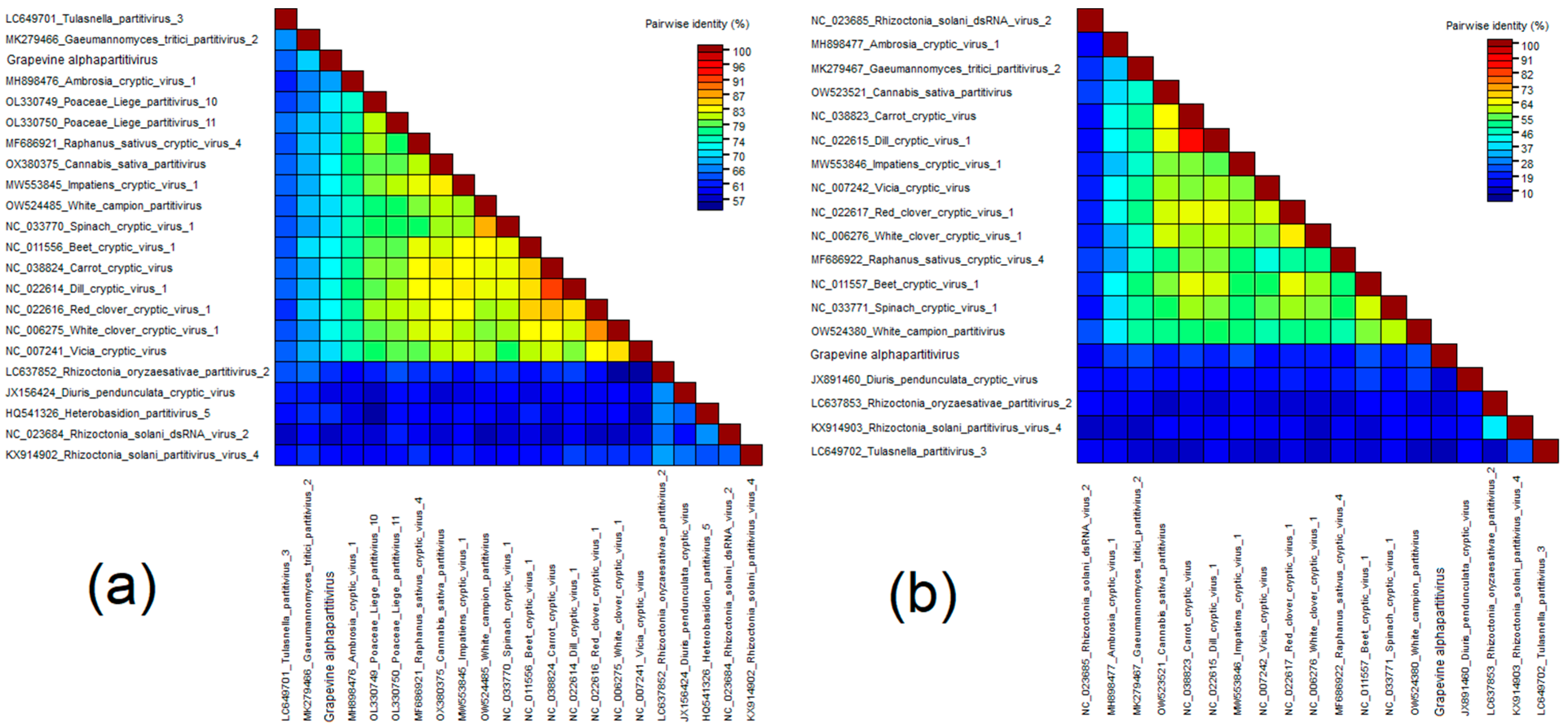
Grapevine Alphapartitivirus (GAPV)
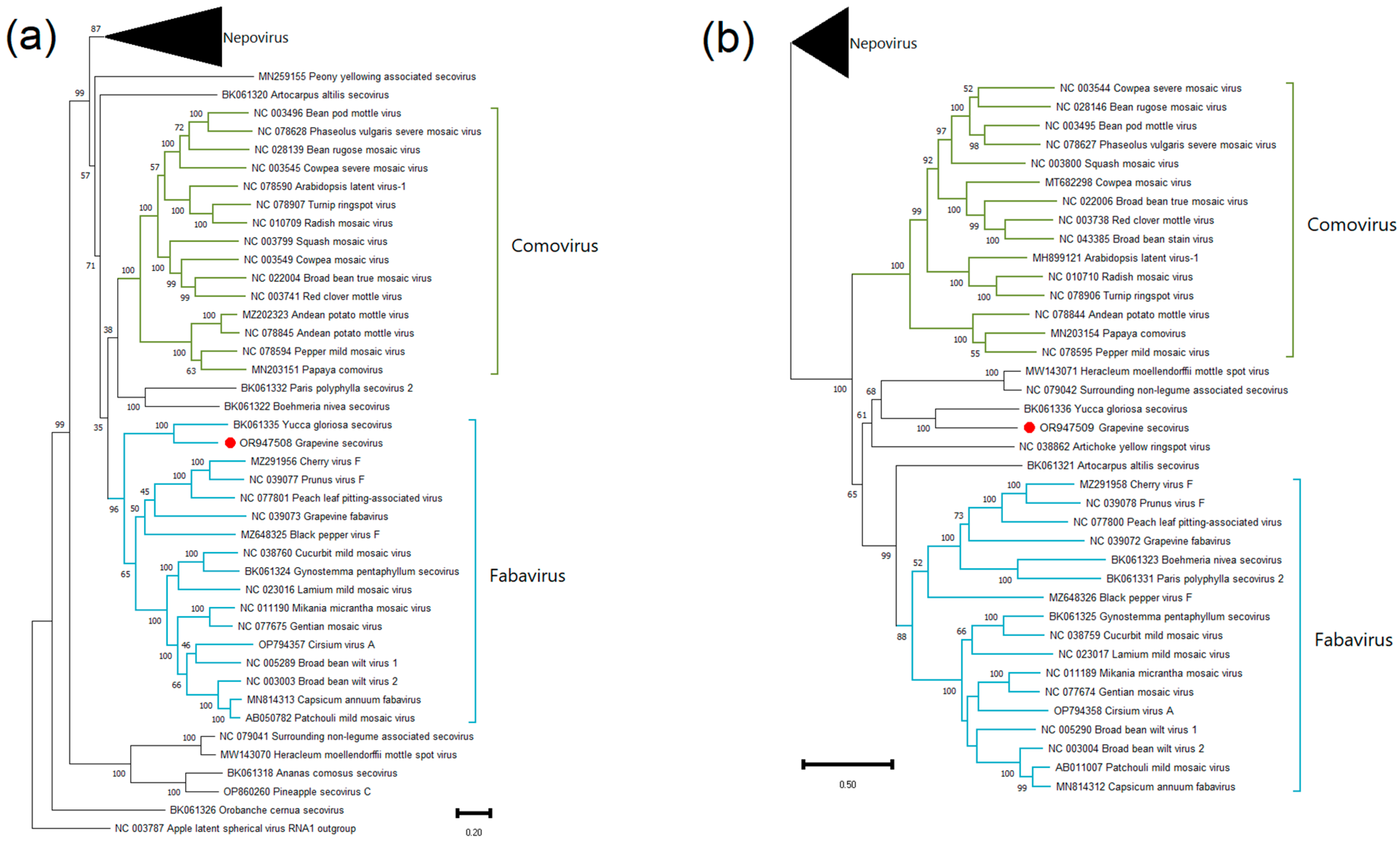
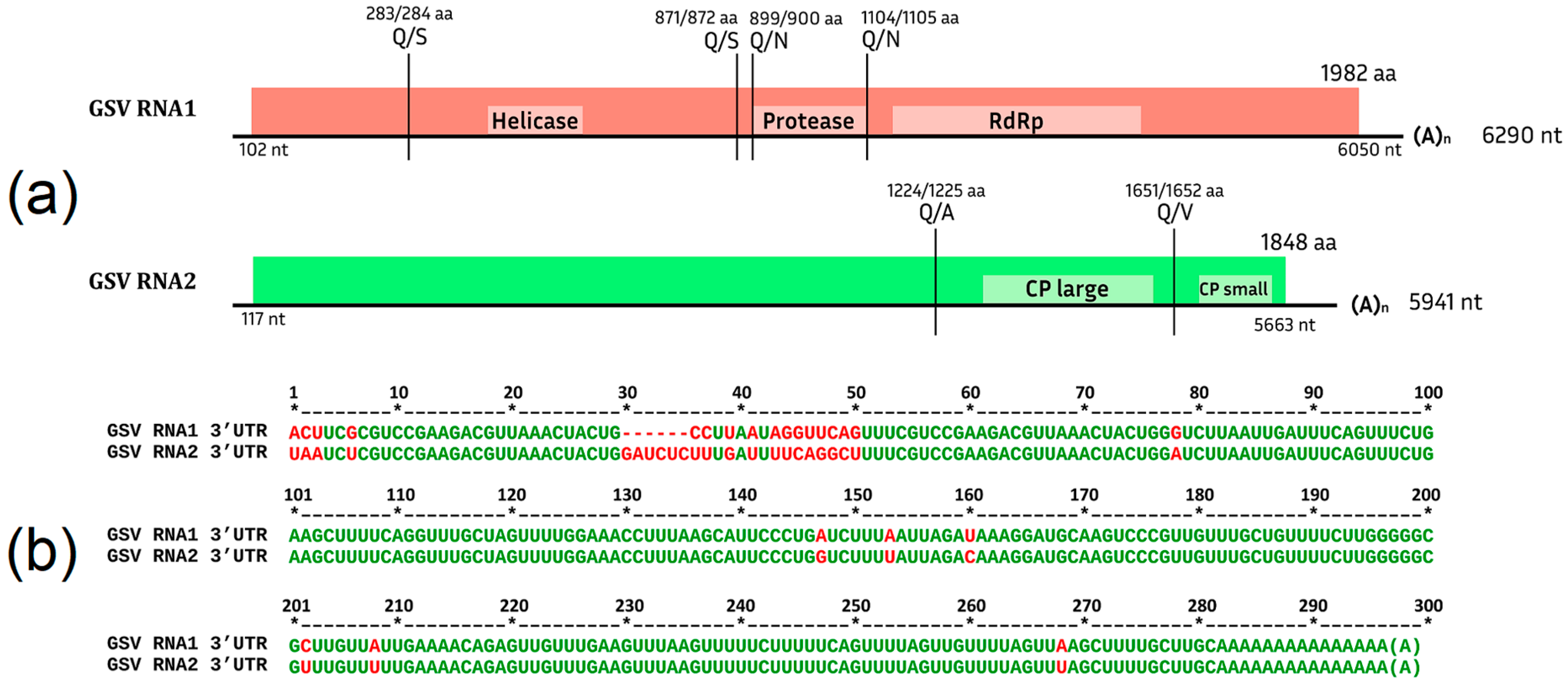
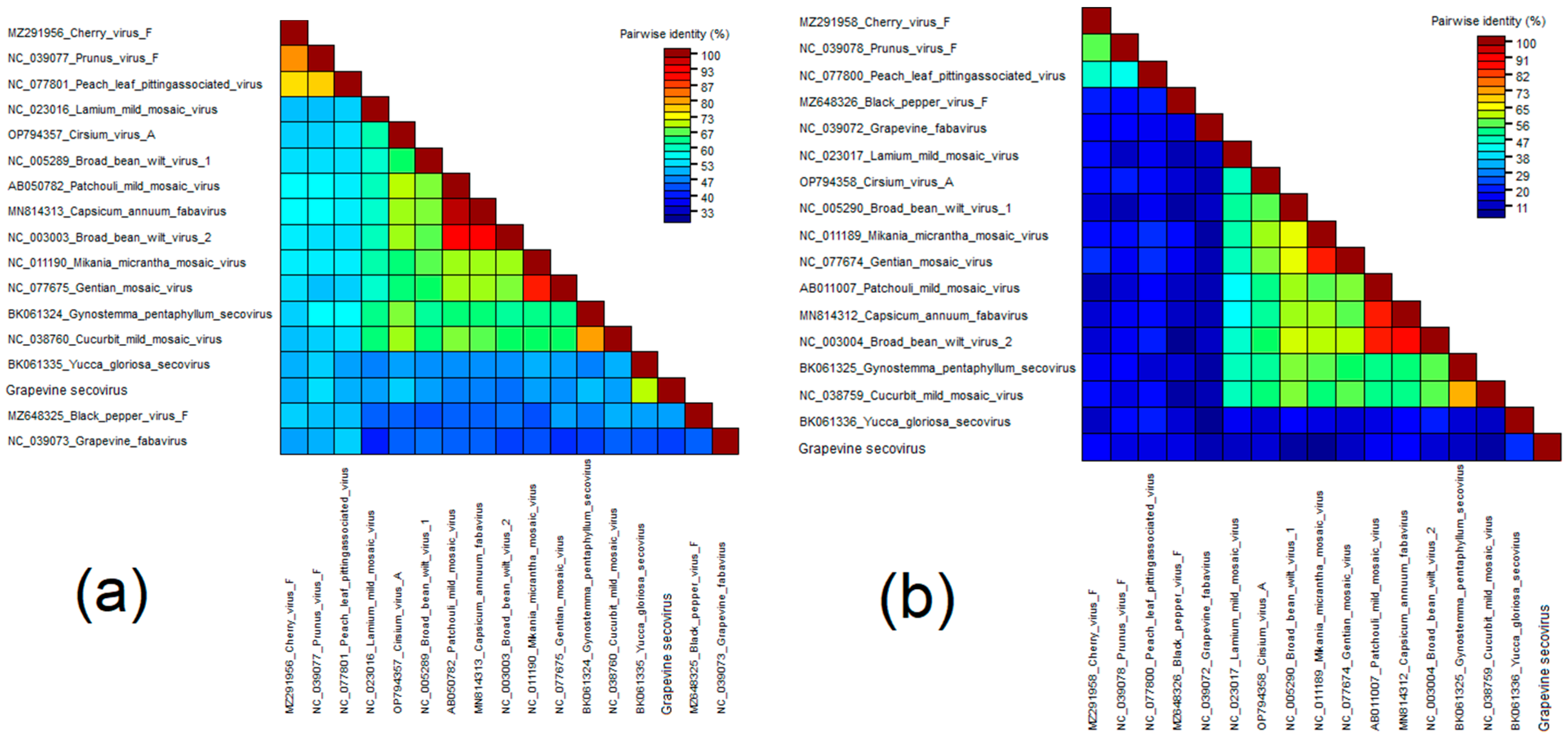
Grapevine Secovirus (GSV)
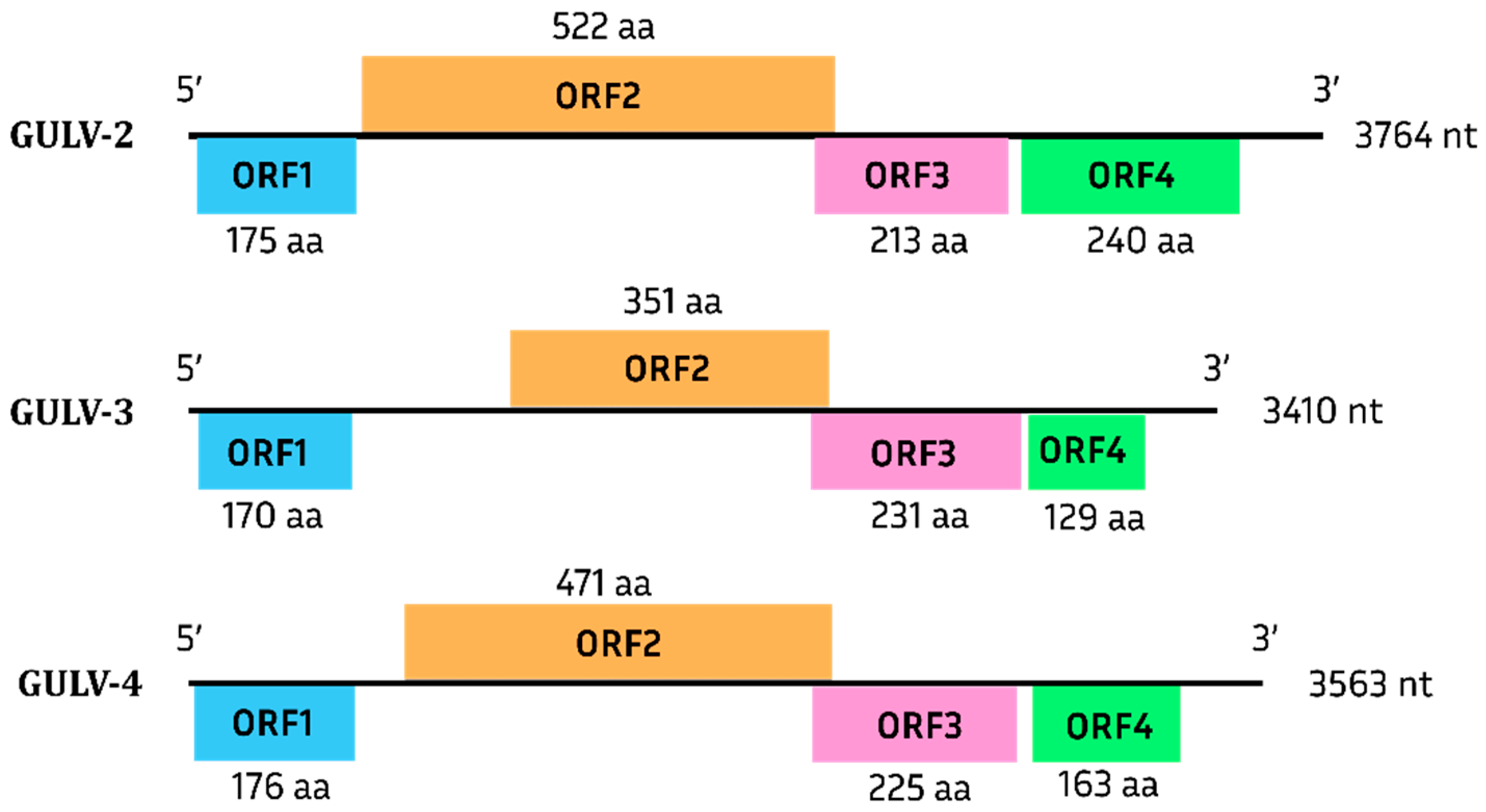
Grapevine Umbra-like Viruses
3.4. Analysis of Mixed Infections
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- International Organisation of Vine and Wine. World Statistics. Available online: https://www.oiv.int/ru/what-we-do/global-report?oiv (accessed on 16 November 2023).
- International Organisation of Vine and Wine. Country Statistics. Available online: https://www.oiv.int/what-we-do/country-report?oiv= (accessed on 16 November 2023).
- Fuchs, M. Grapevine Viruses: A Multitude of Diverse Species with Simple but Overall Poorly Adopted Management Solutions in the Vineyard. J. Plant Pathol. 2020, 102, 643–653. [Google Scholar] [CrossRef]
- Chiapello, M.; Rodríguez-Romero, J.; Nerva, L.; Forgia, M.; Chitarra, W.; Ayllón, M.A.; Turina, M. Putative New Plant Viruses Associated with Plasmopara viticola-infected Grapevine Samples. Ann. Appl. Biol. 2020, 176, 180–191. [Google Scholar] [CrossRef]
- Hu, R.; Dias, N.P.; Soltani, N.; Vargas-Asencio, J.; Hensley, D.D.; Perry, K.L.; Domier, L.L.; Hajimorad, M.R. Cultivated and Wild Grapevines in Tennessee Possess Overlapping but Distinct Virus Populations. Plant Dis. 2021, 105, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Lara, A.; Stevens, K.; Aguilar-Molina, V.H.; Fernández-Cortés, J.M.; Chabacano León, V.M.; De Donato, M.; Sharma, A.; Erickson, T.M.; Al Rwahnih, M. High-Throughput Sequencing of Grapevine in Mexico Reveals a High Incidence of Viruses Including a New Member of the Genus Enamovirus. Viruses 2023, 15, 1561. [Google Scholar] [CrossRef] [PubMed]
- Bertazzon, N.; Chitarra, W.; Angelini, E.; Nerva, L. Two New Putative Plant Viruses from Wood Metagenomics Analysis of an Esca Diseased Vineyard. Plants 2020, 9, 835. [Google Scholar] [CrossRef]
- Katsarou, K.; Andronis, C.; James, A.; Euthymiou, K.; Kryovrysanaki, N.; Pappi, P.G.; Kalantidis, K. Complete Genome Sequence of a Carlavirus Identified in Grapevine (Vitis sp.) in Greece. Arch. Virol. 2023, 168, 172. [Google Scholar] [CrossRef]
- Al Rwahnih, M.; Alabi, O.J.; Hwang, M.S.; Tian, T.; Mollov, D.; Golino, D. Characterization of a New Nepovirus Infecting Grapevine. Plant Dis. 2021, 105, 1432–1439. [Google Scholar] [CrossRef]
- Dahan, J.; Orellana, G.E.; Lee, J.; Karasev, A.V. Grapevine Endophyte Endornavirus and Two New Endornaviruses Found Associated with Grapevines (Vitis vinifera L.) in Idaho, USA. Viruses 2023, 15, 1347. [Google Scholar] [CrossRef]
- Reynard, J.-S.; Brodard, J.; Remoliff, E.; Lefebvre, M.; Schumpp, O.; Candresse, T. A Novel Foveavirus Identified in Wild Grapevine (Vitis vinifera subsp. Sylvestris). Arch. Virol. 2020, 165, 2999–3002. [Google Scholar] [CrossRef]
- Read, D.A.; Thompson, G.D.; Cordeur, N.L.; Swanevelder, D.; Pietersen, G. Genomic Characterization of Grapevine Viruses N and O: Novel Vitiviruses from South Africa. Arch. Virol. 2022, 167, 611–614. [Google Scholar] [CrossRef]
- Fan, X.; Zhang, Z.; Li, C.; Ren, F.; Hu, G.; Zhang, B.; Dong, Y. High-Throughput Sequencing Indicates a Novel Marafivirus in Grapevine Showing Vein-Clearing Symptoms. Plants 2021, 10, 1487. [Google Scholar] [CrossRef]
- Basso, M.F.; Fajardo, T.V.M.; Saldarelli, P. Grapevine Virus Diseases: Economic Impact and Current Advances in Viral Prospection and Management. Rev. Bras. Frutic. 2017, 39, 1–12. [Google Scholar] [CrossRef]
- Fraile, A.; McLeish, M.J.; Pagán, I.; González-Jara, P.; Piñero, D.; García-Arenal, F. Environmental Heterogeneity and the Evolution of Plant-Virus Interactions: Viruses in Wild Pepper Populations. Virus Res. 2017, 241, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Roossinck, M.J. Deep Sequencing for Discovery and Evolutionary Analysis of Plant Viruses. Virus Res. 2017, 239, 82–86. [Google Scholar] [CrossRef] [PubMed]
- Massart, S.; Adams, I.; Al Rwahnih, M.; Baeyen, S.; Bilodeau, G.J.; Blouin, A.G.; Boonham, N.; Candresse, T.; Chandellier, A.; De Jonghe, K.; et al. Guidelines for the Reliable Use of High Throughput Sequencing Technologies to Detect Plant Pathogens and Pests. Peer Community J. 2022, 2, e62. [Google Scholar] [CrossRef]
- Tarquini, G.; Dall’Ara, M.; Ermacora, P.; Ratti, C. Traditional Approaches and Emerging Biotechnologies in Grapevine Virology. Viruses 2023, 15, 826. [Google Scholar] [CrossRef]
- Aou-Ouad, H.E.; Montero, R.; Baraza, E.; Bota, J. Recovering Ancient Grapevine Cultivars in the Balearic Islands: Sanitary Status Evaluation and Virus Elimination. Plants 2022, 11, 1754. [Google Scholar] [CrossRef] [PubMed]
- Read, D.A.; Thompson, G.D.; Swanevelder, D.Z.H.; Pietersen, G. Metaviromic Characterization of Betaflexivirus Populations Associated with a Vitis Cultivar Collection in South Africa. Viruses 2023, 15, 1474. [Google Scholar] [CrossRef]
- Shvets, D.; Porotikova, E.; Sandomirsky, K.; Vinogradova, S. Virome of Grapevine Germplasm from the Anapa Ampelographic Collection (Russia). Viruses 2022, 14, 1314. [Google Scholar] [CrossRef]
- Shvets, D.; Sandomirsky, K.; Porotikova, E.; Vinogradova, S. Metagenomic Analysis of Ampelographic Collections of Dagestan Revealed the Presence of Two Novel Grapevine Viruses. Viruses 2022, 14, 2623. [Google Scholar] [CrossRef]
- Maliogka, V.I.; Martelli, G.P.; Fuchs, M.; Katis, N.I. Control of Viruses Infecting Grapevine. In Advances in Virus Research; Academic Press Inc.: Cambridge, MA, USA, 2015; Volume 91, pp. 175–227. [Google Scholar]
- Don Ampelographic Collection Named after Ya. I. Potapenko. Available online: https://rusvine.ru/ (accessed on 16 November 2023).
- Ganich, V.; Naumova, L. Autochthonous Georgian and Dagestan Grapevine Varieties on the Collection in the Rostov Region. In Proceedings of the E3S Web of Conferences, Divnomorskoe, Russia, 4 December 2020; EDP Sciences: Les Ulis, France, 2020; Volume 210. [Google Scholar] [CrossRef]
- Naumova, L.G.; Ganich, V.A.; Matveeva, N.V. Uvological Evaluation of Don Aborigenous Grapes Varieties at the Don Ampelographic Collection Named After Ya. I. Potapenko. Pomic. Small Fruits Cult. Russ. 2019, 59, 152–161. [Google Scholar] [CrossRef]
- Morante-Carriel, J.; Sellés-Marchart, S.; Martínez-Márquez, A.; Martínez-Esteso, M.J.; Luque, I.; Bru-Martínez, R. RNA Isolation from Loquat and Other Recalcitrant Woody Plants with High Quality and Yield. Anal. Biochem. 2014, 452, 46–53. [Google Scholar] [CrossRef]
- Casali, N.; Preston, A.; Swords, W.E. Chemical Transformation of E. Coli. In E. coli Plasmid Vectors: Methods and Applications, Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2003; pp. 49–53. [Google Scholar]
- Paysan-Lafosse, T.; Blum, M.; Chuguransky, S.; Grego, T.; Pinto, B.L.; Salazar, G.A.; Bileschi, M.L.; Bork, P.; Bridge, A.; Colwell, L.; et al. InterPro in 2022. Nucleic Acids Res. 2023, 51, D418–D427. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Muhire, B.M.; Varsani, A.; Martin, D.P. SDT: A Virus Classification Tool Based on Pairwise Sequence Alignment and Identity Calculation. PLoS ONE 2014, 9, e108277. [Google Scholar] [CrossRef]
- Martelli, G.P. An Overview on Grapevine Viruses, Viroids, and the Diseases They Cause. In Grapevine Viruses: Molecular Biology, Diagnostics and Management; Springer International Publishing: Berlin/Heidelberg, Germany, 2017; pp. 31–46. ISBN 9783319577067. [Google Scholar]
- Naidu, R.A. Grapevine Leafroll-Associated Virus 1. In Grapevine Viruses: Molecular Biology, Diagnostics and Management; Springer International Publishing: Berlin/Heidelberg, Germany, 2017; pp. 127–139. ISBN 9783319577067. [Google Scholar]
- Reynard, J.S.; Brodard, J.; Zufferey, V.; Rienth, M.; Gugerli, P.; Schumpp, O.; Blouin, A.G. Nuances of Responses to Two Sources of Grapevine Leafroll Disease on Pinot Noir Grown in the Field for 17 Years. Viruses 2022, 14, 1333. [Google Scholar] [CrossRef]
- Alabi, O.J.; Rwahnih, M.A.; Karthikeyan, G.; Poojari, S.; Fuchs, M.; Rowhani, A.; Naidu, R.A. Grapevine Leafroll-Associated Virus 1 Occurs as Genetically Diverse Populations. Phytopathology 2011, 101, 1446–1456. [Google Scholar] [CrossRef] [PubMed]
- Sabella, E.; Pierro, R.; Luvisi, A.; Panattoni, A.; D’Onofrio, C.; Scalabrelli, G.; Nutricati, E.; Aprile, A.; De Bellis, L.; Materazzi, A. Phylogenetic Analysis of Viruses in Tuscan Vitis vinifera sylvestris (Gmeli) Hegi. PLoS ONE 2018, 13, e0200875. [Google Scholar] [CrossRef]
- Caruso, A.G.; Bertacca, S.; Ragona, A.; Matić, S.; Davino, S.; Panno, S. Epidemiological Survey of Grapevine LeafrollAssociated Virus 1 and 3 in Sicily (Italy): Genetic Structure and Molecular Variability. Agriculture 2022, 12, 647. [Google Scholar] [CrossRef]
- Morán, F.; Olmos, A.; Glasa, M.; Silva, M.B.D.; Maliogka, V.; Wetzel, T.; Ruiz-García, A.B. A Novel and Highly Inclusive Quantitative Real-Time RT-PCR Method for the Broad and Efficient Detection of Grapevine Leafroll-Associated Virus 1. Plants 2023, 12, 876. [Google Scholar] [CrossRef]
- Angelini, E.; Aboughanem-Sabanadzovic, N.; Dolja, V.V.; Meng, B. Grapevine Leafroll-Associated Virus 2. In Grapevine Viruses: Molecular Biology, Diagnostics and Management; Springer International Publishing: Berlin/Heidelberg, Germany, 2017; pp. 141–165. ISBN 9783319577067. [Google Scholar]
- Habili, N.; Wu, Q.; Rinaldo, A.; Constable, F. A Chronological Study on Grapevine Leafroll-Associated Virus 2 in Australia. Viruses 2023, 15, 1105. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Dong, Y.F.; Zhang, Z.P.; Ren, F.; Hu, G.; Zhou, J. Detection and Sequence Analysis of Grapevine Leafroll-Associated Virus 2 Isolates from China. J. Phytopathol. 2015, 163, 978–986. [Google Scholar] [CrossRef]
- Bertazzon, N.; Borgo, M.; Vanin, S.; Angelini, E. Genetic Variability and Pathological Properties of Grapevine Leafroll-Associated Virus 2 Isolates. Eur. J. Plant Pathol. 2010, 127, 185–197. [Google Scholar] [CrossRef]
- Bertazzon, N.; Borgo, M.; Angelini, E. The Complete Genome Sequence of the BD Variant of Grapevine Leafroll-Associated Virus 2. Arch. Virol. 2010, 155, 1717–1719. [Google Scholar] [CrossRef] [PubMed]
- Poojari, S.; Alabi, O.J.; Naidu, R.A. Molecular Characterization and Impacts of a Strain of Grapevine Leafroll-Associated Virus 2 Causing Asymptomatic Infection in a Wine Grape Cultivar. Virol. J. 2013, 10, 324. [Google Scholar] [CrossRef]
- Song, Y.; Hanner, R.H.; Meng, B. Transcriptomic Analyses of Grapevine Leafroll-Associated Virus 3 Infection in Leaves and Berries of ‘Cabernet Franc’. Viruses 2022, 14, 1831. [Google Scholar] [CrossRef] [PubMed]
- Habili, N.; Little, A.; Essling, M.; Rinaldo, A. Grapevine Leafroll-Associated Virus 3 and Its Management Strategies in Vineyards. Wine Vitic. J. 2022, 38, 34–40. [Google Scholar]
- Naidu, R.A.; Maree, H.J.; Burger, J.T. Grapevine Leafroll Disease and Associated Viruses: A Unique Pathosystem. Annu. Rev. Phytopathol. 2015, 53, 613–634. [Google Scholar] [CrossRef]
- Gouveia, P.; Santos, M.T.; Eiras-Dias, J.E.; Nolasco, G. Five Phylogenetic Groups Identified in the Coat Protein Gene of Grapevine Leafroll-Associated Virus 3 Obtained from Portuguese Grapevine Varieties. Arch. Virol. 2011, 156, 413–420. [Google Scholar] [CrossRef]
- Maree, H.J.; Pirie, M.D.; Oosthuizen, K.; Bester, R.; Jasper, D.; Rees, G.; Burger, J.T. Phylogenomic Analysis Reveals Deep Divergence and Recombination in an Economically Important Grapevine Virus. PLoS ONE 2015, 10, e0126819. [Google Scholar] [CrossRef]
- Diaz-Lara, A.; Klaassen, V.; Stevens, K.; Sudarshana, M.R.; Rowhani, A.; Maree, H.J.; Chooi, K.M.; Blouin, A.G.; Habili, N.; Song, Y.; et al. Characterization of Grapevine Leafroll-Associated Virus 3 Genetic Variants and Application towards RT-QPCR Assay Design. PLoS ONE 2018, 13, e0208862. [Google Scholar] [CrossRef] [PubMed]
- Thompson, B.D.; Dahan, J.; Lee, J.; Martin, R.R.; Karasev, A.V. A Novel Genetic Variant of Grapevine Leafroll-Associated Virus-3 (GLRaV-3) from Idaho Grapevines. Plant Dis. 2019, 103, 509–518. [Google Scholar] [CrossRef] [PubMed]
- Navrotskaya, E.; Porotikova, E.; Yurchenko, E.; Galbacs, Z.N.; Varallyay, E.; Vinogradova, S. High-Throughput Sequencing of Small RNAs for Diagnostics of Grapevine Viruses and Viroids in Russia. Viruses 2021, 13, 2432. [Google Scholar] [CrossRef] [PubMed]
- Kubina, J.; Hily, J.M.; Mustin, P.; Komar, V.; Garcia, S.; Martin, I.R.; Poulicard, N.; Velt, A.; Bonnet, V.; Mercier, L.; et al. Characterization of Grapevine Fanleaf Virus Isolates in ‘Chardonnay’ Vines Exhibiting Severe and Mild Symptoms in Two Vineyards. Viruses 2022, 14, 2303. [Google Scholar] [CrossRef] [PubMed]
- Andret-Link, P.; Schmitt-Keichinger, C.; Demangeat, G.; Komar, V.; Fuchs, M. The Specific Transmission of Grapevine Fanleaf Virus by Its Nematode Vector Xiphinema Index Is Solely Determined by the Viral Coat Protein. Virology 2004, 320, 12–22. [Google Scholar] [CrossRef]
- Roy, B.G.; DeBlasio, S.; Yang, Y.; Thannhauser, T.; Heck, M.; Fuchs, M. Profiling Plant Proteome and Transcriptome Changes during Grapevine Fanleaf Virus Infection. J. Proteome Res. 2023, 22, 1997–2017. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, M.; Lemaire, O. Novel Approaches for Viral Disease Management. In Grapevine Viruses: Molecular Biology, Diagnostics and Management; Springer International Publishing: Cham, Switzerland, 2017; pp. 599–621. [Google Scholar]
- Djennane, S.; Prado, E.; Dumas, V.; Demangeat, G.; Gersch, S.; Alais, A.; Gertz, C.; Beuve, M.; Lemaire, O.; Merdinoglu, D. A Single Resistance Factor to Solve Vineyard Degeneration Due to Grapevine Fanleaf Virus. Commun. Biol. 2021, 4, 637. [Google Scholar] [CrossRef]
- Giampetruzzi, A.; Roumi, V.; Roberto, R.; Malossini, U.; Yoshikawa, N.; La Notte, P.; Terlizzi, F.; Credi, R.; Saldarelli, P. A New Grapevine Virus Discovered by Deep Sequencing of Virus- and Viroid-Derived Small RNAs in Cv Pinot Gris. Virus Res. 2012, 163, 262–268. [Google Scholar] [CrossRef]
- Bianchi, G.L.; De Amicis, F.; De Sabbata, L.; Di Bernardo, N.; Governatori, G.; Nonino, F.; Prete, G.; Marrazzo, T.; Versolatto, S.; Frausin, C. Occurrence of Grapevine Pinot Gris Virus in Friuli Venezia Giulia (Italy): Field Monitoring and Virus Quantification by Real-Time RT-PCR. EPPO Bull. 2015, 45, 22–32. [Google Scholar] [CrossRef]
- Shvets, D.; Vinogradova, S. Occurrence and Genetic Characterization of Grapevine Pinot Gris Virus in Russia. Plants 2022, 11, 1061. [Google Scholar] [CrossRef]
- Vu, M.; Vemulapati, B.M.; McFadden-Smith, W.; Fall, M.L.; Úrbez-Torres, J.R.; Moreau, D.L.; Poojari, S. Phylogenetic and Evolutionary Studies of Grapevine Pinot Gris Virus Isolates from Canada. Viruses 2023, 15, 735. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Habili, N.; Constable, F.; Al Rwahnih, M.; Goszczynski, D.E.; Wang, Y.; Pagay, V. Virus Pathogens in Australian Vineyards with an Emphasis on Shiraz Disease. Viruses 2020, 12, 818. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Habili, N.; Kinoti, W.M.; Tyerman, S.D.; Rinaldo, A.; Zheng, L.; Constable, F.E. A Metagenomic Investigation of the Viruses Associated with Shiraz Disease in Australia. Viruses 2023, 15, 774. [Google Scholar] [CrossRef] [PubMed]
- Goszczynski, D.E. Single-Strand Conformation Polymorphism (SSCP), Cloning and Sequencing Reveal a Close Association between Related Molecular Variants of Grapevine Virus A (GVA) and Shiraz Disease in South Africa. Plant Pathol. 2007, 56, 755–762. [Google Scholar] [CrossRef]
- Goszczynski, D.E.; du Preez, J.; Burger, J.T. Molecular Divergence of Grapevine Virus A (GVA) Variants Associated with Shiraz Disease in South Africa. Virus Res. 2008, 138, 105–110. [Google Scholar] [CrossRef]
- Goszczynski, D.E. Complete Genome Sequence of a Natural Mutant of Grapevine Virus A (GVA). Arch. Virol. 2014, 159, 2523–2528. [Google Scholar] [CrossRef]
- Alabi, O.J.; Al Rwahnih, M.; Mekuria, T.A.; Naidu, R.A. Genetic Diversity of Grapevine Virus a in Washington and California Vineyards. Phytopathology 2014, 104, 548–560. [Google Scholar] [CrossRef]
- Goszczynski, D.E. Divergent Molecular Variants of Grapevine Virus B (GVB) from Corky Bark (CB)-Affected and CB-Negative LN33 Hybrid Grapevines. Virus Genes 2010, 41, 273–281. [Google Scholar] [CrossRef]
- Chitarra, W.; Cuozzo, D.; Ferrandino, A.; Secchi, F.; Palmano, S.; Perrone, I.; Boccacci, P.; Pagliarani, C.; Gribaudo, I.; Mannini, F.; et al. Dissecting Interplays between Vitis vinifera L. and Grapevine Virus B (GVB) under Field Conditions. Mol. Plant Pathol. 2018, 19, 2651–2666. [Google Scholar] [CrossRef]
- Hu, G.J.; Dong, Y.F.; Zhang, Z.P.; Fan, X.D.; Fang, R.; Zhu, H.J. Detection and Sequence Analysis of Grapevine Virus B Isolates from China. Acta Virol. 2014, 58, 180–184. [Google Scholar] [CrossRef]
- Fonseca, F.; Duarte, V.; Teixeira Santos, M.; Brazão, J.; Eiras-Dias, E. First Molecular Characterization of Grapevine Virus B (GVB) in Portuguese Grapevine Cultivars and Improvement of the RT-PCR Detection Assay. Arch. Virol. 2016, 161, 3535–3540. [Google Scholar] [CrossRef] [PubMed]
- Goszczynski, D.E. The Identification of a New Genetic Variant of Grapevine Virus B. J. Plant Pathol. 2018, 100, 105–109. [Google Scholar] [CrossRef]
- Al Rwahnih, M.; Sudarshana, M.R.; Uyemoto, J.K.; Rowhani, A. Complete Genome Sequence of a Novel Vitivirus Isolated from Grapevine. J. Virol. 2012, 86, 9545. [Google Scholar] [CrossRef]
- Jo, Y.; Song, M.K.; Choi, H.; Park, J.S.; Lee, J.W.; Lian, S.; Lee, B.C.; Cho, W.K. Genome Sequence of Grapevine Virus T, a Novel Foveavirus Infecting Grapevine. Genome Announc. 2017, 5, e00995-17. [Google Scholar] [CrossRef] [PubMed]
- Zarghani, S.N.; Hily, J.M.; Glasa, M.; Marais, A.; Wetzel, T.; Faure, C.; Vigne, E.; Velt, A.; Lemaire, O.; Boursiquot, J.M.; et al. Grapevine Virus T Diversity as Revealed by Full-Length Genome Sequences Assembled from High-Throughput Sequence Data. PLoS ONE 2018, 13, e0206010. [Google Scholar] [CrossRef]
- Demian, E.; Holczbauer, A.; Galbacs, Z.N.; Jaksa-Czotter, N.; Turcsan, M.; Olah, R.; Varallyay, E. Variable Populations of Grapevine Virus t Are Present in Vineyards of Hungary. Viruses 2021, 13, 1119. [Google Scholar] [CrossRef] [PubMed]
- Meng, B.; Martelli, G.P.; Golino, D.A.; Fuchs, M. Grapevine Rupestris Stem Pittingassociated Virus. In Grapevine Viruses: Molecular Biology, Diagnostics and Management; Springer International Publishing: Berlin/Heidelberg, Germany, 2017; pp. 257–287. ISBN 9783319577067. [Google Scholar]
- Hily, J.M.; Beuve, M.; Vigne, E.; Demangeat, G.; Candresse, T.; Lemaire, O. A Genome-Wide Diversity Study of Grapevine Rupestris Stem Pitting-Associated Virus. Arch. Virol. 2018, 163, 3105–3111. [Google Scholar] [CrossRef]
- Alabi, O.J.; Martin, R.R.; Naidu, R.A. Sequence Diversity, Population Genetics and Potential Recombination Events in Grapevine Rupestris Stem Pitting-Associated Virus in Pacific North-West Vineyards. J. Gen. Virol. 2010, 91, 265–276. [Google Scholar] [CrossRef]
- Porotikova, E.; Terehova, U.; Volodin, V.; Yurchenko, E.; Vinogradova, S. Distribution and Genetic Diversity of Grapevine Viruses in Russia. Plants 2021, 10, 1080. [Google Scholar] [CrossRef]
- Beuve, M.; Moury, B.; Spilmont, A.S.; Sempé-Ignatovic, L.; Hemmer, C.; Lemaire, O. Viral Sanitary Status of Declining Grapevine Syrah Clones and Genetic Diversity of Grapevine Rupestris Stem Pitting-Associated Virus. Eur. J. Plant Pathol. 2013, 135, 439–452. [Google Scholar] [CrossRef]
- Hu, G.J.; Dong, Y.F.; Zhu, H.J.; Zhang, Z.P.; Fan, X.D.; Ren, F.; Zhou, J. Molecular Characterizations of Two Grapevine Rupestris Stem Pitting-Associated Virus Isolates from China. Arch. Virol. 2015, 160, 2641–2645. [Google Scholar] [CrossRef]
- Mostert, I.; Burger, J.T.; Maree, H.J. Genetic Diversity and Identification of Putative Recombination Events in Grapevine Rupestris Stem Pitting-Associated Virus. Arch. Virol. 2018, 163, 2491–2496. [Google Scholar] [CrossRef] [PubMed]
- Sabanadzovic, S.; Aboughanem-Sabanadzovic, N.; Martelli, G.P. Grapevine Fleck and Similar Viruses. In Grapevine Viruses: Molecular Biology, Diagnostics and Management; Springer International Publishing: Berlin/Heidelberg, Germany, 2017; pp. 331–349. ISBN 9783319577067. [Google Scholar]
- Candresse, T.; Faure, C.; Marais, A. First Report of Grapevine Red Globe Virus (GRGV) and Grapevine Rupestris Vein Feathering Virus (GRVFV) Infecting Grapevine (Vitis vinifera) in Portugal. Plant Dis. 2023, 107, 974. [Google Scholar] [CrossRef] [PubMed]
- Cretazzo, E.; Velasco, L. High-Throughput Sequencing Allowed the Completion of the Genome of Grapevine Red Globe Virus and Revealed Recurring Co-Infection with Other Tymoviruses in Grapevine. Plant Pathol. 2017, 66, 1202–1213. [Google Scholar] [CrossRef]
- Al Rwahnih, M.; Daubert, S.; Golino, D.; Rowhani, A. Deep Sequencing Analysis of RNAs from a Grapevine Showing Syrah Decline Symptoms Reveals a Multiple Virus Infection That Includes a Novel Virus. Virology 2009, 387, 395–401. [Google Scholar] [CrossRef] [PubMed]
- Glasa, M.; Predajňa, L.; Šoltys, K.; Sabanadzovic, S.; Olmos, A. Detection and Molecular Characterisation of Grapevine Syrah Virus-1 Isolates from Central Europe. Virus Genes 2015, 51, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.R.; Gomez, A.L.; Younas, A.; González-Tobón, J.; Cha, A.; Perry, K.L. Grapevine Asteroid Mosaic-Associated Virus Is Resident and Prevalent in Wild, Noncultivated Grapevine of New York State. Plant Dis. 2021, 105, 758–763. [Google Scholar] [CrossRef] [PubMed]
- Hily, J.M.; Candresse, T.; Garcia, S.; Vigne, E.; Tannière, M.; Komar, V.; Barnabé, G.; Alliaume, A.; Gilg, S.; Hommay, G.; et al. High-Throughput Sequencing and the Viromic Study of Grapevine Leaves: From the Detection of Grapevine-Infecting Viruses to the Description of a New Environmental Tymovirales Member. Front. Microbiol. 2018, 9, 1782. [Google Scholar] [CrossRef]
- Dahan, J.; Orellana, G.E.; Lee, J.; Karasev, A.V. Occurrence of grapevine-associated tymo-like virus in wine grapes in the United States. Plant Dis. 2023, 107, 2. [Google Scholar] [CrossRef]
- Vu, M.; McFadden-Smith, W.; Poojari, S. Monitoring the Spread of Grapevine Viruses in Vineyards of Contrasting Agronomic Practices: A Metagenomic Investigation. Biology 2023, 12, 1279. [Google Scholar] [CrossRef]
- Nabeshima, T.; Abe, J. High-Throughput Sequencing Indicates Novel Varicosavirus, Emaravirus, and Deltapartitivirus Infections in Vitis Coignetiae. Viruses 2021, 13, 827. [Google Scholar] [CrossRef]
- Fan, X.D.; Dong, Y.F.; Zhang, Z.P.; Ren, F.; Hu, G.J. First Report of Vitis Cryptic Virus from Grapevines in China. Plant Dis. 2022, 106, 3006. [Google Scholar] [CrossRef]
- Rwahnih, M.A.; Daubert, S.; Sudarshana, M.R.; Rowhani, A. Gene from a Novel Plant Virus Satellite from Grapevine Identifies a Viral Satellite Lineage. Virus Genes 2013, 47, 114–118. [Google Scholar] [CrossRef]
- Candresse, T.; Marais, A.; Theil, S.; Faure, C.; Lacombe, T.; Boursiquot, J.M. Complete Nucleotide Sequence of an Isolate of Grapevine Satellite Virus and Evidence for the Presence of Multimeric Forms in an Infected Grapevine. Genome Announc. 2017, 5, e01703-16. [Google Scholar] [CrossRef] [PubMed]
- Czotter, N.; Molnar, J.; Szabó, E.; Demian, E.; Kontra, L.; Baksa, I.; Szittya, G.; Kocsis, L.; Deak, T.; Bisztray, G.; et al. NGS of Virus-Derived Small RNAs as a Diagnostic Method Used to Determine Viromes of Hungarian Vineyards. Front. Microbiol. 2018, 9, 122. [Google Scholar] [CrossRef]
- Miljanić, V.; Jakše, J.; Beber, A.; Rusjan, D.; Škvarč, A.; Štajner, N. First Report of Grapevine Satellite Virus in Slovenia. J. Plant Pathol. 2021, 103, 1329–1330. [Google Scholar] [CrossRef]
- Maddahian, M.; Massumi, H.; Heydarnejad, J.; Hosseinipour, A.; Khezri, A.; Sano, T. Biological and Molecular Characterization of Hop Stunt Viroid Variants from Pistachio Trees in Iran. J. Phytopathol. 2019, 167, 163–173. [Google Scholar] [CrossRef]
- Alaxin, P.; Predajňa, L.; Achs, A.; Šubr, Z.; Mrkvová, M.; Glasa, M. Analysis of Hop Stunt Viroid Diversity in Grapevine (Vitis vinifera L.) in Slovakia: Coexistence of Two Particular Genetic Groups. Pathogens 2023, 12, 205. [Google Scholar] [CrossRef] [PubMed]
- Marquez-Molins, J.; Gomez, G.; Pallas, V. Hop Stunt Viroid: A Polyphagous Pathogenic RNA That Has Shed Light on Viroid–Host Interactions. Mol. Plant Pathol. 2021, 22, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi-Ito, Y.; Li, S.F.; Tagawa, M.; Araki, H.; Goshono, M.; Yamamoto, S.; Tanaka, M.; Narita, M.; Tanaka, K.; Liu, S.X.; et al. Cultivated Grapevines Represent a Symptomless Reservoir for the Transmission of Hop Stunt Viroid to Hop Crops: 15 Years of Evolutionary Analysis. PLoS ONE 2009, 4, e8386. [Google Scholar] [CrossRef]
- Di Serio, F.; Izadpanah, K.; Hajizadeh, M.; Navarro, B. Viroids Infecting the Grapevine. In Grapevine Viruses: Molecular Biology, Diagnostics and Management; Springer International Publishing: Cham, Switzerland, 2017; pp. 373–392. [Google Scholar]
- Gambino, G.; Navarro, B.; Torchetti, E.M.; La Notte, P.; Schneider, A.; Mannini, F.; Di Serio, F. Survey on Viroids Infecting Grapevine in Italy: Identification and Characterization of Australian Grapevine Viroid and Grapevine Yellow Speckle Viroid 2. Eur. J. Plant Pathol. 2014, 140, 199–205. [Google Scholar] [CrossRef]
- Salman, T.M.; Habili, N.; Shi, B. Effect of Temperature on Symptom Expression and Sequence Polymorphism of Grapevine Yellow Speckle Viroid 1 in Grapevine. Virus Res. 2014, 189, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Fajardo, T.V.M.; Eiras, M.; Nickel, O. Detection and Molecular Characterization of Grapevine Yellow Speckle Viroid 1 Isolates Infecting Grapevines in Brazil. Trop. Plant Pathol. 2016, 41, 246–253. [Google Scholar] [CrossRef]
- Rezaian, M.A.; Koltunow, A.M.; Krake, L.R. Isolation of Three Viroids and a Circular RNA from Grapevines. J. Gen. Virol. 1988, 69 Pt 2, 413–422. [Google Scholar] [CrossRef]
- Blawid, R.; Stephan, D.; Maiss, E. Molecular Characterization and Detection of Vicia Cryptic Virus in Different Vicia Faba Cultivars. Arch. Virol. 2007, 152, 1477–1488. [Google Scholar] [CrossRef] [PubMed]
- Sahin, E.; Keskin, E.; Akata, I. Molecular Characterization of the Complete Genome of a Novel Partitivirus Hosted by the Saprobic Mushroom Leucocybe Candicans. Arch. Microbiol. 2021, 203, 5825–5830. [Google Scholar] [CrossRef] [PubMed]
- Nibert, M.L.; Ghabrial, S.A.; Maiss, E.; Lesker, T.; Vainio, E.J.; Jiang, D.; Suzuki, N. Taxonomic Reorganization of Family Partitiviridae and Other Recent Progress in Partitivirus Research. Virus Res. 2014, 188, 128–141. [Google Scholar] [CrossRef]
- Vainio, E.J.; Chiba, S.; Ghabrial, S.A.; Maiss, E.; Roossinck, M.; Sabanadzovic, S.; Suzuki, N.; Xie, J.; Nibert, M. ICTV Virus Taxonomy Profile: Partitiviridae. J. Gen. Virol. 2018, 99, 17–18. [Google Scholar] [CrossRef]
- Nerva, L.; Varese, G.C.; Falk, B.W.; Turina, M. Mycoviruses of an Endophytic Fungus Can Replicate in Plant Cells: Evolutionary Implications. Sci. Rep. 2017, 7, 1908. [Google Scholar] [CrossRef]
- Nakatsukasa-Akune, M.; Yamashita, K.; Shimoda, Y.; Uchiumi, T.; Abe, M.; Aoki, T.; Kamizawa, A.; Ayabe, S.; Higashi, S.; Suzuki, A. Suppression of Root Nodule Formation by Artificial Expression of the TrEnodDR1 (Coat Protein of White Clover Cryptic Virus 1) Gene in Lotus japonicus. Mol. Plant-Microbe Interact. 2005, 18, 1069–1080. [Google Scholar] [CrossRef]
- Roossinck, M.J. A New Look at Plant Viruses and Their Potential Beneficial Roles in Crops. Mol. Plant Pathol. 2015, 16, 331–333. [Google Scholar] [CrossRef]
- Roossinck, M.J.; Bazán, E.R. Symbiosis: Viruses as Intimate Partners. Annu. Rev. Virol. 2017, 4, 123–139. [Google Scholar] [CrossRef] [PubMed]
- Byrne, M.; Kashyap, A.; Esquirol, L.; Ranson, N.; Sainsbury, F. The Structure of a Plant-Specific Partitivirus Capsid Reveals a Unique Coat Protein Domain Architecture with an Intrinsically Disordered Protrusion. Commun. Biol. 2021, 4, 1–8. [Google Scholar] [CrossRef]
- Sidharthan, V.K.; Rajeswari, V.; Baranwal, V.K. Analysis of Public Domain Plant Transcriptomes Expands the Phylogenetic Diversity of the Family Secoviridae. Virus Genes 2022, 58, 598–604. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, M.; Hily, J.M.; Petrzik, K.; Sanfaçon, H.; Thompson, J.R.; van der Vlugt, R.; Wetzel, T. ICTV Virus Taxonomy Profile: Secoviridae 2022. J. Gen. Virol. 2022, 103, 001807. [Google Scholar] [CrossRef]
- Sanfaçon, H. Secoviridae: A Family of Plant Picorna-Like Viruses with Monopartite or Bipartite Genomes. In eLS; Wiley: Hoboken, NJ, USA, 2015; pp. 1–14. [Google Scholar]
- Sanfaçon, H.; Wellink, J.; Le Gall, O.; Karasev, A.; Van Der Vlugt, R.; Wetzel, T. Secoviridae: A Proposed Family of Plant Viruses within the Order Picornavirales That Combines the Families Sequiviridae and Comoviridae, the Unassigned Genera Cheravirus and Sadwavirus, and the Proposed Genus Torradovirus. Arch. Virol. 2009, 154, 899–907. [Google Scholar] [CrossRef]
- Thompson, J.R.; Kamath, N.; Perry, K.L. An Evolutionary Analysis of the Secoviridae Family of Viruses. PLoS ONE 2014, 9, e106305. [Google Scholar] [CrossRef] [PubMed]
- Rangel, E.A.; Ferriol, I.; Panno, S.; Davino, S.; Olmos, A.; Rubio, L. The Complete Genome Sequence of Lamium Mild Mosaic Virus, a Member of the Genus Fabavirus. Arch. Virol. 2013, 158, 2405–2408. [Google Scholar] [CrossRef]
- He, Y.; Cai, L.; Zhou, L.; Yang, Z.; Hong, N.; Wang, G.; Li, S.; Xu, W. Deep Sequencing Reveals the First Fabavirus Infecting Peach. Sci. Rep. 2017, 7, 11329. [Google Scholar] [CrossRef]
- Villamor, D.E.V.; Pillai, S.S.; Eastwell, K.C. High Throughput Sequencing Reveals a Novel Fabavirus Infecting Sweet Cherry. Arch. Virol. 2017, 162, 811–816. [Google Scholar] [CrossRef]
- Ma, Y.; Xing, F.; Che, H.; Gao, S.; Lin, Y.; Li, S. The Virome of Piper Nigrum: Identification, Genomic Characterization, Prevalence, and Transmission of Three New Viruses of Black Pepper in China. Plant Dis. 2022, 106, 2082–2089. [Google Scholar] [CrossRef]
- Igori, D.; Shin, A.Y.; Hwang, U.S.; Choi, E.K.; Kwon, S.Y.; Moon, J.S. Genome Sequence and Characterization of a Novel Fabavirus Infecting Cirsium Setidens (Gondre) in South Korea. Arch. Virol. 2023, 168, 1–5. [Google Scholar] [CrossRef]
- Ryabov, E.; Taliansky, M.E.; Robinson, D.; Waterhouse Peter Murant, A.F.; de Zoeten, G.; Falk, B.; Vetten, H.; Gibbs, M. Genus Umbravirus. In Virus Taxonomy Classification and Nomenclature of Viruses Ninth Report of the International Committee on Taxonomy of Viruses; King, A.M.Q., Adams, M.J., Carstens, E., Lefkowitz, E.J., Eds.; Academic Press: London, UK, 2011; Volume 9, pp. 1191–1195. ISBN 9780128096338. [Google Scholar]
- Liu, J.; Simon, A.E. Identification of Novel 5′ and 3′ Translation Enhancers in Umbravirus-Like Coat Protein-Deficient RNA Replicons. J. Virol. 2022, 96, e0173621. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Carino, E.; Bera, S.; Gao, F.; May, J.P.; Simon, A.E. Structural Analysis and Whole Genome Mapping of a New Type of Plant Virus Subviral Rna: Umbravirus-like Associated Rnas. Viruses 2021, 13, 646. [Google Scholar] [CrossRef]
- Ryabov, E.V.; Taliansky, M.E. Umbraviruses (Tombusviridae). In Encyclopedia of Virology: Volume 1–5, 4th ed.; Elsevier: Amsterdam, The Netherlands, 2020; Volume 1–5, pp. 827–832. ISBN 9780128145166. [Google Scholar]
- Sõmera, M.; Fargette, D.; Hébrard, E.; Sarmiento, C. ICTV Virus Taxonomy Profile: Solemoviridae 2021. J. Gen. Virol. 2021, 102, 001707. [Google Scholar] [CrossRef]
- Redila, C.D.; Prakash, V.; Nouri, S. Metagenomics Analysis of the Wheat Virome Identifies Novel Plant and Fungal-Associated Viral Sequences. Viruses 2021, 13, 2457. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Olmedo-Velarde, A.; Larrea-Sarmiento, A.; Simon, A.E.; Kong, A.; Borth, W.; Suzuki, J.Y.; Wall, M.M.; Hu, J.; Melzer, M. Genome Characterization of Fig Umbra-like Virus. Virus Genes 2021, 57, 566–570. [Google Scholar] [CrossRef]
- Quito-Avila, D.F.; Reyes, E.; Mendoza, A.; Margaria, P.; Menzel, W.; Bera, S.; Simon, A. Two New Umbravirus-like Associated RNAs (UlaRNAs) Discovered in Maize and Johnsongrass from Ecuador. Arch. Virol. 2022, 167, 2093–2098. [Google Scholar] [CrossRef] [PubMed]
- Lappe, R.R.; Elmore, M.G.; Lozier, Z.R.; Jander, G.; Miller, W.A.; Whitham, S.A. Metagenomic Identification of Novel Viruses of Maize and Teosinte in North America. BMC Genom. 2022, 23, 767. [Google Scholar] [CrossRef]
- Cimino, P.A.; Nicholson, B.L.; Wu, B.; Xu, W.; White, K.A. Multifaceted Regulation of Translational Readthrough by RNA Replication Elements in a Tombusvirus. PLoS Pathog. 2011, 7, e1002423. [Google Scholar] [CrossRef]
- Mikkelsen, A.A.; Gao, F.; Carino, E.; Bera, S.; Simon, A.E. -1 Programmed Ribosomal Frameshifting in Class 2 Umbravirus-like RNAs Uses Multiple Long-Distance Interactions to Shift between Active and Inactive Structures and Destabilize the Frameshift Stimulating Element. Nucleic Acids Res. 2023, 51, 10700–10718. [Google Scholar] [CrossRef] [PubMed]










 .
.
.
.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GULV-2 | GULV-3 | |
|---|---|---|
| GULV-3 | 44.5 | 100 |
| GULV-4 | 48.4 | 44.3 |
| Group | Sequence | GULV-2 | GULV-3 | GULV-4 |
|---|---|---|---|---|
| Umbraviruses | Genome | 47.2–49.1 | 46.9–49.1 | 46.6–49.5 |
| RdRp | 49.8–52.7 | 48.4–51.7 | 48.6–52.9 | |
| UlaRNAs | Genome | 43.5–50.0 | 43.7–49.1 | 44.2–49.9 |
| RdRp | 47.4–50.6 | 47.6–50.5 | 47.6–51.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Belkina, D.; Karpova, D.; Porotikova, E.; Lifanov, I.; Vinogradova, S. Grapevine Virome of the Don Ampelographic Collection in Russia Has Concealed Five Novel Viruses. Viruses 2023, 15, 2429. https://doi.org/10.3390/v15122429
Belkina D, Karpova D, Porotikova E, Lifanov I, Vinogradova S. Grapevine Virome of the Don Ampelographic Collection in Russia Has Concealed Five Novel Viruses. Viruses. 2023; 15(12):2429. https://doi.org/10.3390/v15122429
Chicago/Turabian StyleBelkina, Daria, Daria Karpova, Elena Porotikova, Ilya Lifanov, and Svetlana Vinogradova. 2023. "Grapevine Virome of the Don Ampelographic Collection in Russia Has Concealed Five Novel Viruses" Viruses 15, no. 12: 2429. https://doi.org/10.3390/v15122429
APA StyleBelkina, D., Karpova, D., Porotikova, E., Lifanov, I., & Vinogradova, S. (2023). Grapevine Virome of the Don Ampelographic Collection in Russia Has Concealed Five Novel Viruses. Viruses, 15(12), 2429. https://doi.org/10.3390/v15122429