HTLV-1 and Innate Immunity


Abstract
:1. Introduction
1.1. Innate Immune Response
1.2. HTLV-1
2. HTLV-1 Infects Cells that Play a Major Role in the Innate Immune Response

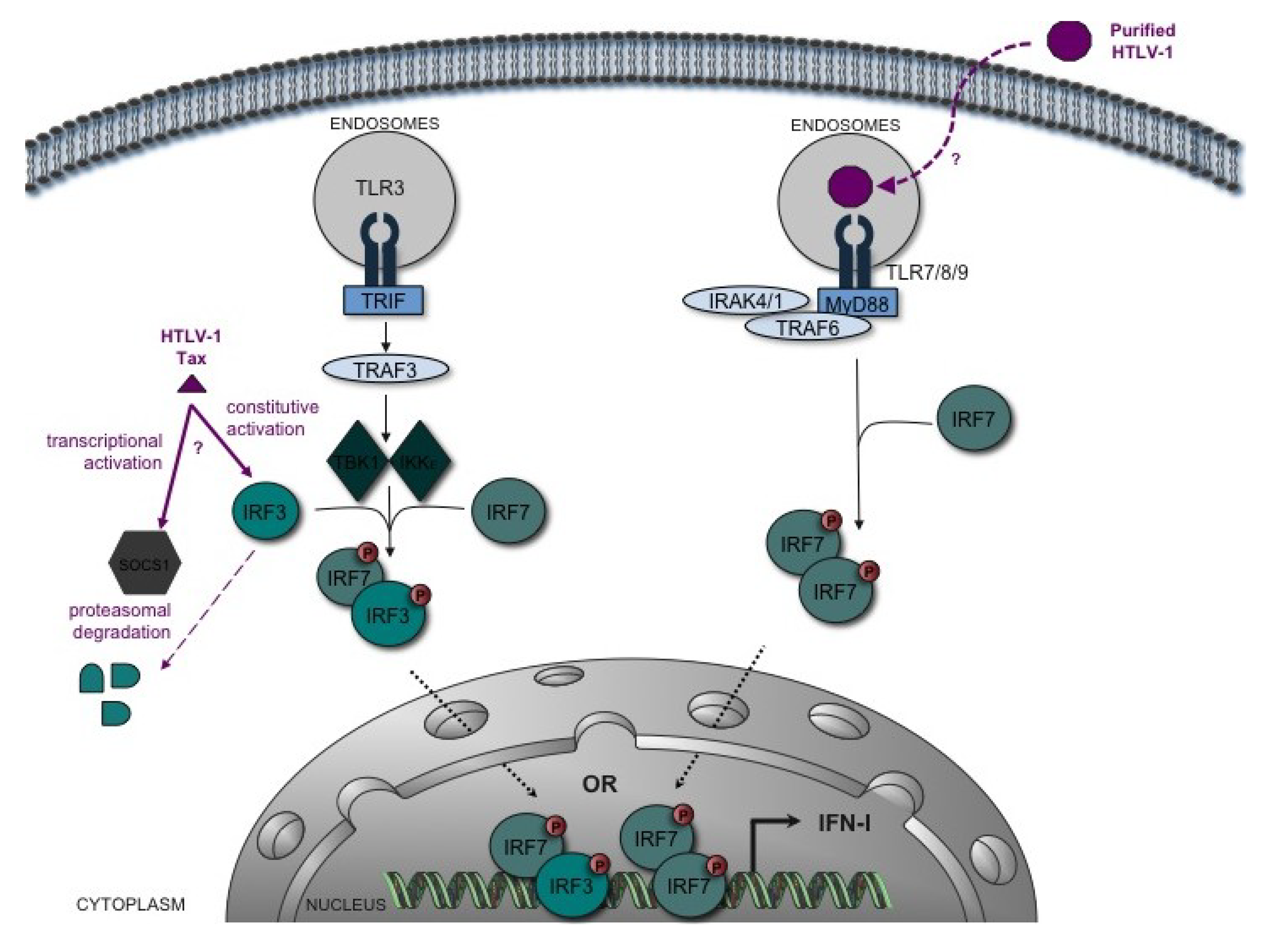{kind=link}
{kind=link}
| Infection by HTLV-1 | Control of HTLV-1 Replication | Infection-Induced Alteration of Cell Numbers in vivo | Infection-Induced Alteration of Function | ||
|---|---|---|---|---|---|
| Monocytes / Macrophages | Demonstrated in vitro and in vivo [39,43,44] | ND | ND | Alteration of the in vitro differentiation into functional dendritic cells (DCs) [56,57] | |
| Dendritic Cells | myDCs | Demonstrated in vitro and in vivo [45]1 [46,48] | - Secretion of IFN-α upon in vitro HTLV-1 sensing [58]2 | Decreased (ATLL and HAM/TSP patients) [46,47] | ND |
| pDCs | Demonstrated in vitro and in vivo [46,47,48] | - Secretion of IFN-α and maturation into killer pDCs upon in vitro HTLV-1 sensing [59] - Inverse correlation between the PVL and the ability of ex vivo-stimulated cells to produce IFN-α (ACs) [46] | Decreased (ATLL and HAM/TSP patients) [46,47] | Alteration of the ability to secrete IFN-α upon ex vivo stimulation (ACs) [46] | |
| Natural Killer Cells | Demonstrated only in vitro, on activated cells [55] | Inverse correlation between infected cell lines sensitivity to NK-mediated cell lysis and tumorigenicity in SCID mice [60] | Decreased (ATLL and HAM/TSP patients) [47,61] | High rate of ex vivo proliferation (ACs and HAM/TSP patients) [62] | |
| Invariant Natural Killer T Cells | Demonstrated in vivo [47] | Inverse correlation between the PVL and iNKT cells frequency (ATLL and HAM/TSP patients) [47] | Decreased (ATLL and HAM/TSP patients) [47,63] | Low rate of ex vivo proliferation and perforin production in infected individuals [47] | |
3. Do Innate Immune Cells Control HTLV-1 Replication?
3.1. DCs

3.2. NK Cells
3.3. iNKT Cells
4. Does HTLV Alter the Innate Response?
4.1. Alteration of Innate Immune Cell Phenotype, Function and Abundance in the Context of HTLV-1 Infection
4.1.1. Monocytes, Macrophages and DCs
4.1.2. NK Cells
4.2. Alteration of Molecular Pathways by HTLV-1
4.2.1. Modulation of IFN-I Production
4.2.2. Modulation of IFN-I Signaling
4.2.3. Modulation of Cell Susceptibility to NK-Mediated Cell Lysis
5. Treating HTLV-1 ATLL or HAM/TSP Patients with IFN-I
6. In vitro IFN-α or -β Treatment of HTLV-1-Infected Cells
7. Conclusions
Acknowledgments
Conflict of Interest
References and Notes
- Isaacs, A.; Lindenmann, J.; Valentine, R.C. Virus interference. II. Some properties of interferon. Proc. R. Soc. Lond. B Biol. Sci. 1957, 147, 268–273. [Google Scholar] [PubMed]
- Kawai, T.; Akira, S. Antiviral signaling through pattern recognition receptors. J. Biochem. 2007, 141, 137–145. [Google Scholar] [CrossRef]
- Thompson, A.J.; Locarnini, S.A. Toll-like receptors, RIG-I-like RNA helicases and the antiviral innate immune response. Immunol. Cell Biol. 2007, 85, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Hiscott, J. Triggering the innate antiviral response through IRF-3 activation. J. Biol. Chem. 2007, 282, 15325–15329. [Google Scholar] [CrossRef] [PubMed]
- Honda, K.; Yanai, H.; Negishi, H.; Asagiri, M.; Sato, M.; Mizutani, T.; Shimada, N.; Ohba, Y.; Takaoka, A.; Yoshida, N.; et al. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature 2005, 434, 772–777. [Google Scholar] [CrossRef] [PubMed]
- Platanias, L.C. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 2005, 5, 375–386. [Google Scholar] [CrossRef]
- Williams, B.R. PKR; a sentinel kinase for cellular stress. Oncogene 1999, 18, 6112–6120. [Google Scholar] [CrossRef]
- Ju, X.; Clark, G.; Hart, D.N. Review of human DC subtypes. Methods Mol. Biol. 2010, 595, 3–20. [Google Scholar]
- Douville, R.N.; Hiscott, J. The interface between the innate interferon response and expression of host retroviral restriction factors. Cytokine 2010, 52, 108–115. [Google Scholar] [CrossRef]
- Verdonck, K.; Gonzalez, E.; Van Dooren, S.; Vandamme, A.M.; Vanham, G.; Gotuzzo, E. Human T-lymphotropic virus 1: recent knowledge about an ancient infection. Lancet Infect. Dis. 2007, 7, 266–281. [Google Scholar] [CrossRef]
- Mueller, N. The epidemiology of HTLV-I infection. Canc. Causes Contr. 1991, 2, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Ureta-Vidal, A.; Angelin-Duclos, C.; Tortevoye, P.; Murphy, E.; Lepere, J.F.; Buigues, R.P.; Jolly, N.; Joubert, M.; Carles, G.; Pouliquen, J.F.; et al. Mother-to-child transmission of human T-cell-leukemia/lymphoma virus type I: Implication of high antiviral antibody titer and high proviral load in carrier mothers. Int. J. Cancer 1999, 82, 832–836. [Google Scholar] [CrossRef]
- Murphy, E.L.; Figueroa, J.P.; Gibbs, W.N.; Brathwaite, A.; Holding-Cobham, M.; Waters, D.; Cranston, B.; Hanchard, B.; Blattner, W.A. Sexual transmission of human T-lymphotropic virus type I (HTLV-I). Ann. Intern. Med. 1989, 111, 555–560. [Google Scholar] [CrossRef]
- Manns, A.; Wilks, R.J.; Murphy, E.L.; Haynes, G.; Figueroa, J.P.; Barnett, M.; Hanchard, B.; Blattner, W.A. A prospective study of transmission by transfusion of HTLV-I and risk factors associated with seroconversion. Int. J. Cancer 1992, 51, 886–891. [Google Scholar] [CrossRef] [PubMed]
- Uchiyama, T.; Yodoi, J.; Sagawa, K.; Takatsuki, K.; Uchino, H. Adult T-cell leukemia: clinical and hematologic features of 16 cases. Blood 1977, 50, 481–492. [Google Scholar] [CrossRef]
- Shimoyama, M. Diagnostic criteria and classification of clinical subtypes of adult T-cell leukaemia-lymphoma. A report from the Lymphoma Study Group (1984–87). Br. J. Haematol. 1991, 79, 428–437. [Google Scholar] [CrossRef] [PubMed]
- Bangham, C.R.; Osame, M. Cellular immune response to HTLV-1. Oncogene 2005, 24, 6035–6046. [Google Scholar] [CrossRef]
- Takenouchi, N.; Yao, K.; Jacobson, S. Immunopathogensis of HTLV-I associated neurologic disease: Molecular, histopathologic, and immunologic approaches. Front. Biosci. 2004, 9, 2527–2539. [Google Scholar] [CrossRef]
- Araujo, A.Q.; Silva, M.T. The HTLV-1 neurological complex. Lancet Neurol. 2006, 5, 1068–1076. [Google Scholar] [CrossRef]
- Hoger, T.A.; Jacobson, S.; Kawanishi, T.; Kato, T.; Nishioka, K.; Yamamoto, K. Accumulation of human T lymphotropic virus (HTLV)-I-specific T cell clones in HTLV-I-associated myelopathy/tropical spastic paraparesis patients. J. Immunol. 1997, 159, 2042–2048. [Google Scholar] [CrossRef]
- Gout, O.; Baulac, M.; Gessain, A.; Semah, F.; Saal, F.; Peries, J.; Cabrol, C.; Foucault-Fretz, C.; Laplane, D.; Sigaux, F.; et al. Rapid development of myelopathy after HTLV-I infection acquired by transfusion during cardiac transplantation. N. Engl. J. Med. 1990, 322, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Bartholomew, C.; Jack, N.; Edwards, J.; Charles, W.; Corbin, D.; Cleghorn, F.R.; Blattner, W.A. HTLV-I serostatus of mothers of patients with adult T-cell leukemia and HTLV-I-associated myelopathy/tropical spastic paraparesis. J. Hum. Virol. 1998, 1, 302–305. [Google Scholar] [PubMed]
- Pombo-de-Oliveira, M.S.; Carvalho, S.M.; Borducchi, D.; Dobbin, J.; Salvador, J.; Correa, R.B.; Moellman, A.; Loureiro, P.; Chiattone, C.; Rios, M. Adult T-cell leukemia/lymphoma and cluster of HTLV-I associated diseases in Brazilian settings. Leuk Lymphoma 2001, 42, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Heyden, N.V.; Ratner, L. Alpha interferon inhibits human T-cell leukemia virus type 1 assembly by preventing Gag interaction with rafts. J. Virol. 2003, 77, 13389–13395. [Google Scholar] [CrossRef]
- Feng, X.; Ratner, L. Human T-cell leukemia virus type 1 blunts signaling by interferon alpha. Virology 2008, 374, 210–216. [Google Scholar] [CrossRef]
- Smith, D.; Buckle, G.J.; Hafler, D.A.; Frank, D.A.; Hollsberg, P. HTLV-I-infected T cells evade the antiproliferative action of IFN-beta. Virology 1999, 257, 314–321. [Google Scholar] [CrossRef]
- Zhang, J.; Yamada, O.; Kawagishi, K.; Araki, H.; Yamaoka, S.; Hattori, T.; Shimotohno, K. Human T-cell leukemia virus type 1 Tax modulates interferon-alpha signal transduction through competitive usage of the coactivator CBP/p300. Virology 2008, 379, 306–313. [Google Scholar] [CrossRef]
- Bazarbachi, A.; Nasr, R.; El-Sabban, M.E.; Mahe, A.; Mahieux, R.; Gessain, A.; Darwiche, N.; Dbaibo, G.; Kersual, J.; Zermati, Y.; et al. Evidence against a direct cytotoxic effect of alpha interferon and zidovudine in HTLV-I associated adult T cell leukemia/lymphoma. Leukemia 2000, 14, 716–721. [Google Scholar] [CrossRef]
- Bazarbachi, A.; Plumelle, Y.; Carlos Ramos, J.; Tortevoye, P.; Otrock, Z.; Taylor, G.; Gessain, A.; Harrington, W.; Panelatti, G.; Hermine, O. Meta-analysis on the use of zidovudine and interferon-alfa in adult T-cell leukemia/lymphoma showing improved survival in the leukemic subtypes. J. Clin. Oncol. 2010, 28, 4177–4183. [Google Scholar] [CrossRef]
- Hermine, O.; Wattel, E.; Gessain, A.; Bazarbachi, A. Adult T cell leukaemia: A review of established and new treatments. BioDrugs 1998, 10, 447–462. [Google Scholar] [CrossRef]
- Bazarbachi, A.; El-Sabban, M.E.; Nasr, R.; Quignon, F.; Awaraji, C.; Kersual, J.; Dianoux, L.; Zermati, Y.; Haidar, J.H.; Hermine, O.; et al. Arsenic trioxide and interferon-alpha synergize to induce cell cycle arrest and apoptosis in human T-cell lymphotropic virus type I-transformed cells. Blood 1999, 93, 278–283. [Google Scholar] [CrossRef]
- El Hajj, H.; El-Sabban, M.; Hasegawa, H.; Zaatari, G.; Ablain, J.; Saab, S.T.; Janin, A.; Mahfouz, R.; Nasr, R.; Kfoury, Y.; et al. Therapy-induced selective loss of leukemia-initiating activity in murine adult T cell leukemia. J. Exp. Med. 2010, 207, 2785–2792. [Google Scholar] [CrossRef] [PubMed]
- El-Sabban, M.E.; Nasr, R.; Dbaibo, G.; Hermine, O.; Abboushi, N.; Quignon, F.; Ameisen, J.C.; Bex, F.; de The, H.; Bazarbachi, A. Arsenic-interferon-alpha-triggered apoptosis in HTLV-I transformed cells is associated with tax down-regulation and reversal of NF-kappa B activation. Blood 2000, 96, 2849–2855. [Google Scholar]
- Hermine, O.; Dombret, H.; Poupon, J.; Arnulf, B.; Lefrere, F.; Rousselot, P.; Damaj, G.; Delarue, R.; Fermand, J.P.; Brouet, J.C.; et al. Phase II trial of arsenic trioxide and alpha interferon in patients with relapsed/refractory adult T-cell leukemia/lymphoma. Hematol. J. 2004, 5, 130–134. [Google Scholar] [CrossRef]
- Kchour, G.; Tarhini, M.; Kooshyar, M.M.; El Hajj, H.; Wattel, E.; Mahmoudi, M.; Hatoum, H.; Rahimi, H.; Maleki, M.; Rafatpanah, H.; et al. Phase 2 study of the efficacy and safety of the combination of arsenic trioxide, interferon alpha, and zidovudine in newly diagnosed chronic adult T-cell leukemia/lymphoma (ATL). Blood 2009, 113, 6528–6532. [Google Scholar] [CrossRef]
- Mahieux, R.; Pise-Masison, C.; Gessain, A.; Brady, J.N.; Olivier, R.; Perret, E.; Misteli, T.; Nicot, C. Arsenic trioxide induces apoptosis in human T-cell leukemia virus type 1- and type 2-infected cells by a caspase-3-dependent mechanism involving Bcl-2 cleavage. Blood 2001, 98, 3762–3769. [Google Scholar] [CrossRef] [PubMed]
- Nasr, R.; Rosenwald, A.; El-Sabban, M.E.; Arnulf, B.; Zalloua, P.; Lepelletier, Y.; Bex, F.; Hermine, O.; Staudt, L.; de The, H.; et al. Arsenic/interferon specifically reverses 2 distinct gene networks critical for the survival of HTLV-1-infected leukemic cells. Blood 2003, 101, 4576–4582. [Google Scholar] [CrossRef] [PubMed]
- Richardson, J.H.; Edwards, A.J.; Cruickshank, J.K.; Rudge, P.; Dalgleish, A.G. In vivo cellular tropism of human T-cell leukemia virus type 1. J. Virol. 1990, 64, 5682–5687. [Google Scholar] [CrossRef]
- Koyanagi, Y.; Itoyama, Y.; Nakamura, N.; Takamatsu, K.; Kira, J.; Iwamasa, T.; Goto, I.; Yamamoto, N. In vivo infection of human T-cell leukemia virus type I in non-T cells. Virology 1993, 196, 25–33. [Google Scholar] [CrossRef]
- Hanon, E.; Stinchcombe, J.C.; Saito, M.; Asquith, B.E.; Taylor, G.P.; Tanaka, Y.; Weber, J.N.; Griffiths, G.M.; Bangham, C.R. Fratricide among CD8(+) T lymphocytes naturally infected with human T cell lymphotropic virus type I. Immunity 2000, 13, 657–664. [Google Scholar] [CrossRef]
- Cho, I.; Sugimoto, M.; Mita, S.; Tokunaga, M.; Imamura, F.; Ando, M. In vivo proviral burden and viral RNA expression in T cell subsets of patients with human T lymphotropic virus type-1-associated myelopathy/tropical spastic paraparesis. Am. J. Trop. Med. Hyg. 1995, 53, 412–418. [Google Scholar] [CrossRef] [PubMed]
- Franchini, G.; Mann, D.L.; Popovic, M.; Zicht, R.R.; Gallo, R.C.; Wong-Staal, F. HTLV-I infection of T and B cells of a patient with adult T-cell leukemia-lymphoma (ATLL) and transmission of HTLV-I from B cells to normal T cells. Leuk Res. 1985, 9, 1305–1314. [Google Scholar] [CrossRef] [PubMed]
- Koralnik, I.J.; Lemp, J.F., Jr.; Gallo, R.C.; Franchini, G. In vitro infection of human macrophages by human T-cell leukemia/lymphotropic virus type I (HTLV-I). AIDS Res. Hum. Retroviruses 1992, 8, 1845–1849. [Google Scholar] [CrossRef] [PubMed]
- de Revel, T.; Mabondzo, A.; Gras, G.; Delord, B.; Roques, P.; Boussin, F.; Neveux, Y.; Bahuau, M.; Fleury, H.J.; Dormont, D. In vitro infection of human macrophages with human T-cell leukemia virus type 1. Blood 1993, 81, 1598–1606. [Google Scholar] [CrossRef] [PubMed]
- Macatonia, S.E.; Cruickshank, J.K.; Rudge, P.; Knight, S.C. Dendritic cells from patients with tropical spastic paraparesis are infected with HTLV-1 and stimulate autologous lymphocyte proliferation. AIDS Res. Hum. Retroviruses 1992, 8, 1699–1706. [Google Scholar] [CrossRef]
- Hishizawa, M.; Imada, K.; Kitawaki, T.; Ueda, M.; Kadowaki, N.; Uchiyama, T. Depletion and impaired interferon-alpha-producing capacity of blood plasmacytoid dendritic cells in human T-cell leukaemia virus type I-infected individuals. Br. J. Haematol. 2004, 125, 568–575. [Google Scholar] [CrossRef]
- Azakami, K.; Sato, T.; Araya, N.; Utsunomiya, A.; Kubota, R.; Suzuki, K.; Hasegawa, D.; Izumi, T.; Fujita, H.; Aratani, S.; et al. Severe loss of invariant NKT cells exhibiting anti-HTLV-1 activity in patients with HTLV-1-associated disorders. Blood 2009, 114, 3208–3215. [Google Scholar] [CrossRef]
- Jones, K.S.; Petrow-Sadowski, C.; Huang, Y.K.; Bertolette, D.C.; Ruscetti, F.W. Cell-free HTLV-1 infects dendritic cells leading to transmission and transformation of CD4(+) T cells. Nat. Med. 2008, 14, 429–436. [Google Scholar] [CrossRef]
- Dube, D.K.; Dube, S.; Erensoy, S.; Jones, B.; Bryz-Gornia, V.; Spicer, T.; Love, J.; Saksena, N.; Lechat, M.F.; Shrager, D.I.; et al. Serological and nucleic acid analyses for HIV and HTLV infection on archival human plasma samples from Zaire. Virology 1994, 202, 379–389. [Google Scholar] [CrossRef]
- Jain, P.; Manuel, S.L.; Khan, Z.K.; Ahuja, J.; Quann, K.; Wigdahl, B. DC-SIGN mediates cell-free infection and transmission of human T-cell lymphotropic virus type 1 by dendritic cells. J. Virol. 2009, 83, 10908–10921. [Google Scholar] [CrossRef]
- Svajger, U.; Anderluh, M.; Jeras, M.; Obermajer, N. C-type lectin DC-SIGN: An adhesion, signalling and antigen-uptake molecule that guides dendritic cells in immunity. Cell Signal 2010, 22, 1397–1405. [Google Scholar] [CrossRef] [PubMed]
- Valeri, V.W.; Hryniewicz, A.; Andresen, V.; Jones, K.; Fenizia, C.; Bialuk, I.; Chung, H.K.; Fukumoto, R.; Washington Parks, R.; Ferrari, M.G.; et al. Requirement of the human T-cell leukemia virus p12 and p30 genes for infectivity of human dendritic cells and macaques but not rabbits. Blood 2010, 116, 3809–3817. [Google Scholar] [CrossRef] [PubMed]
- Grant, C.; Jain, P.; Nonnemacher, M.; Flaig, K.E.; Irish, B.; Ahuja, J.; Alexaki, A.; Alefantis, T.; Wigdahl, B. AP-1-directed human T cell leukemia virus type 1 viral gene expression during monocytic differentiation. J. Leukoc. Biol. 2006, 80, 640–650. [Google Scholar] [CrossRef]
- Takeuchi, H.; Takahashi, M.; Norose, Y.; Takeshita, T.; Fukunaga, Y.; Takahashi, H. Transformation of breast milk macrophages by HTLV-I: Implications for HTLV-I transmission via breastfeeding. Biomed. Res. 2010, 31, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Lo, K.M.; Vivier, E.; Rochet, N.; Dehni, G.; Levine, H.; Haseltine, W.A.; Anderson, P. Infection of human natural killer (NK) cells with replication-defective human T cell leukemia virus type I provirus. Increased proliferative capacity and prolonged survival of functionally competent NK cells. J. Immunol. 1992, 149, 4101–4108. [Google Scholar] [CrossRef] [PubMed]
- Makino, M.; Wakamatsu, S.; Shimokubo, S.; Arima, N.; Baba, M. Production of functionally deficient dendritic cells from HTLV-I-infected monocytes: implications for the dendritic cell defect in adult T cell leukemia. Virology 2000, 274, 140–148. [Google Scholar] [CrossRef]
- Nascimento, C.R.; Lima, M.A.; de Andrada Serpa, M.J.; Espindola, O.; Leite, A.C.; Echevarria-Lima, J. Monocytes from HTLV-1-infected patients are unable to fully mature into dendritic cells. Blood 2011, 117, 489–499. [Google Scholar] [CrossRef]
- Rahman, S.; Khan, Z.K.; Wigdahl, B.; Jennings, S.R.; Tangy, F.; Jain, P. Murine FLT3 ligand-derived dendritic cell-mediated early immune responses are critical to controlling cell-free human T cell leukemia virus type 1 infection. J. Immunol. 2011, 186, 390–402. [Google Scholar] [CrossRef]
- Colisson, R.; Barblu, L.; Gras, C.; Raynaud, F.; Hadj-Slimane, R.; Pique, C.; Hermine, O.; Lepelletier, Y.; Herbeuval, J.P. Free HTLV-1 induces TLR7-dependent innate immune response and TRAIL relocalization in killer plasmacytoid dendritic cells. Blood 2010, 115, 2177–2185. [Google Scholar] [CrossRef] [PubMed]
- Stewart, S.A.; Feuer, G.; Jewett, A.; Lee, F.V.; Bonavida, B.; Chen, I.S. HTLV-1 gene expression in adult T-cell leukemia cells elicits an NK cell response in vitro and correlates with cell rejection in SCID mice. Virology 1996, 226, 167–175. [Google Scholar] [CrossRef]
- Yu, F.; Itoyama, Y.; Fujihara, K.; Goto, I. Natural killer (NK) cells in HTLV-I-associated myelopathy/tropical spastic paraparesis-decrease in NK cell subset populations and activity in HTLV-I seropositive individuals. J. Neuroimmunol. 1991, 33, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Norris, P.J.; Hirschkorn, D.F.; DeVita, D.A.; Lee, T.H.; Murphy, E.L. Human T cell leukemia virus type 1 infection drives spontaneous proliferation of natural killer cells. Virulence 2010, 1, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Ndhlovu, L.C.; Snyder-Cappione, J.E.; Carvalho, K.I.; Leal, F.E.; Loo, C.P.; Bruno, F.R.; Jha, A.R.; Devita, D.; Hasenkrug, A.M.; Barbosa, H.M.; et al. Lower numbers of circulating Natural Killer T (NK T) cells in individuals with human T lymphotropic virus type 1 (HTLV-1) associated neurological disease. Clin. Exp. Immunol. 2009, 158, 294–299. [Google Scholar] [CrossRef]
- Al-Dahoodi, Z.M.; Takemoto, S.; Kataoka, S.; Taguchi, H. Dysfunction of dendritic and T cells as the cause of immune suppression in HTLV-I infected individuals. J. Clin. Exp. Hematopathol. 2003, 43, 43–48. [Google Scholar] [CrossRef]
- Mascarenhas, R.E.; Brodskyn, C.; Barbosa, G.; Clarencio, J.; Andrade-Filho, A.S.; Figueiroa, F.; Galvao-Castro, B.; Grassi, F. Peripheral blood mononuclear cells from individuals infected with human T-cell lymphotropic virus type 1 have a reduced capacity to respond to recall antigens. Clin. Vaccine Immunol. 2006, 13, 547–552. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, H.; Miyoshi, I. Immune suppression in HTLV-I carriers: A predictive sign of adult T-cell leukemia. Acta Med. Okayama 1989, 43, 317–321. [Google Scholar]
- Oliere, S.; Hernandez, E.; Lezin, A.; Arguello, M.; Douville, R.; Nguyen, T.L.; Olindo, S.; Panelatti, G.; Kazanji, M.; Wilkinson, P.; et al. HTLV-1 evades type I interferon antiviral signaling by inducing the suppressor of cytokine signaling 1 (SOCS1). PLoS Pathog. 2010, 6, e1001177. [Google Scholar] [CrossRef]
- Suzuki, S.; Zhou, Y.; Refaat, A.; Takasaki, I.; Koizumi, K.; Yamaoka, S.; Tabuchi, Y.; Saiki, I.; Sakurai, H. Human T cell lymphotropic virus 1 manipulates interferon regulatory signals by controlling the TAK1-IRF3 and IRF4 pathways. J. Biol. Chem. 2010, 285, 4441–4446. [Google Scholar] [CrossRef]
- Banerjee, P.; Feuer, G.; Barker, E. Human T-cell leukemia virus type 1 (HTLV-1) p12I down-modulates ICAM-1 and -2 and reduces adherence of natural killer cells, thereby protecting HTLV-1-infected primary CD4+ T cells from autologous natural killer cell-mediated cytotoxicity despite the reduction of major histocompatibility complex class I molecules on infected cells. J. Virol. 2007, 81, 9707–9717. [Google Scholar]
- Ali, A.; Patterson, S.; Cruickshank, K.; Rudge, P.; Dalgleish, A.G.; Knight, S.C. Dendritic cells infected in vitro with human T cell leukaemia/lymphoma virus type-1 (HTLV-1); enhanced lymphocytic proliferation and tropical spastic paraparesis. Clin. Exp. Immunol. 1993, 94, 32–37. [Google Scholar] [CrossRef]
- Arima, N.; Daitoku, Y.; Hidaka, S.; Yamamoto, Y.; Fujimoto, K.; Matsushita, K.; Ohtsubo, H.; Fukumori, J.; Tanaka, H.; Onoue, K. Interleukin-2 production by primary adult T cell leukemia tumor cells is macrophage dependent. Am. J. Hematol. 1992, 41, 258–263. [Google Scholar] [CrossRef] [PubMed]
- Fujihara, K.; Itoyama, Y.; Yu, F.; Kubo, C.; Goto, I. Cellular immune surveillance against HTLV-I infected T lymphocytes in HTLV-I associated myelopathy/tropical spastic paraparesis (HAM/TSP). J. Neurol. Sci. 1991, 105, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Fenner, J.E.; Starr, R.; Cornish, A.L.; Zhang, J.G.; Metcalf, D.; Schreiber, R.D.; Sheehan, K.; Hilton, D.J.; Alexander, W.S.; Hertzog, P.J. Suppressor of cytokine signaling 1 regulates the immune response to infection by a unique inhibition of type I interferon activity. Nat. Immunol. 2006, 7, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Charoenthongtrakul, S.; Zhou, Q.; Shembade, N.; Harhaj, N.S.; Harhaj, E.W. HTLV-I Tax inhibits innate antiviral signaling via NF-{kappa}B-dependent induction of SOCS1. J. Virol. 2011. [Google Scholar] [CrossRef]
- Datta, A.; Sinha-Datta, U.; Dhillon, N.K.; Buch, S.; Nicot, C. The HTLV-I p30 interferes with TLR4 signaling and modulates the release of pro- and anti-inflammatory cytokines from human macrophages. J. Biol. Chem. 2006, 281, 23414–23424. [Google Scholar] [CrossRef]
- Johnson, J.M.; Nicot, C.; Fullen, J.; Ciminale, V.; Casareto, L.; Mulloy, J.C.; Jacobson, S.; Franchini, G. Free major histocompatibility complex class I heavy chain is preferentially targeted for degradation by human T-cell leukemia/lymphotropic virus type 1 p12(I) protein. J. Virol. 2001, 75, 6086–6094. [Google Scholar] [CrossRef]
- Arimura, K.; Nakagawa, M.; Izumo, S.; Usuku, K.; Itoyama, Y.; Kira, J.; Osame, M. Safety and efficacy of interferon-alpha in 167 patients with human T-cell lymphotropic virus type 1-associated myelopathy. J. Neurovirol. 2007, 13, 364–372. [Google Scholar] [CrossRef]
- Izumo, S.; Goto, I.; Itoyama, Y.; Okajima, T.; Watanabe, S.; Kuroda, Y.; Araki, S.; Mori, M.; Nagataki, S.; Matsukura, S.; et al. Interferon-alpha is effective in HTLV-I-associated myelopathy: a multicenter, randomized, double-blind, controlled trial. Neurology 1996, 46, 1016–1021. [Google Scholar] [CrossRef]
- Kuroda, Y.; Kurohara, K.; Fujiyama, F.; Takashima, H.; Endo, C.; Matsui, M.; Neshige, R.; Kakigi, R. Systemic interferon-alpha in the treatment of HTLV-I-associated myelopathy. Acta Neurol. Scand. 1992, 86, 82–86. [Google Scholar] [CrossRef]
- Nakagawa, M.; Nakahara, K.; Maruyama, Y.; Kawabata, M.; Higuchi, I.; Kubota, H.; Izumo, S.; Arimura, K.; Osame, M. Therapeutic trials in 200 patients with HTLV-I-associated myelopathy/ tropical spastic paraparesis. J. Neurovirol. 1996, 2, 345–355. [Google Scholar] [CrossRef]
- Nakamura, T.; Shibayama, K.; Nagasato, K.; Matsuo, H.; Tsujihata, M.; Nagataki, S. The efficacy of interferon-alpha treatment in human T-lymphotropic virus type-I-associated myelopathy. Jpn. J. Med. 1990, 29, 362–367. [Google Scholar] [CrossRef] [PubMed]
- Saito, M.; Nakagawa, M.; Kaseda, S.; Matsuzaki, T.; Jonosono, M.; Eiraku, N.; Kubota, R.; Takenouchi, N.; Nagai, M.; Furukawa, Y.; et al. Decreased human T lymphotropic virus type I (HTLV-I) provirus load and alteration in T cell phenotype after interferon-alpha therapy for HTLV-I-associated myelopathy/tropical spastic paraparesis. J. Infect. Dis. 2004, 189, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, K.; Kira, J.; Koyanagi, Y.; Kawano, Y.; Miyano-Kurosaki, N.; Nakamura, M.; Baba, E.; Suzuki, J.; Yamamoto, A.; Yamamoto, N.; et al. Long-term, high dose interferon-alpha treatment in HTLV-I-associated myelopathy/tropical spastic paraparesis: a combined clinical, virological and immunological study. J. Neurol. Sci. 1997, 147, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Oh, U.; Yamano, Y.; Mora, C.A.; Ohayon, J.; Bagnato, F.; Butman, J.A.; Dambrosia, J.; Leist, T.P.; McFarland, H.; Jacobson, S. Interferon-beta1a therapy in human T-lymphotropic virus type I-associated neurologic disease. Ann. Neurol. 2005, 57, 526–534. [Google Scholar] [CrossRef] [PubMed]
- Datta, A.; Bellon, M.; Sinha-Datta, U.; Bazarbachi, A.; Lepelletier, Y.; Canioni, D.; Waldmann, T.A.; Hermine, O.; Nicot, C. Persistent inhibition of telomerase reprograms adult T-cell leukemia to p53-dependent senescence. Blood 2006, 108, 1021–1029. [Google Scholar] [CrossRef]
- Evans, D.T.; Serra-Moreno, R.; Singh, R.K.; Guatelli, J.C. BST-2/tetherin: A new component of the innate immune response to enveloped viruses. Trends Microbiol. 2010, 18, 388–396. [Google Scholar] [CrossRef]
- Jouvenet, N.; Neil, S.J.; Zhadina, M.; Zang, T.; Kratovac, Z.; Lee, Y.; McNatt, M.; Hatziioannou, T.; Bieniasz, P.D. Broad-spectrum inhibition of retroviral and filoviral particle release by tetherin. J. Virol. 2009, 83, 1837–1844. [Google Scholar] [CrossRef]
- Kinpara, S.; Hasegawa, A.; Utsunomiya, A.; Nishitsuji, H.; Furukawa, H.; Masuda, T.; Kannagi, M. Stromal cell-mediated suppression of human T-cell leukemia virus type 1 expression in vitro and in vivo by type I interferon. J. Virol. 2009, 83, 5101–5108. [Google Scholar] [CrossRef]

© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Journo, C.; Mahieux, R. HTLV-1 and Innate Immunity. Viruses 2011, 3, 1374-1394. https://doi.org/10.3390/v3081374
Journo C, Mahieux R. HTLV-1 and Innate Immunity. Viruses. 2011; 3(8):1374-1394. https://doi.org/10.3390/v3081374
Chicago/Turabian StyleJourno, Chloé, and Renaud Mahieux. 2011. "HTLV-1 and Innate Immunity" Viruses 3, no. 8: 1374-1394. https://doi.org/10.3390/v3081374
APA StyleJourno, C., & Mahieux, R. (2011). HTLV-1 and Innate Immunity. Viruses, 3(8), 1374-1394. https://doi.org/10.3390/v3081374