
Successful Whole Genome Nanopore Sequencing of Swine Influenza A Virus (swIAV) Directly from Oral Fluids Collected in Polish Pig Herds

 ,
,  , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Collection of Oral Fluids from Polish Herds
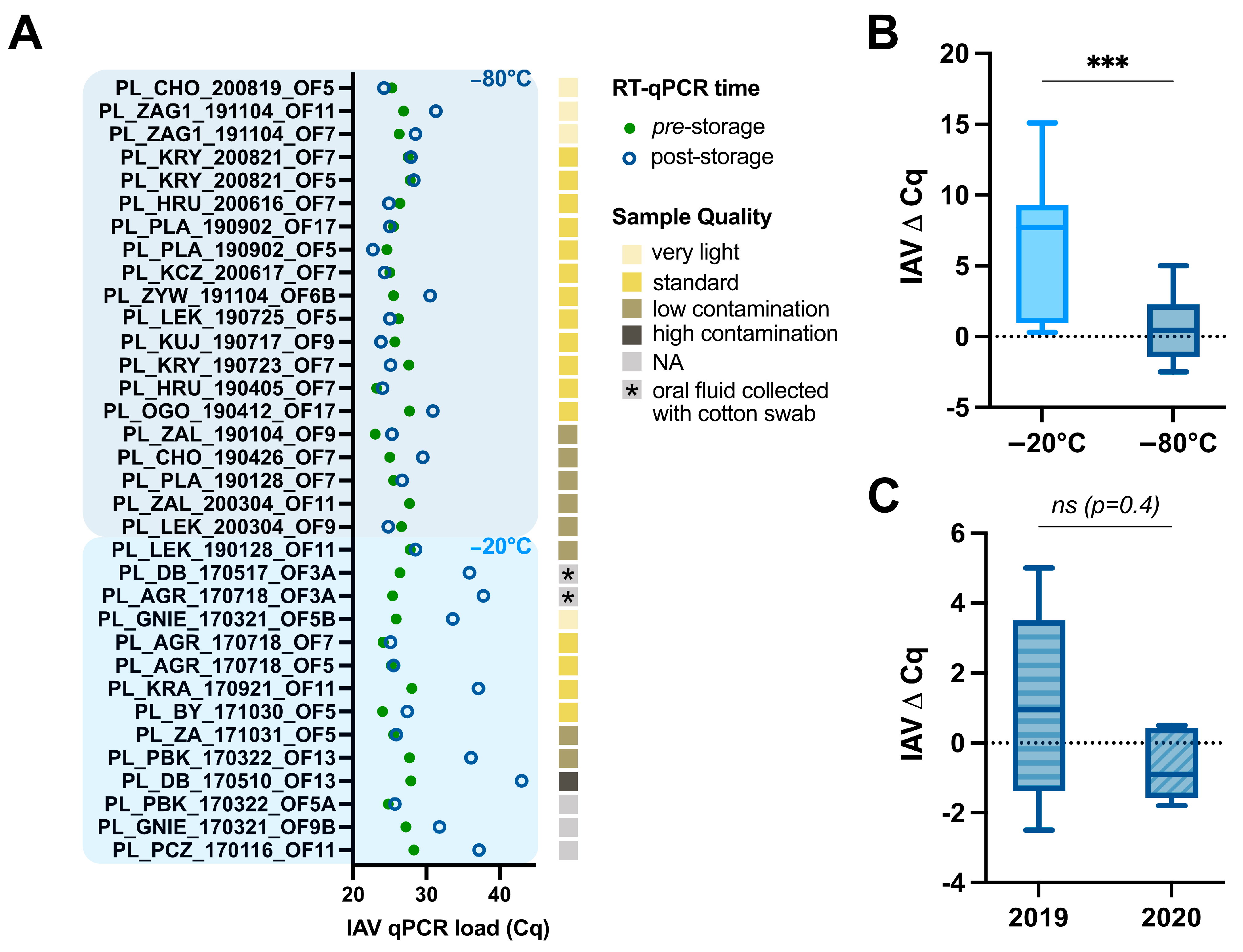
2.2. Assessment of swIAV Detection Using RT-qPCR Pre- and Post-Storage
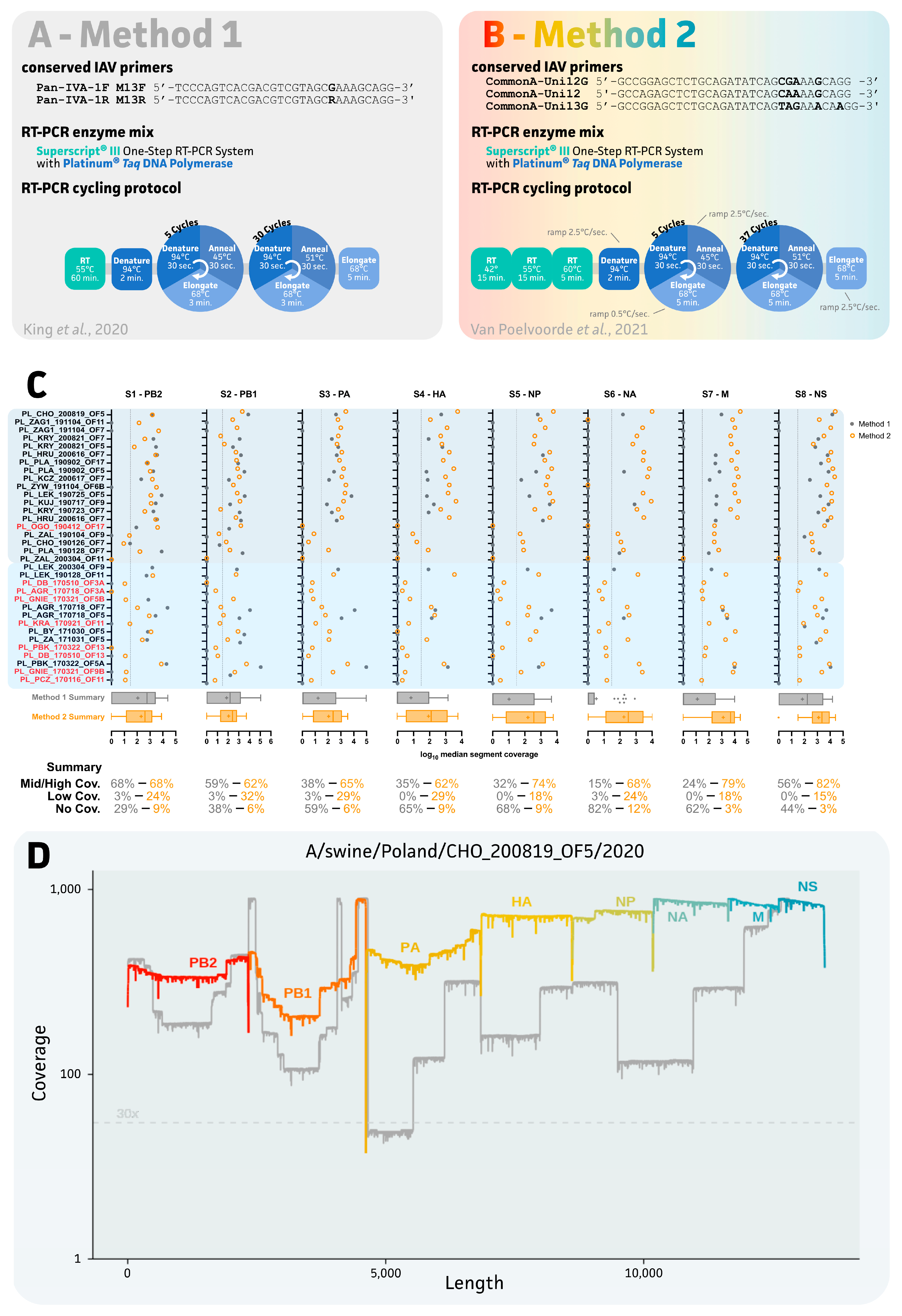
2.3. Evaluation of Two IAV Sequencing Protocols for Oral Fluids
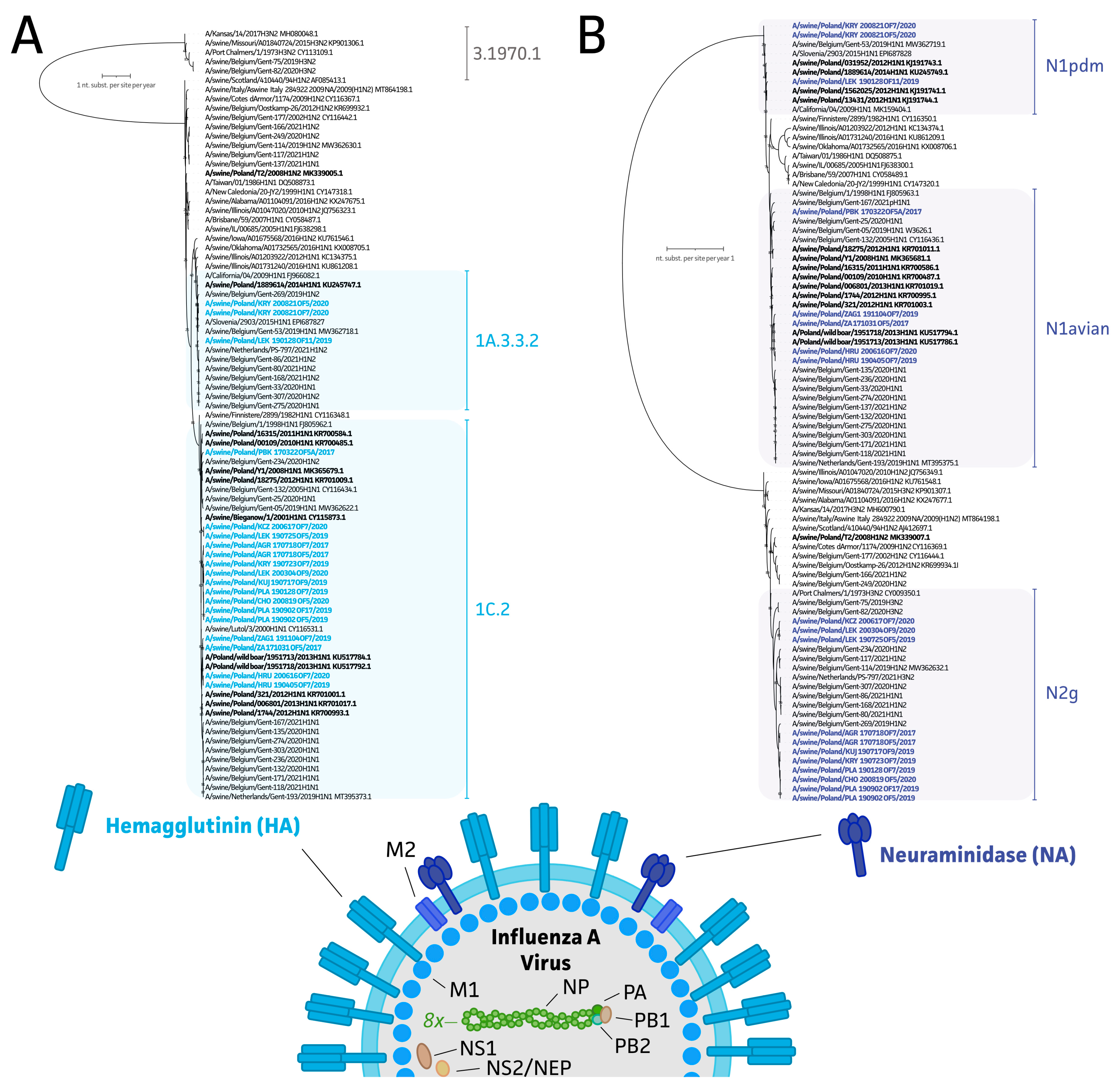
2.4. Genome Assembly and Epidemiological Analysis of Polish swIAV Segment Sequences
2.5. Statistical Analysis
3. Results
3.1. Impact of Storage Temperature, Time, and Oral Fluid Sample Quality on swIAV Detection
3.2. Impact of IAV Sequencing Protocol on Sequencing Success of the Eight IAV Gene Segments
3.3. Genetic Diversity of swIAV Strains in Polish Farms between 2017 and 2020
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Van Reeth, K.; Vincent, A.L. Influenza Viruses. In Diseases of Swine; Wiley: Hoboken, NJ, USA, 2019; pp. 576–593. [Google Scholar]
- Brown, I.H. The epidemiology and evolution of influenza viruses in pigs. Vet. Microbiol. 2000, 74, 29–46. [Google Scholar] [CrossRef] [PubMed]
- Sanjuán, R.; Nebot, M.R.; Chirico, N.; Mansky, L.M.; Belshaw, R. Viral Mutation Rates. J. Virol. 2010, 84, 9733–9748. [Google Scholar] [CrossRef] [PubMed]
- Anderson, T.K.; Macken, C.A.; Lewis, N.S.; Scheuermann, R.H.; Van Reeth, K.; Brown, I.H.; Swenson, S.L.; Simon, G.; Saito, T.; Berhane, Y.; et al. A Phylogeny-Based Global Nomenclature System and Automated Annotation Tool for H1 Hemagglutinin Genes from Swine Influenza A Viruses. mSphere 2016, 1, e00275-16. [Google Scholar] [CrossRef]
- Watson, S.J.; Langat, P.; Reid, S.M.; Lam, T.T.-Y.; Cotten, M.; Kelly, M.; Van Reeth, K.; Qiu, Y.; Simon, G.; Bonin, E.; et al. Molecular Epidemiology and Evolution of Influenza Viruses Circulating within European Swine between 2009 and 2013. J. Virol. 2015, 89, 9920–9931. [Google Scholar] [CrossRef] [PubMed]
- Henritzi, D.; Petric, P.P.; Lewis, N.S.; Graaf, A.; Pessia, A.; Starick, E.; Breithaupt, A.; Strebelow, G.; Luttermann, C.; Parker, L.M.K.; et al. Surveillance of European Domestic Pig Populations Identifies an Emerging Reservoir of Potentially Zoonotic Swine Influenza A Viruses. Cell Host Microbe 2020, 28, 614–627.e6. [Google Scholar] [CrossRef]
- Henritzi, D.; Zhao, N.; Starick, E.; Simon, G.; Krog, J.S.; Larsen, L.E.; Reid, S.M.; Brown, I.H.; Chiapponi, C.; Foni, E.; et al. Rapid detection and subtyping of European swine influenza viruses in porcine clinical samples by haemagglutinin- and neuraminidase-specific tetra- and triplex real-time RT-PCRs. Influenza Other Respi. Viruses 2016, 10, 504–517. [Google Scholar] [CrossRef]
- Chauhan, R.P.; Gordon, M.L. Review of genome sequencing technologies in molecular characterization of influenza A viruses in swine. J. Vet. Diagn. Investig. 2022, 34, 177–189. [Google Scholar] [CrossRef]
- Beato, M.S.; Tassoni, L.; Milani, A.; Salviato, A.; Di Martino, G.; Mion, M.; Bonfanti, L.; Monne, I.; Watson, S.J.; Fusaro, A. Circulation of multiple genotypes of H1N2 viruses in a swine farm in Italy over a two-month period. Vet. Microbiol. 2016, 195, 25–29. [Google Scholar] [CrossRef]
- Decorte, I.; Steensels, M.; Lambrecht, B.; Cay, A.B.; Regge, N. De Detection and isolation of swine influenza a virus in spiked oral fluid and samples from individually housed, experimentally infected pigs: Potential role of porcine oral fluid in active influenza a virus surveillance in swine. PLoS ONE 2015, 10, e0139586. [Google Scholar] [CrossRef]
- Biernacka, K.; Karbowiak, P.; Wróbel, P.; Charęza, T.; Czopowicz, M.; Balka, G.; Goodell, C.; Rauh, R.; Stadejek, T. Detection of porcine reproductive and respiratory syndrome virus (PRRSV) and influenza A virus (IAV) in oral fluid of pigs. Res. Vet. Sci. 2016, 109, 74–80. [Google Scholar] [CrossRef]
- Woźniak, A.; Miłek, D.; Stadejek, T. Wide range of the prevalence and viral loads of porcine circovirus type 3 (PCV3) in different clinical materials from 21 Polish pig farms. Pathogens 2020, 9, 411. [Google Scholar] [CrossRef] [PubMed]
- Woźniak, A.; Miłek, D.; Matyba, P.; Stadejek, T. Real-time PCR detection patterns of porcine circovirus type 2 (PCV2) in Polish farms with different status of vaccination against PCV2. Viruses 2019, 11, 1135. [Google Scholar] [CrossRef]
- Zhang, J.; Harmon, K.M. RNA Extraction from Swine Samples and Detection of Influenza A Virus in Swine by Real-Time RT-PCR. In Springer Protocols-Methods in Molecular Biology; Springer: Berlin/Heidelberg, Germany, 2020; pp. 295–310. ISBN 9781071603451. [Google Scholar]
- Chittick, W.A.; Stensland, W.R.; Prickett, J.R.; Strait, E.L.; Harmon, K.; Yoon, K.J.; Wang, C.; Zimmerman, J.J. Comparison of RNA extraction and real-time reverse transcription polymerase chain reaction methods for the detection of porcine reproductive and respiratory syndrome virus in porcine oral fluid specimens. J. Vet. Diagn. Investig. 2011, 23, 248–253. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Li, X.; Deng, R.; Zhang, G. Detection and genetic characteristics of porcine circovirus 3 based on oral fluids from asymptomatic pigs in central China. BMC Vet. Res. 2019, 15, 200. [Google Scholar] [CrossRef]
- Zhang, J.; Zheng, Y.; Xia, X.Q.; Chen, Q.; Bade, S.A.; Yoon, K.J.; Harmon, K.M.; Gauger, P.C.; Main, R.G.; Li, G. High-throughput whole genome sequencing of Porcine reproductive and respiratory syndrome virus from cell culture materials and clinical specimens using next-generation sequencing technology. J. Vet. Diagn. Investig. 2017, 29, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Flores, C.; Ariyama, N.; Bennett, B.; Mena, J.; Verdugo, C.; Mor, S.; Brito, B.; Ramírez-Toloza, G.; Neira, V. Case Report: First Report and Phylogenetic Analysis of Porcine Astroviruses in Chile. Front. Vet. Sci. 2021, 8, 764837. [Google Scholar] [CrossRef]
- Gagnon, C.A.; Lalonde, C.; Provost, C. Porcine reproductive and respiratory syndrome virus whole-genome sequencing efficacy with field clinical samples using a poly(A)-tail viral genome purification method. J. Vet. Diagn. Investig. 2021, 33, 216–226. [Google Scholar] [CrossRef]
- Miłek, D.; Woźniak, A.; Guzowska, M.; Stadejek, T. Detection patterns of porcine parvovirus (PPV) and novel porcine parvoviruses 2 through 6 (PPV2–PPV6) in Polish swine farms. Viruses 2019, 11, 474. [Google Scholar] [CrossRef]
- Hoffmann, B.; Harder, T.; Lange, E.; Kalthoff, D.; Reimann, I.; Grund, C.; Oehme, R.; Vahlenkamp, T.W.; Beer, M. New real-time reverse transcriptase polymerase chain reactions facilitate detection and differentiation of novel A/H1N1 influenza virus in porcine and human samples. Berl. Munch. Tierarztl. Wochenschr. 2016, 123, 286–292. [Google Scholar] [CrossRef]
- King, J.; Harder, T.; Beer, M.; Pohlmann, A. Rapid multiplex MinION nanopore sequencing workflow for Influenza A viruses. BMC Infect. Dis. 2020, 20, 648. [Google Scholar] [CrossRef]
- Van Poelvoorde, L.A.E.; Bogaerts, B.; Fu, Q.; De Keersmaecker, S.C.J.; Thomas, I.; Van Goethem, N.; Van Gucht, S.; Winand, R.; Saelens, X.; Roosens, N.; et al. Whole-genome-based phylogenomic analysis of the Belgian 2016–2017 influenza A(H3N2) outbreak season allows improved surveillance. Microb. Genom. 2021, 7, 000643. [Google Scholar] [CrossRef] [PubMed]
- De Coster, W.; D’Hert, S.; Schultz, D.T.; Cruts, M.; Van Broeckhoven, C. NanoPack: Visualizing and processing long-read sequencing data. Bioinformatics 2018, 34, 2666–2669. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef] [PubMed]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive κ-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; Von Haeseler, A.; Lanfear, R.; Teeling, E. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Chepkwony, S.; Parys, A.; Vandoorn, E.; Stadejek, W.; Xie, J.; King, J.; Graaf, A.; Pohlmann, A.; Beer, M.; Harder, T.; et al. Genetic and antigenic evolution of H1 swine influenza A viruses isolated in Belgium and the Netherlands from 2014 through 2019. Sci. Rep. 2021, 11, 11276. [Google Scholar] [CrossRef]
- Parys, A.; Vereecke, N.; Vandoorn, E.; Theuns, S.; Van Reeth, K. Virological Surveillance and Genomic Characterization of Influenza A and D Viruses in Swine in Belgium and the Netherlands from 2019 through 2021. Emerg. Infect. Dis. 2023, 29, 3. [Google Scholar]
- Letunic, I.; Bork, P. Interactive tree of life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Henao-Diaz, A.; Giménez-Lirola, L.; Baum, D.H.; Zimmerman, J. Guidelines for oral fluid-based surveillance of viral pathogens in swine. Porc. Health Manag. 2020, 6, 28. [Google Scholar] [CrossRef] [PubMed]
- Forster, J.L.; Harkin, V.B.; Graham, D.A.; McCullough, S.J. The effect of sample type, temperature and RNAlater on the stability of avian influenza virus RNA. J. Virol. Methods 2008, 149, 190–194. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz Gulec, E.; Cesur, N.P.; Yesilyurt Fazlioğlu, G.; Kazezoğlu, C. Effect of different storage conditions on COVID-19 RT-PCR results. J. Med. Virol. 2021, 93, 6575–6581. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Kwon, A.; Roh, E.Y.; Yoon, J.H.; Han, M.S.; Park, S.-W.; Park, H.; Shin, S. Effects of Storage Temperature and Media/Buffer for SARS-CoV-2 Nucleic Acid Detection. Am. J. Clin. Pathol. 2021, 155, 280–285. [Google Scholar] [CrossRef] [PubMed]
- Bal, A.; Pichon, M.; Picard, C.; Casalegno, J.S.; Valette, M.; Schuffenecker, I.; Billard, L.; Vallet, S.; Vilchez, G.; Cheynet, V.; et al. Quality control implementation for universal characterization of DNA and RNA viruses in clinical respiratory samples using single metagenomic next-generation sequencing workflow. BMC Infect. Dis. 2018, 18, 537. [Google Scholar] [CrossRef]
- Hou, N.; Wang, K.; Zhang, H.; Bai, M.; Chen, H.; Song, W.; Jia, F.; Zhang, Y.; Han, S.; Xie, B. Comparison of detection rate of 16 sampling methods for respiratory viruses: A Bayesian network meta-analysis of clinical data and systematic review. BMJ Glob. Health 2020, 5, e003053. [Google Scholar] [CrossRef]
- Tan, S.; Dvorak, C.M.T.; Murtaugh, M.P. Rapid, Unbiased PRRSV Strain Detection Using MinION Direct RNA Sequencing and Bioinformatics Tools. Viruses 2019, 11, 1132. [Google Scholar] [CrossRef]
- Umar, S.; Anderson, B.D.; Chen, K.; Wang, G.L.; Ma, M.J.; Gray, G.C. Metagenomic analysis of endemic viruses in oral secretions from Chinese pigs. Vet. Med. Sci. 2022, 8, 1982–1992. [Google Scholar] [CrossRef]
- Franco-Martínez, L.; Ortín-Bustillo, A.; Rubio, C.P.; Escribano, D.; López-Arjona, M.; García-Manzanilla, E.; Cerón, J.J.; Martínez-Subiela, S.; Tvarijonaviciute, A.; Tecles, F. Effects of pen faeces and feed contamination in biomarkers determination in oral fluid of pigs. Res. Vet. Sci. 2022, 152, 403–409. [Google Scholar] [CrossRef]
- Theuns, S.; Vanmechelen, B.; Bernaert, Q.; Deboutte, W.; Vandenhole, M.; Beller, L.; Matthijnssens, J.; Maes, P.; Nauwynck, H.J. Nanopore sequencing as a revolutionary diagnostic tool for porcine viral enteric disease complexes identifies porcine kobuvirus as an important enteric virus. Sci. Rep. 2018, 8, 9830. [Google Scholar] [CrossRef]
- Bokma, J.; Vereecke, N.; Pas, M.L.; Chantillon, L.; Vahl, M.; Weesendorp, E.; Deurenberg, R.H.; Nauwynck, H.; Haesebrouck, F.; Theuns, S.; et al. Evaluation of Nanopore Sequencing as a Diagnostic Tool for the Rapid Identification of Mycoplasma bovis from Individual and Pooled Respiratory Tract Samples. J. Clin. Microbiol. 2021, 59, e0111021. [Google Scholar] [CrossRef] [PubMed]
- Höper, D.; Hoffmann, B.; Beer, M. A comprehensive deep sequencing strategy for full-length genomes of influenza A. PLoS ONE 2011, 6, e19075. [Google Scholar] [CrossRef] [PubMed]
- Watson, S.J.; Welkers, M.R.A.; Depledge, D.P.; Coulter, E.; Breuer, J.M.; de Jong, M.D.; Kellam, P. Viral population analysis and minority-variant detection using short read next-generation sequencing. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2013, 368, 20120205. [Google Scholar] [CrossRef] [PubMed]
- Van den Hoecke, S.; Verhelst, J.; Vuylsteke, M.; Saelens, X. Analysis of the genetic diversity of influenza A viruses using next-generation DNA sequencing. BMC Genom. 2015, 16, 79. [Google Scholar] [CrossRef] [PubMed]
- Jonges, M.; Welkers, M.R.A.; Jeeninga, R.E.; Meijer, A.; Schneeberger, P.; Fouchier, R.A.M.; de Jong, M.D.; Koopmans, M. Emergence of the virulence-associated PB2 E627K substitution in a fatal human case of highly pathogenic avian influenza virus A(H7N7) infection as determined by Illumina ultra-deep sequencing. J. Virol. 2014, 88, 1694–1702. [Google Scholar] [CrossRef] [PubMed]
- Crossley, B.M.; Rejmanek, D.; Baroch, J.; Stanton, J.B.; Young, K.T.; Killian, M.L.; Torchetti, M.K.; Hietala, S.K. Nanopore sequencing as a rapid tool for identification and pathotyping of avian influenza A viruses. J. Vet. Diagn. Investig. 2021, 33, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Rambo-Martin, B.L.; Keller, M.W.; Wilson, M.M.; Nolting, J.M.; Anderson, T.K.; Vincent, A.L.; Bagal, U.R.; Jang, Y.; Neuhaus, E.B.; Davis, C.T.; et al. Influenza A Virus Field Surveillance at a Swine-Human Interface. mSphere 2020, 5, e00822-19. [Google Scholar] [CrossRef] [PubMed]
- Thai, P.Q.; Mai, L.Q.; Welkers, M.R.A.; Hang, N.L.K.; Thanh, L.T.; Dung, V.T.V.; Yen, N.T.T.; Duong, T.N.; Hoa, L.N.M.; Thoang, D.D.; et al. Pandemic H1N1 virus transmission and shedding dynamics in index case households of a prospective Vietnamese cohort. J. Infect. 2014, 68, 581–590. [Google Scholar] [CrossRef]
- Phyu, W.W.; Saito, R.; Kyaw, Y.; Lin, N.; Win, S.M.K.; Win, N.C.; Ja, L.D.; Htwe, K.T.Z.; Aung, T.Z.; Tin, H.H.; et al. Evolutionary Dynamics of Whole-Genome Influenza A/H3N2 Viruses Isolated in Myanmar from 2015 to 2019. Viruses 2022, 14, 2414. [Google Scholar] [CrossRef]
- Malboeuf, C.M.; Isaacs, S.J.; Tran, N.H.; Kim, B. Thermal effects on reverse transcription: Improvement of accuracy and processivity in cDNA synthesis. Biotechniques 2001, 30, 1074–8, 1080, 1082, passim. [Google Scholar] [CrossRef]
- Invitrogen Corporate. Greater Yields from More Targets. 2003, 44. Available online: https://assets.fishersci.com/TFS-Assets/LSG/brochures/711_032688_B_SS3_OneStepRT_bro.pdf (accessed on 21 December 2022).
- Derendinger, B.; de Vos, M.; Nathavitharana, R.R.; Dolby, T.; Simpson, J.A.; van Helden, P.D.; Warren, R.M.; Theron, G. Widespread use of incorrect PCR ramp rate negatively impacts multidrug-resistant tuberculosis diagnosis (MTBDRplus). Sci. Rep. 2018, 8, 3206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frey, U.H.; Bachmann, H.S.; Peters, J.; Siffert, W. PCR-amplification of GC-rich regions: “slowdown PCR”. Nat. Protoc. 2008, 3, 1312–1317. [Google Scholar] [CrossRef] [PubMed]
- Simon, G.; Larsen, L.E.; Dürrwald, R.; Foni, E.; Harder, T.; Van Reeth, K.; Markowska-Daniel, I.; Reid, S.M.; Dan, A.; Maldonado, J.; et al. European surveillance network for influenza in pigs: Surveillance programs, diagnostic tools and swine influenza virus subtypes identified in 14 European countries from 2010 to 2013. PLoS ONE 2014, 9, e0115815. [Google Scholar] [CrossRef] [PubMed]
- Lewis, N.S.; Russell, C.A.; Langat, P.; Anderson, T.K.; Berger, K.; Bielejec, F.; Burke, D.F.; Dudas, G.; Fonville, J.M.; Fouchier, R.A.M.; et al. The global antigenic diversity of swine influenza A viruses. eLife 2016, 5, e12217. [Google Scholar] [CrossRef]
- Alleweldt, F.; Kara, Ş.; Best, K.; Aarestrup, F.M.; Beer, M.; Bestebroer, T.M.; Campos, J.; Casadei, G.; Chinen, I.; Van Domselaar, G.; et al. Economic evaluation of whole genome sequencing for pathogen identification and surveillance—Results of case studies in Europe and the Americas 2016 to 2019. Euro Surveill. 2021, 26, 1900606. [Google Scholar] [CrossRef]
- Jones, T.H.; Muehlhauser, V. Effect of handling and storage conditions and stabilizing agent on the recovery of viral RNA from oral fluid of pigs. J. Virol. Methods 2014, 198, 26–31. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Surface Genes | Internal Genes | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Farm | Year | Subtype | Genotype | S4 HA | S6 NA | S1 PB2 | S2 PB1 | S3 PA | S5 NP | S7 M | S8 NS | ||
| A/swine/Poland/BY_171030_OF5/2017 | BY | 2017 | HxNx | Und. | * | ||||||||
| A/swine/Poland/GNI_170321_OF9B/2017 | GNI | HxN1 | Und. | ||||||||||
| A/swine/Poland/PBK_170322_OF5A/2017 | PBK | H1N1 | A | ||||||||||
| A/swine/Poland/AGR_170718_OF5_2017 | AGR | H1N2 | T | ||||||||||
| A/swine/Poland/AGR_170718_OF7/2017 | H1N2 | T | |||||||||||
| A/swine/Poland/ZA_171031_OF5/2017 | ZA | H1N1 | U | ||||||||||
| A/swine/Poland/HRU_190405_OF7/2019 | HRU | 2019 | H1N1 | U | |||||||||
| A/swine/Poland/HRU_200616_OF7/2020 | 2020 | H1N1 | U | ||||||||||
| A/swine/Poland/KRY_190723_OF5/2019 | KRY | 2019 | H1N2 | T | |||||||||
| A/swine/Poland/KRY_200821_OF5/2020 | 2020 | H1N1 | P | * | * | ||||||||
| A/swine/Poland/KRY_200821_OF7/2020 | H1N1 | P | |||||||||||
| A/swine/Poland/KUJ_190717_OF9/2019 | KUJ | 2019 | H1N2 | T | |||||||||
| A/swine/Poland/LEK_190725_OF5_2019 | LEK | H1N2 | T | ||||||||||
| A/swine/Poland/LEK_190128_OF11/2019 | H1N1 | P | |||||||||||
| A/swine/Poland/LEK_200304_OF9/2020 | 2020 | H1N2 | T | ||||||||||
| A/swine/Poland/ZAG1_191104_OF7/2019 | ZAG1 | 2019 | H1N1 | U | |||||||||
| A/swine/Poland/ZAG1_191104_OF11/2019 | H1Nx | Und. | |||||||||||
| A/swine/Poland/ZYW_191104_OF6b/2019 | ZYW | H1Nx | Und. | ||||||||||
| A/swine/Poland/PLA_190902_OF5/2019 | PLA | H1N2 | T | ||||||||||
| A/swine/Poland/PLA_190902_OF17/2019 | H1N2 | T | |||||||||||
| A/swine/Poland/PLA_190128_OF7/2019 | H1N2 | T | * | ||||||||||
| A/swine/Poland/CHO_200819_OF5/2020 | CHO | 2020 | H1N2 | T | |||||||||
| A/swine/Poland/KCZ_200617_OF7/2020 | KCZ | H1N2 | T | ||||||||||
| European avian-like H1N1 | |||||||||||||
| 2009 pandemic H1N1 | |||||||||||||
| Human-like H3N2 | |||||||||||||
| Genetic information not available | |||||||||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vereecke, N.; Woźniak, A.; Pauwels, M.; Coppens, S.; Nauwynck, H.; Cybulski, P.; Theuns, S.; Stadejek, T. Successful Whole Genome Nanopore Sequencing of Swine Influenza A Virus (swIAV) Directly from Oral Fluids Collected in Polish Pig Herds. Viruses 2023, 15, 435. https://doi.org/10.3390/v15020435
Vereecke N, Woźniak A, Pauwels M, Coppens S, Nauwynck H, Cybulski P, Theuns S, Stadejek T. Successful Whole Genome Nanopore Sequencing of Swine Influenza A Virus (swIAV) Directly from Oral Fluids Collected in Polish Pig Herds. Viruses. 2023; 15(2):435. https://doi.org/10.3390/v15020435
Chicago/Turabian StyleVereecke, Nick, Aleksandra Woźniak, Marthe Pauwels, Sieglinde Coppens, Hans Nauwynck, Piotr Cybulski, Sebastiaan Theuns, and Tomasz Stadejek. 2023. "Successful Whole Genome Nanopore Sequencing of Swine Influenza A Virus (swIAV) Directly from Oral Fluids Collected in Polish Pig Herds" Viruses 15, no. 2: 435. https://doi.org/10.3390/v15020435
APA StyleVereecke, N., Woźniak, A., Pauwels, M., Coppens, S., Nauwynck, H., Cybulski, P., Theuns, S., & Stadejek, T. (2023). Successful Whole Genome Nanopore Sequencing of Swine Influenza A Virus (swIAV) Directly from Oral Fluids Collected in Polish Pig Herds. Viruses, 15(2), 435. https://doi.org/10.3390/v15020435