Viral Determinants of FeLV Infection and Pathogenesis: Lessons Learned from Analysis of a Natural Cohort

Abstract
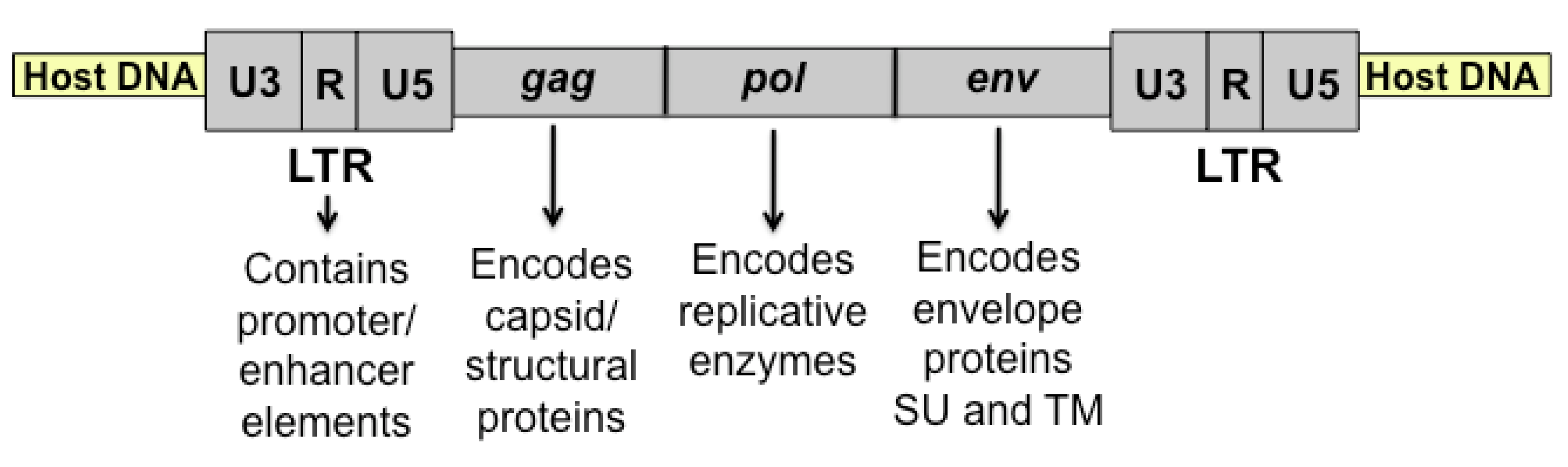
:1. Introduction
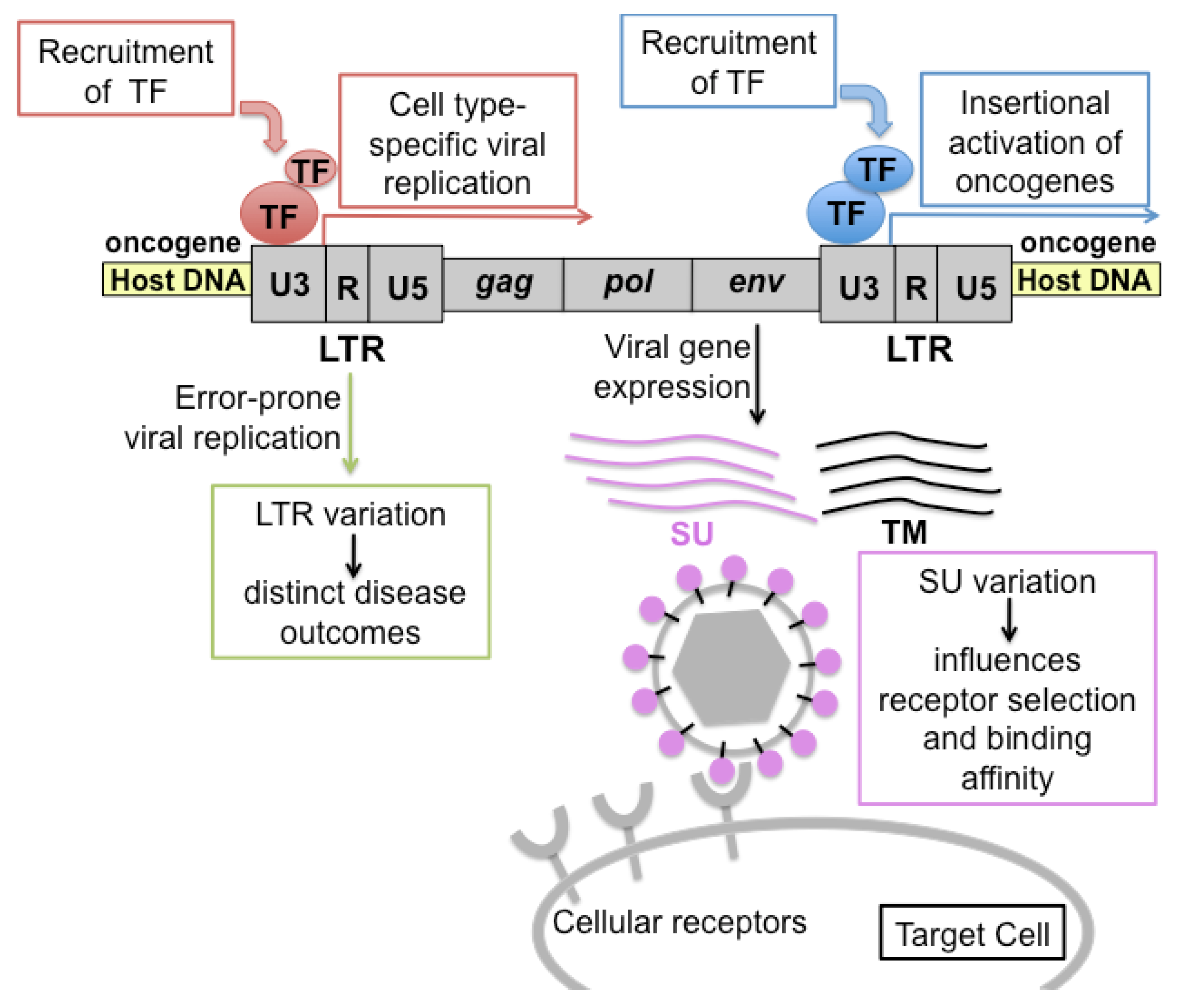
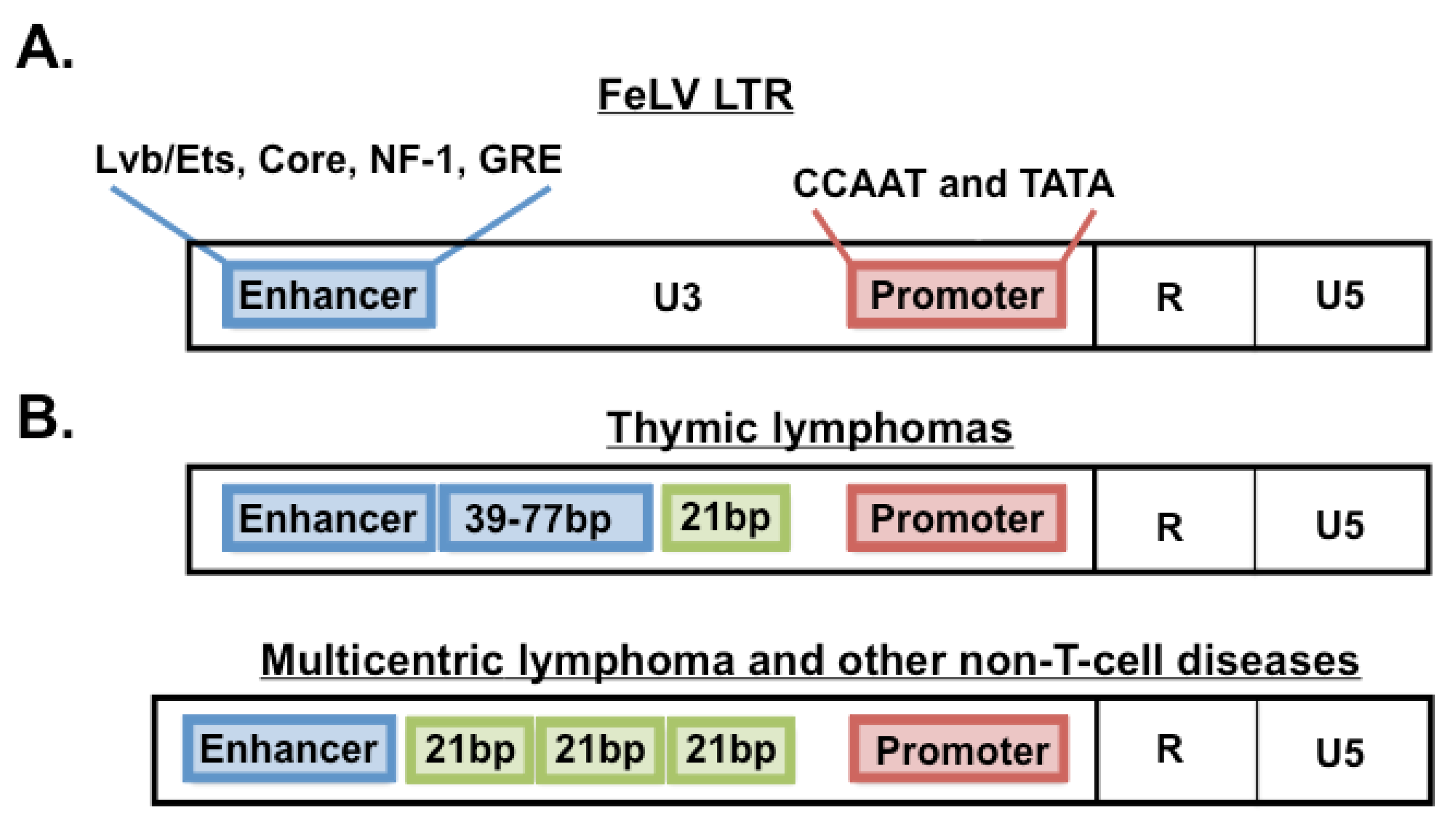
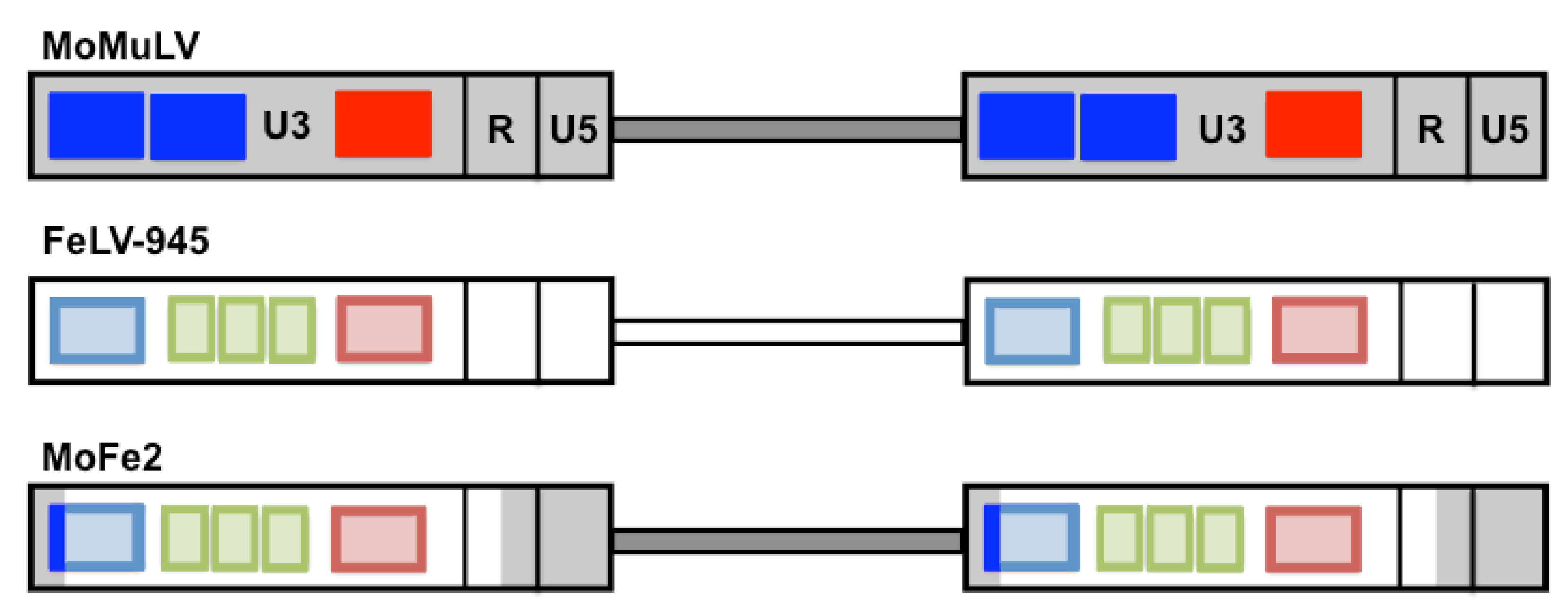
2. The FeLV Long Terminal Repeat (LTR) as a Determinant of Pathogenesis in the Cohort
3. Viral Surface Glycoprotein (SU) as a Determinant of Pathogenesis in the Cohort
4. Oncogene Activation in FeLV-Mediated Lymphomas in the Cohort
4.1. Oncogene Activation in Thymic Lymphomas
4.2. Oncogene Activation in Multicentric, Non-T-Cell Tumors
5. Conclusions
Acknowledgments
Conflict of Interest
References and Notes
- Hoover, E.A.; Mullins, J.I. Feline leukemia virus infection and diseases. J. Am. Vet. Med. Assoc. 1991, 199, 1287–1297. [Google Scholar] [PubMed]
- Neil, J.C.; Fulton, R.; Rigby, M.; Stewart, M. Feline leukaemia virus: Generation of pathogenic and oncogenic variants. Curr. Top. Microbiol. Immunol. 1991, 171, 67–93. [Google Scholar] [PubMed]
- Overbaugh, J.; Bangham, C.R. Selection forces and constraints on retroviral sequence variation. Science 2001, 292, 1106–1109. [Google Scholar] [CrossRef]
- Cotter, S.M. Feline leukemia virus: Pathophysiology, prevention, and treatment. Cancer Invest. 1992, 10, 173–181. [Google Scholar] [CrossRef]
- Athas, G.B.; Choi, B.; Prabhu, S.; Lobelle-Rich, P.A.; Levy, L.S. Genetic determinants of feline leukemia virus-induced multicentric lymphomas. Virology 1995, 214, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Fan, H. Leukemogenesis by moloney murine leukemia virus: A multistep process. Trends Microbiol. 1997, 5, 74–82. [Google Scholar] [CrossRef]
- Fulton, R.; Plumb, M.; Shield, L.; Neil, J.C. Structural diversity and nuclear protein binding sites in the long terminal repeats of feline leukemia virus. J. Virol. 1990, 64, 1675–1682. [Google Scholar] [CrossRef]
- Helfer-Hungerbuehler, A.K.; Cattori, V.; Boretti, F.S.; Ossent, P.; Grest, P.; Reinacher, M.; Henrich, M.; Bauer, E.; Bauer-Pham, K.; Niederer, E.; et al. Dominance of highly divergent feline leukemia virus a progeny variants in a cat with recurrent viremia and fatal lymphoma. Retrovirology 2010, 7, 14. [Google Scholar] [CrossRef]
- Hisasue, M.; Nagashima, N.; Nishigaki, K.; Fukuzawa, I.; Ura, S.; Katae, H.; Tsuchiya, R.; Yamada, T.; Hasegawa, A.; Tsujimoto, H. Myelodysplastic syndromes and acute myeloid leukemia in cats infected with feline leukemia virus clone33 containing a unique long terminal repeat. Int. J. Cancer 2009, 124, 1133–1141. [Google Scholar] [CrossRef]
- Nishigaki, K.; Hanson, C.; Thompson, D.; Yugawa, T.; Hisasue, M.; Tsujimoto, H.; Ruscetti, S. Analysis of the disease potential of a recombinant retrovirus containing friend murine leukemia virus sequences and a unique long terminal repeat from feline leukemia virus. J. Virol. 2002, 76, 1527–1532. [Google Scholar] [CrossRef]
- Nishigaki, K.; Okuda, M.; Endo, Y.; Watari, T.; Tsujimoto, H.; Hasegawa, A. Structure and function of the long terminal repeats of feline leukemia viruses derived from naturally occurring acute myeloid leukemias in cats. J. Virol. 1997, 71, 9823–9827. [Google Scholar] [CrossRef] [PubMed]
- Chandhasin, C.; Lobelle-Rich, P.; Levy, L.S. Feline leukaemia virus ltr variation and disease association in a geographical and temporal cluster. J. Gen. Virol. 2004, 85, 2937–2942. [Google Scholar] [CrossRef] [PubMed]
- Pantginis, J.; Beaty, R.M.; Levy, L.S.; Lenz, J. The feline leukemia virus long terminal repeat contains a potent genetic determinant of t-cell lymphomagenicity. J. Virol. 1997, 71, 9786–9791. [Google Scholar] [CrossRef]
- Levesque, K.S.; Bonham, L.; Levy, L.S. Flvi-1, a common integration domain of feline leukemia virus in naturally occurring lymphomas of a particular type. J. Virol. 1990, 64, 3455–3462. [Google Scholar] [CrossRef] [PubMed]
- Athas, G.B.; Lobelle-Rich, P.; Levy, L.S. Function of a unique sequence motif in the long terminal repeat of feline leukemia virus isolated from an unusual set of naturally occurring tumors. J. Virol. 1995, 69, 3324–3332. [Google Scholar] [CrossRef]
- Prabhu, S.; Lobelle-Rich, P.A.; Levy, L.S. The felv-945 ltr confers a replicative advantage dependent on the presence of a tandem triplication. Virology 1999, 263, 460–470. [Google Scholar] [CrossRef]
- Coffin, J.M. Genetic diversity and evolution of retroviruses. Curr. Top. Microbiol. Immunol. 1992, 176, 143–164. [Google Scholar]
- Rhodes, D.; Klug, A. Helical periodicity of DNA determined by enzyme digestion. Nature 1980, 286, 573–578. [Google Scholar] [CrossRef]
- Tullius, T.D.; Dombroski, B.A. Iron(ii) edta used to measure the helical twist along any DNA molecule. Science 1985, 230, 679–681. [Google Scholar] [CrossRef]
- Finstad, S.L.; Prabhu, S.; Rulli, K.R.; Levy, L.S. Regulation of felv-945 by c-myb binding and cbp recruitment to the ltr. Virol. J. 2004, 1, 3. [Google Scholar] [CrossRef]
- Starkey, C.R.; Lobelle-Rich, P.A.; Granger, S.; Brightman, B.K.; Fan, H.; Levy, L.S. Tumorigenic potential of a recombinant retrovirus containing sequences from moloney murine leukemia virus and feline leukemia virus. J. Virol. 1998, 72, 1078–1084. [Google Scholar] [CrossRef] [PubMed]
- Battini, J.L.; Danos, O.; Heard, J.M. Receptor-binding domain of murine leukemia virus envelope glycoproteins. J. Virol. 1995, 69, 713–719. [Google Scholar] [CrossRef] [PubMed]
- Battini, J.L.; Heard, J.M.; Danos, O. Receptor choice determinants in the envelope glycoproteins of amphotropic, xenotropic, and polytropic murine leukemia viruses. J. Virol. 1992, 66, 1468–1475. [Google Scholar] [CrossRef] [PubMed]
- Boomer, S.; Eiden, M.; Burns, C.C.; Overbaugh, J. Three distinct envelope domains, variably present in subgroup b feline leukemia virus recombinants, mediate pit1 and pit2 receptor recognition. J. Virol. 1997, 71, 8116–8123. [Google Scholar] [CrossRef]
- MacKrell, A.J.; Soong, N.W.; Curtis, C.M.; Anderson, W.F. Identification of a subdomain in the moloney murine leukemia virus envelope protein involved in receptor binding. J. Virol. 1996, 70, 1768–1774. [Google Scholar] [CrossRef]
- Morgan, R.A.; Nussbaum, O.; Muenchau, D.D.; Shu, L.; Couture, L.; Anderson, W.F. Analysis of the functional and host range-determining regions of the murine ectropic and amphotropic retrovirus envelope proteins. J. Virol. 1993, 67, 4712–4721. [Google Scholar] [CrossRef]
- Rigby, M.A.; Rojko, J.L.; Stewart, M.A.; Kociba, G.J.; Cheney, C.M.; Rezanka, L.J.; Mathes, L.E.; Hartke, J.R.; Jarrett, O.; Neil, J.C. Partial dissociation of subgroup c phenotype and in vivo behaviour in feline leukaemia viruses with chimeric envelope genes. J. Gen. Virol. 1992, 73, 2839–2847. [Google Scholar] [CrossRef]
- Tailor, C.S.; Kabat, D. Variable regions a and b in the envelope glycoproteins of feline leukemia virus subgroup b and amphotropic murine leukemia virus interact with discrete receptor domains. J. Virol. 1997, 71, 9383–9391. [Google Scholar] [CrossRef]
- Lavillette, D.; Maurice, M.; Roche, C.; Russell, S.J.; Sitbon, M.; Cosset, F.L. A proline-rich motif downstream of the receptor binding domain modulates conformation and fusogenicity of murine retroviral envelopes. J. Virol. 1998, 72, 9955–9965. [Google Scholar] [CrossRef]
- Faix, P.H.; Feldman, S.A.; Overbaugh, J.; Eiden, M.V. Host range and receptor binding properties of vectors bearing feline leukemia virus subgroup b envelopes can be modulated by envelope sequences outside of the receptor binding domain. J. Virol. 2002, 76, 12369–12375. [Google Scholar] [CrossRef]
- Sugai, J.; Eiden, M.; Anderson, M.M.; Van Hoeven, N.; Meiering, C.D.; Overbaugh, J. Identification of envelope determinants of feline leukemia virus subgroup b that permit infection and gene transfer to cells expressing human pit1 or pit2. J. Virol. 2001, 75, 6841–6849. [Google Scholar] [CrossRef] [PubMed]
- Donahue, P.R.; Quackenbush, S.L.; Gallo, M.V.; deNoronha, C.M.; Overbaugh, J.; Hoover, E.A.; Mullins, J.I. Viral genetic determinants of t-cell killing and immunodeficiency disease induction by the feline leukemia virus felv-faids. J. Virol. 1991, 65, 4461–4469. [Google Scholar] [CrossRef] [PubMed]
- Rezanka, L.J.; Rojko, J.L.; Neil, J.C. Feline leukemia virus: Pathogenesis of neoplastic disease. Cancer Invest. 1992, 10, 371–389. [Google Scholar] [CrossRef] [PubMed]
- Rohn, J.L.; Moser, M.S.; Gwynn, S.R.; Baldwin, D.N.; Overbaugh, J. In vivo evolution of a novel, syncytium-inducing and cytopathic feline leukemia virus variant. J. Virol. 1998, 72, 2686–2696. [Google Scholar] [CrossRef]
- Mendoza, R.; Anderson, M.M.; Overbaugh, J. A putative thiamine transport protein is a receptor for feline leukemia virus subgroup a. J. Virol. 2006, 80, 3378–3385. [Google Scholar] [CrossRef]
- Overbaugh, J.; Miller, A.D.; Eiden, M.V. Receptors and entry cofactors for retroviruses include single and multiple transmembrane-spanning proteins as well as newly described glycophosphatidylinositol-anchored and secreted proteins. Microbiol. Mol. Biol. Rev. 2001, 65, 371–389, table of contents. [Google Scholar] [CrossRef]
- Chandhasin, C.; Coan, P.N.; Levy, L.S. Subtle mutational changes in the su protein of a natural feline leukemia virus subgroup a isolate alter disease spectrum. J. Virol. 2005, 79, 1351–1360. [Google Scholar] [CrossRef]
- Donahue, P.R.; Hoover, E.A.; Beltz, G.A.; Riedel, N.; Hirsch, V.M.; Overbaugh, J.; Mullins, J.I. Strong sequence conservation among horizontally transmissible, minimally pathogenic feline leukemia viruses. J. Virol. 1988, 62, 722–731. [Google Scholar] [CrossRef]
- Ahmad, S.; Levy, L.S. The frequency of occurrence and nature of recombinant feline leukemia viruses in the induction of multicentric lymphoma by infection of the domestic cat with felv-945. Virology 2010, 403, 103–110. [Google Scholar] [CrossRef]
- Jarrett, O. Natural occurrence of subgroups of feline leukemia virus. In Viruses in Naturally Occurring Cancers; Essex, M., Todaro, G., zur Hausen, H., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1980; pp. 603–611. [Google Scholar]
- Jarrett, O. Pathogenicity of feline leukemia virus is commonly associated with variant viruses. Leukemia 1992, 6, 153S–154S. [Google Scholar]
- Roy-Burman, P. Endogenous env elements: Partners in generation of pathogenic feline leukemia viruses. Virus Genes 1995, 11, 147–161. [Google Scholar] [CrossRef] [PubMed]
- Sheets, R.L.; Pandey, R.; Jen, W.C.; Roy-Burman, P. Recombinant feline leukemia virus genes detected in naturally occurring feline lymphosarcomas. J. Virol. 1993, 67, 3118–3125. [Google Scholar] [CrossRef] [PubMed]
- Tsatsanis, C.; Fulton, R.; Nishigaki, K.; Tsujimoto, H.; Levy, L.; Terry, A.; Spandidos, D.; Onions, D.; Neil, J.C. Genetic determinants of feline leukemia virus-induced lymphoid tumors: Patterns of proviral insertion and gene rearrangement. J. Virol. 1994, 68, 8296–8303. [Google Scholar] [CrossRef] [PubMed]
- Chandhasin, C.; Coan, P.N.; Pandrea, I.; Grant, C.K.; Lobelle-Rich, P.A.; Puetter, A.; Levy, L.S. Unique long terminal repeat and surface glycoprotein gene sequences of feline leukemia virus as determinants of disease outcome. J. Virol. 2005, 79, 5278–5287. [Google Scholar] [CrossRef]
- Bolin, L.L.; Ahmad, S.; Lobelle-Rich, P.; Levy, L.S. Department of Microbiology and Immunology and Tulane Cancer Center, School of Medicine, Tulane University, New Orleans, LA, USA. FeLV-945 redirects T cell lymphoma to thymus-excluded, non-T cell disease utilizing its unique SU and LTR. Preliminary observation. 2011. [Google Scholar]
- Bolin, L.L.; Chandhasin, C.; Lobelle-Rich, P.A.; Albritton, L.M.; Levy, L.S. Distinctive receptor binding properties of the surface glycoprotein of a natural feline leukemia virus isolate with unusual disease spectrum. Retrovirology 2011, 8, 35. [Google Scholar] [CrossRef]
- Barnett, A.L.; Wensel, D.L.; Li, W.; Fass, D.; Cunningham, J.M. Structure and mechanism of a coreceptor for infection by a pathogenic feline retrovirus. J. Virol. 2003, 77, 2717–2729. [Google Scholar] [CrossRef]
- Hansen, G.M.; Skapura, D.; Justice, M.J. Genetic profile of insertion mutations in mouse leukemias and lymphomas. Genome Res. 2000, 10, 237–243. [Google Scholar] [CrossRef]
- Li, J.; Shen, H.; Himmel, K.L.; Dupuy, A.J.; Largaespada, D.A.; Nakamura, T.; Shaughnessy, J.D., Jr.; Jenkins, N.A.; Copeland, N.G. Leukaemia disease genes: Large-scale cloning and pathway predictions. Nat. Genet. 1999, 23, 348–353. [Google Scholar] [CrossRef]
- Mikkers, H.; Berns, A. Retroviral insertional mutagenesis: Tagging cancer pathways. Adv. Cancer Res. 2003, 88, 53–99. [Google Scholar]
- Levy, L.S.; Gardner, M.B.; Casey, J.W. Isolation of a feline leukaemia provirus containing the oncogene myc from a feline lymphosarcoma. Nature 1984, 308, 853–856. [Google Scholar] [CrossRef]
- Levy, L.S.; Lobelle-Rich, P.A. Insertional mutagenesis of flvi-2 in tumors induced by infection with lc-felv, a myc-containing strain of feline leukemia virus. J. Virol. 1992, 66, 2885–2892. [Google Scholar] [CrossRef] [PubMed]
- Levy, L.S.; Lobelle-Rich, P.A.; Overbaugh, J.; Abkowitz, J.L.; Fulton, R.; Roy-Burman, P. Coincident involvement of flvi-2, c-myc, and novel env genes in natural and experimental lymphosarcomas induced by feline leukemia virus. Virology 1993, 196, 892–895. [Google Scholar] [CrossRef]
- Johnson, C.; Lobelle-Rich, P.A.; Puetter, A.; Levy, L.S. Substitution of feline leukemia virus long terminal repeat sequences into murine leukemia virus alters the pattern of insertional activation and identifies new common insertion sites. J. Virol. 2005, 79, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Levy, L.S.; Starkey, C.R.; Prabhu, S.; Lobelle-Rich, P.A. Cooperating events in lymphomagenesis mediated by feline leukemia virus. Leukemia 1997, 11, 239–241. [Google Scholar]
- Braun, M.J.; Deininger, P.L.; Casey, J.W. Nucleotide sequence of a transduced myc gene from a defective feline leukemia provirus. J. Virol. 1985, 55, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Bonham, L.; Lobelle-Rich, P.A.; Henderson, L.A.; Levy, L.S. Transforming potential of a myc-containing variant of feline leukemia virus in vitro in early-passage feline cells. J. Virol. 1987, 61, 3072–3081. [Google Scholar] [CrossRef]
- Levy, L.S.; Fish, R.E.; Baskin, G.B. Tumorigenic potential of a myc-containing strain of feline leukemia virus in vivo in domestic cats. J. Virol. 1988, 62, 4770–4773. [Google Scholar] [CrossRef]
- Levy, L.S.; Lobelle-Rich, P.A.; Overbaugh, J. Flvi-2, a target of retroviral insertional mutagenesis in feline thymic lymphosarcomas, encodes bmi-1. Oncogene 1993, 8, 1833–1838. [Google Scholar]
- Jiang, L.; Li, J.; Song, L. Bmi-1, stem cells and cancer. Acta Biochim. Biophys. Sin. (Shanghai) 2009, 41, 527–534. [Google Scholar] [CrossRef]
- Raaphorst, F.M. Self-renewal of hematopoietic and leukemic stem cells: A central role for the polycomb-group gene bmi-1. Trends Immunol. 2003, 24, 522–524. [Google Scholar] [CrossRef]
- Tsujimoto, H.; Fulton, R.; Nishigaki, K.; Matsumoto, Y.; Hasegawa, A.; Tsujimoto, A.; Cevario, S.; O'Brien, S.J.; Terry, A.; Onions, D.; et al. A common proviral integration region, fit-1, in t-cell tumors induced by myc-containing feline leukemia viruses. Virology 1993, 196, 845–848. [Google Scholar] [CrossRef] [PubMed]
- Levesque, K.S.; Mattei, M.G.; Levy, L.S. Evolutionary conservation and chromosomal localization of flvi-1. Oncogene 1991, 6, 1377–1379. [Google Scholar] [PubMed]
- Johnson, C.; Marriott, S.J.; Levy, L.S. Overexpression of p101 activates pi3kgamma signaling in t cells and contributes to cell survival. Oncogene 2007, 26, 7049–7057. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CIS | Virus | Tumor Type b | Gene Function | Reference |
|---|---|---|---|---|
| c-Myc | FeLV | Thymic lymphoma | Controls cell proliferation and survival | [44,52] |
| flvi-2 = bmi-1 | FeLV, LC-FeLV | Thymic lymphoma | Controls self-renewal vs. differentiation in hematopoiesis | [44,53,54] |
| pim-1 | FeLV | Thymic lymphoma | serine-threonine kinase; survival signaling | [44] |
| fit-1 | FeLV | Thymic lymphoma | Unknown | [44] |
| flvi-1 | FeLV-945 | Multicentric lymphoma | Unknown | [5,14] |
| p101 | MoFe2 | Thymic lymphoma | Regulatory subunit of PI3Kγ | [55] |
| Rasgrp1 | MoFe2 | Thymic lymphoma | Ras guanyl releasing protein; cell signaling | [55] |
| Jundm2 | MoFe2 | Thymic lymphoma | Jun dimerization protein (predicted); cell signaling | [55] |
| Ahi-1 | MoFe2 | Thymic lymphoma | Abelson helper virus integration site; associated with neurologic and hematologic disorders | [55] |
| Rras2 | MoFe2 | Thymic lymphoma | Ras-related protein; cell signaling | [55] |
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bolin, L.L.; Levy, L.S. Viral Determinants of FeLV Infection and Pathogenesis: Lessons Learned from Analysis of a Natural Cohort. Viruses 2011, 3, 1681-1698. https://doi.org/10.3390/v3091681
Bolin LL, Levy LS. Viral Determinants of FeLV Infection and Pathogenesis: Lessons Learned from Analysis of a Natural Cohort. Viruses. 2011; 3(9):1681-1698. https://doi.org/10.3390/v3091681
Chicago/Turabian StyleBolin, Lisa L., and Laura S. Levy. 2011. "Viral Determinants of FeLV Infection and Pathogenesis: Lessons Learned from Analysis of a Natural Cohort" Viruses 3, no. 9: 1681-1698. https://doi.org/10.3390/v3091681
APA StyleBolin, L. L., & Levy, L. S. (2011). Viral Determinants of FeLV Infection and Pathogenesis: Lessons Learned from Analysis of a Natural Cohort. Viruses, 3(9), 1681-1698. https://doi.org/10.3390/v3091681