Recombination in Eukaryotic Single Stranded DNA Viruses


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Homologous Recombination between ssDNA Virus Genomes
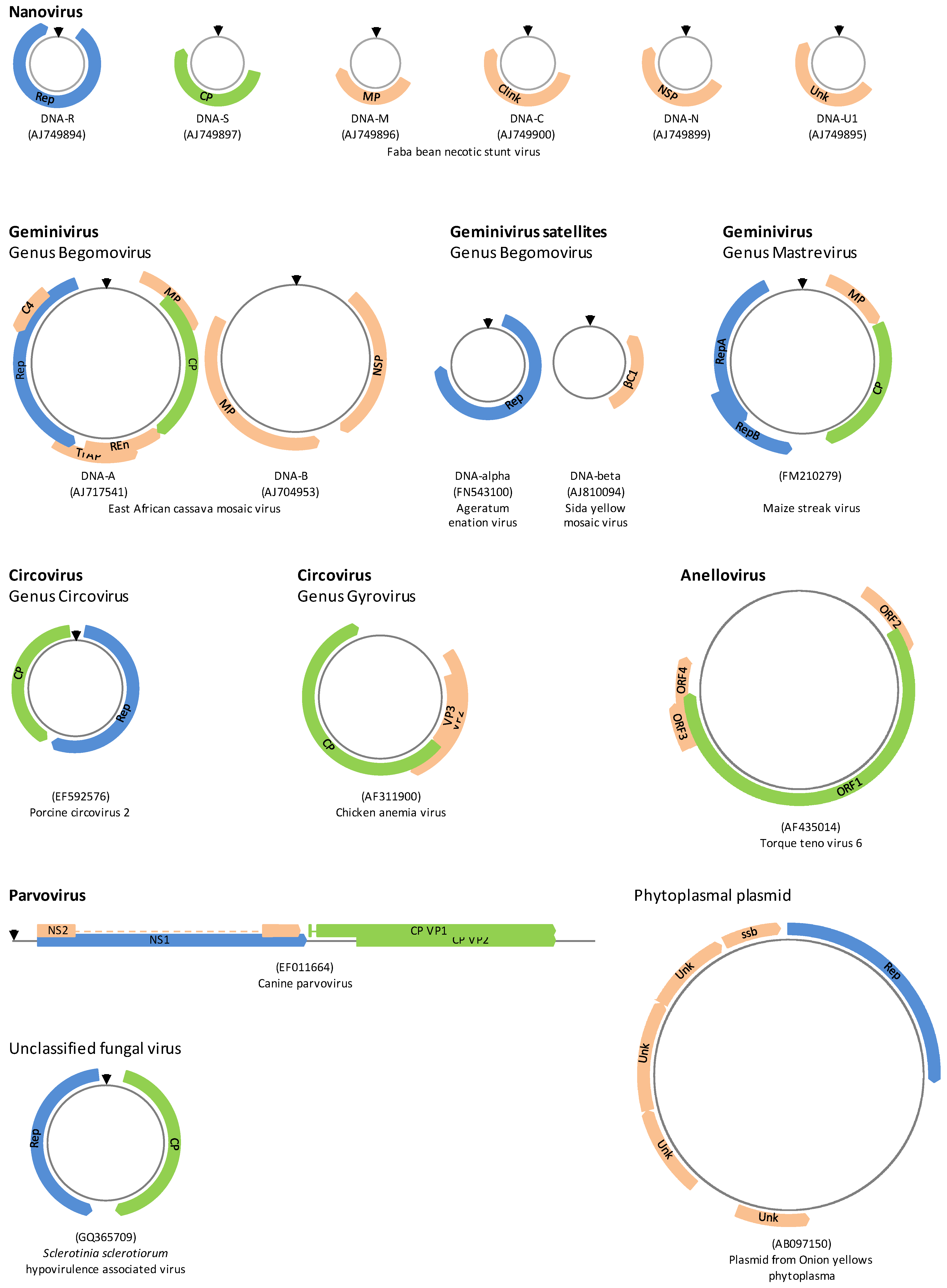
2.1. Replication of ssDNA Viruses
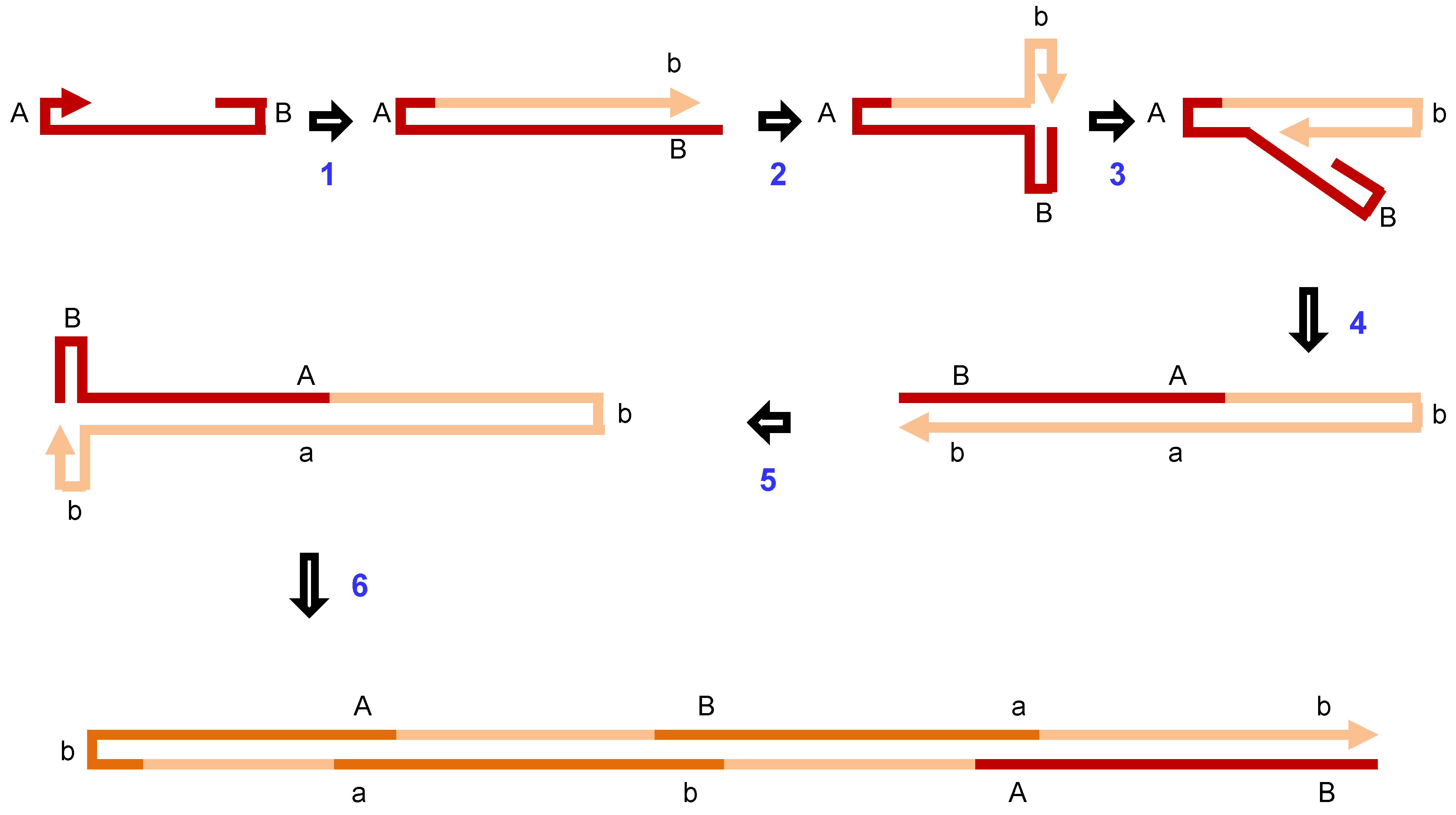
2.2. Mechanisms of Homologous Recombination
3. Component Reassortment
4. Inter-Component Recombination
5. Genome Rearrangement, Insertions and Deletions
6. Recombination between Viral and Host Genomes
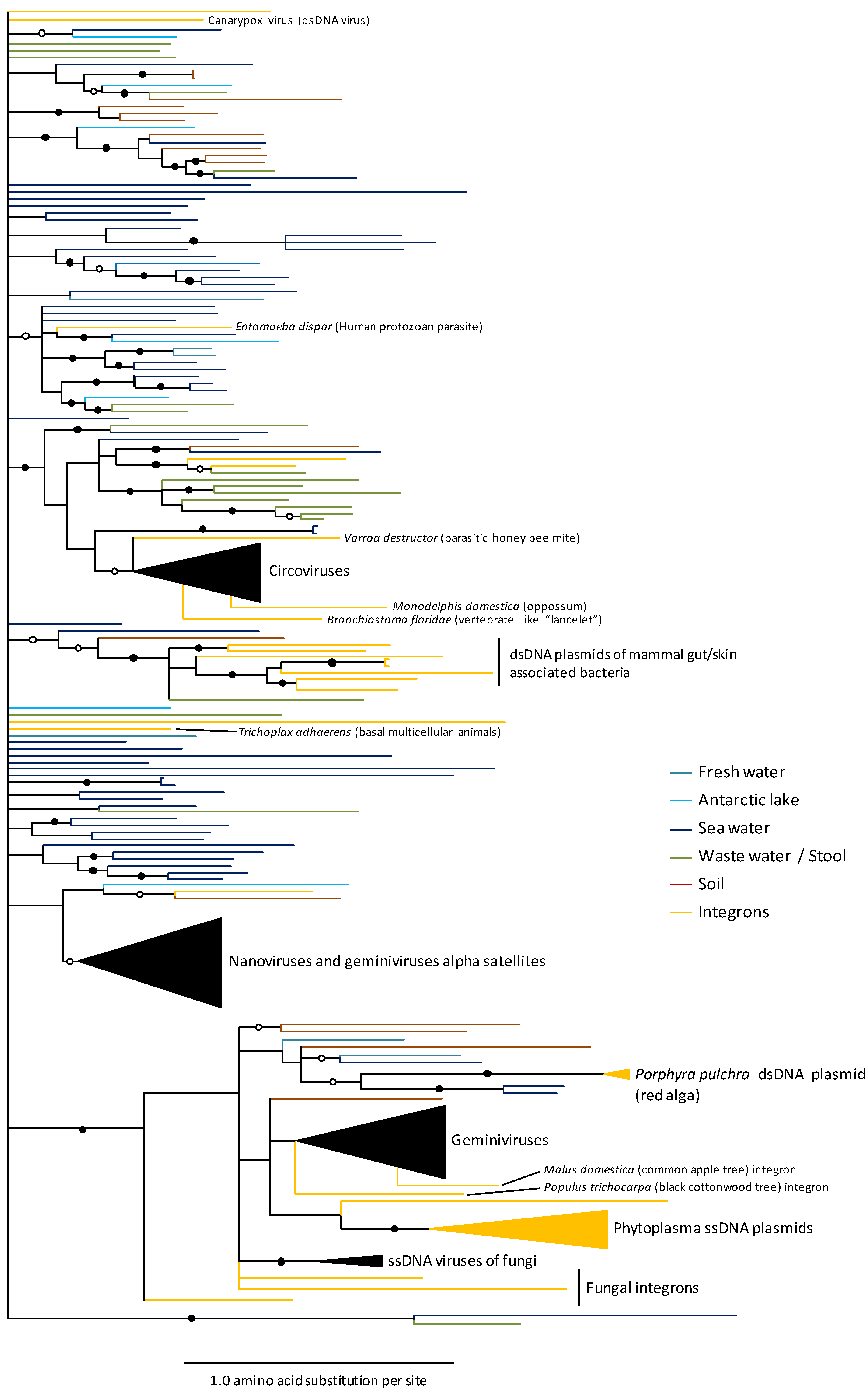
7. Ecological and Epidemiological Influences on Patterns of Recombination between ssDNA Virus Populations
8. Mechanistic Influences on Homologous Recombination Patterns
8.1. Replication Origins
8.2. Sequence Similarity
8.3. ssDNA Secondary Structure
8.4. Transcription-Replication Clashes
8.5. Differential Degrees of ssDNA Exposure within Mini-Chromosomes
9. The Adaptive Value of Recombination in ssDNA Viruses
10. Selective Constraints on the Adaptive Value of Recombination
10.1. Disruption of Long-Range Intra-Genome Interactions
10.2. Disruption of Protein Folding and Oligomerization
10.3. Disruption of Genomic Secondary Structure
11. Conclusions
Acknowledgments
Conflict of Interest
References and Notes
- Blinkova, O.; Rosario, K.; Li, L.; Kapoor, A.; Slikas, B.; Bernardin, F.; Breitbart, M.; Delwart, E. Frequent detection of highly diverse variants of cardiovirus, cosavirus, bocavirus, and circovirus in sewage samples collected in the united states. J. Clin. Microbiol. 2009, 47, 3507–3513. [Google Scholar] [CrossRef]
- Blinkova, O.; Victoria, J.; Li, Y.Y.; Keele, B.F.; Sanz, C.; Ndjango, J.B.N.; Peeters, M.; Travis, D.; Lonsdorf, E.V.; Wilson, M.L.; et al. Novel circular DNA viruses in stool samples of wild living chimpanzees. J. Gen. Virol. 2010, 91, 74–86. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Chang, H.W.; Nam, Y.D.; Roh, S.W.; Kim, M.S.; Sung, Y.; Jeon, C.O.; Oh, H.M.; Bae, J.W. Amplification of uncultured single-stranded DNA viruses from rice paddy soil. Appl. Environ. Microbiol. 2008, 74, 5975–5985. [Google Scholar] [CrossRef] [PubMed]
- Krupovic, M.; Ravantti, J.J.; Bamford, D.H. Geminiviruses: A tale of a plasmid becoming a virus. BMC Evol. Biol. 2009, 9, 112. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Bueno, A.; Tamames, J.; Velazquez, D.; Moya, A.; Quesada, A.; Alcami, A. High diversity of the viral community from an antarctic lake. Science 2009, 326, 858–861. [Google Scholar] [CrossRef]
- Yu, X.; Li, B.; Fu, Y.P.; Jiang, D.H.; Ghabrial, S.A.; Li, G.Q.; Peng, Y.L.; Xie, J.T.; Cheng, J.S.; Huang, J.B.; et al. A geminivirus-related DNA mycovirus that confers hypovirulence to a plant pathogenic fungus. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 8387–8392. [Google Scholar] [CrossRef]
- Rosario, K.; Duffy, S.; Breitbart, M. Diverse circovirus-like genome architectures revealed by environmental metagenomics. J. Gen. Virol. 2009, 90, 2418–2424. [Google Scholar] [CrossRef]
- Nishigawa, H.; Oshima, K.; Kakizawa, S.; Jung, H.; Kuboyama, T.; Miyata, S.; Ugaki, M.; Namba, S. Evidence of intermolecular recombination between extrachromosomal dnas in phytoplasma: A trigger for the biological diversity of phytoplasma? Microbiology 2002, 148, 1389–1396. [Google Scholar] [CrossRef]
- Campos-Olivas, R.; Louis, J.M.; Clerot, D.; Gronenborn, B.; Gronenborn, A.M. The structure of a replication initiator unites diverse aspects of nucleic acid metabolism. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 10310–10315. [Google Scholar] [CrossRef]
- Koonin, E.V.; Ilyina, T.V. Geminivirus replication proteins are related to prokaryotic plasmid rolling circle DNA-replication initiator proteins. J. Gen. Virol. 1992, 73, 2763–2766. [Google Scholar] [CrossRef]
- Ilyina, T.V.; Koonin, E.V. Conserved sequence motifs in the initiator proteins for rolling circle DNA replication encoded by diverse replicons from eubacteria, eucaryotes and archaebacteria. Nucleic Acids Res. 1992, 20, 3279–3285. [Google Scholar] [CrossRef] [PubMed]
- Londono, A.; Riego-Ruiz, L.; Arguello-Astorga, G.R. DNA-binding specificity determinants of replication proteins encoded by eukaryotic ssdna viruses are adjacent to widely separated rcr conserved motifs. Arch. Virol. 2010, 155, 1033–1046. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V. On the origin of cells and viruses primordial virus world scenario. Ann. NY Acad. Sci. 2009, 1178, 47–64. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Senkevich, T.G.; Dolja, V.V. The ancient virus world and evolution of cells. Biol. Direct 2006, 1, 29. [Google Scholar] [CrossRef]
- Shackelton, L.A.; Holmes, E.C. Phylogenetic evidence for the rapid evolution of human b19 erythrovirus. J. Virol. 2006, 80, 3666–3669. [Google Scholar] [CrossRef]
- Duffy, S.; Holmes, E.C. Phylogenetic evidence for rapid rates of molecular evolution in the single-stranded DNA begomovirus tomato yellow leaf curl virus. J. Virol. 2008, 82, 957–965. [Google Scholar] [CrossRef]
- Gallian, P.; Biagini, P.; Attoui, H.; Cantaloube, J.F.; Dussol, B.; Berland, Y.; de Micco, P.; de Lamballerie, X. High genetic diversity revealed by the study of tlmv infection in french hemodialysis patients. J. Med. Virol. 2002, 67, 630–635. [Google Scholar] [CrossRef]
- Ge, L.M.; Zhang, J.T.; Zhou, X.P.; Li, H.Y. Genetic structure and population variability of tomato yellow leaf curl china virus. J. Virol. 2007, 81, 5902–5907. [Google Scholar] [CrossRef]
- Grigoras, I.; Timchenko, T.; Grande-Perez, A.; Katul, L.; Vetten, H.J.; Gronenborn, B. High variability and rapid evolution of a nanovirus. J. Virol. 2010, 84, 9105–9117. [Google Scholar] [CrossRef]
- Isnard, M.; Granier, M.; Frutos, R.; Reynaud, B.; Peterschmitt, M. Quasispecies nature of three maize streak virus isolates obtained through different modes of selection from a population used to assess response to infection of maize cultivars. J. Gen. Virol. 1998, 79, 3091–3099. [Google Scholar] [CrossRef]
- van der Walt, E.; Rybicki, E.P.; Varsani, A.; Polston, J.E.; Billharz, R.; Donaldson, L.; Monjane, A.L.; Martin, D.P. Rapid host adaptation by extensive recombination. J. Gen. Virol. 2009, 90, 734–746. [Google Scholar] [CrossRef] [PubMed]
- Raney, J.L.; Delongchamp, R.R.; Valentine, C.R. Spontaneous mutant frequency and mutation spectrum for gene a of phi x174 grown in E. coli. Environ. Mol. Mutagen. 2004, 44, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Drake, J.W. A constant rate of spontaneous mutation in DNA-based microbes. Proc. Natl. Acad. Sci. U. S. A. 1991, 88, 7160–7164. [Google Scholar] [CrossRef] [PubMed]
- Shackelton, L.A.; Parrish, C.R.; Truyen, U.; Holmes, E.C. High rate of viral evolution associated with the emergence of carnivore parvovirus. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 379–384. [Google Scholar] [CrossRef]
- Gibbs, M.J.; Weiller, G.F. Evidence that a plant virus switched hosts to infect a vertebrate and then recombined with a vertebrate-infecting virus. Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 8022–8027. [Google Scholar] [CrossRef]
- Suchard, M.A.R.; Redelings, B.D. Bali-phy: Simultaneous bayesian inference of alignment and phylogeny. Bioinformatics 2006, 22, 2047–2048. [Google Scholar] [CrossRef]
- Saccardo, F.; Cettul, E.; Palmano, S.; Noris, E.; Firrao, G. On the alleged origin of geminiviruses from extrachromosomal DNAs of phytoplasmas. BMC Evol. Biol. 2011, 11, 185. [Google Scholar] [CrossRef]
- Gibbs, M.J.; Smeianov, V.V.; Steele, J.L.; Upcroft, P.; Efimov, B.A. Two families of rep-like genes that probably originated by interspecies recombination are represented in viral, plasmid, bacterial, and parasitic protozoan genomes. Mol. Biol. Evol. 2006, 23, 1097–1100. [Google Scholar] [CrossRef]
- Klute, K.A.; Nadler, S.A.; Stenger, D.C. Horseradish curly top virus is a distinct subgroup ii geminivirus species with rep and c4 genes derived from a subgroup iii ancestor. J. Gen. Virol. 1996, 77, 1369–1378. [Google Scholar] [CrossRef]
- Briddon, R.W.; Bedford, I.D.; Tsai, J.H.; Markham, P.G. Analysis of the nucleotide sequence of the treehopper-transmitted geminivirus, tomato pseudo-curly top virus, suggests a recombinant origin. Virology 1996, 219, 387–394. [Google Scholar] [CrossRef]
- Varsani, A.; Shepherd, D.N.; Dent, K.; Monjane, A.L.; Rybicki, E.P.; Martin, D.P. A highly divergent south african geminivirus species illuminates the ancient evolutionary history of this family. Virol. J. 2009, 6, 36. [Google Scholar] [CrossRef] [PubMed]
- Yazdi, H.R.B.; Heydarnejad, J.; Massumi, H. Genome characterization and genetic diversity of beet curly top iran virus: A geminivirus with a novel nonanucleotide. Virus Genes 2008, 36, 539–545. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.P.; Liu, Y.L.; Calvert, L.; Munoz, C.; OtimNape, G.W.; Robinson, D.J.; Harrison, B.D. Evidence that DNA-a of a geminivirus associated with severe cassava mosaic disease in uganda has arisen by interspecific recombination. J. Gen. Virol. 1997, 78, 2101–2111. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.P.; Liu, Y.L.; Robinson, D.J.; Harrison, B.D. Four DNA-a variants among pakistani isolates of cotton leaf curl virus and their affinities to DNA-a of geminivirus isolates from okra. J. Gen. Virol. 1998, 79, 915–923. [Google Scholar] [CrossRef]
- Fondong, V.N.; Pita, J.S.; Rey, M.E.C.; de Kochko, A.; Beachy, R.N.; Fauquet, C.M. Evidence of synergism between african cassava mosaic virus and a new double-recombinant geminivirus infecting cassava in cameroon. J. Gen. Virol. 2000, 81, 287–297. [Google Scholar] [CrossRef]
- Padidam, M.; Sawyer, S.; Fauquet, C.M. Possible emergence of new geminiviruses by frequent recombination. Virology 1999, 265, 218–225. [Google Scholar] [CrossRef]
- Varsani, A.; Regnard, G.L.; Bragg, R.; Hitzeroth, I.I.; Rybicki, E.P. Global genetic diversity and geographical and host-species distribution of beak and feather disease virus isolates. J. Gen. Virol. 2011, 92, 752–767. [Google Scholar] [CrossRef]
- Heath, L.; Martin, D.P.; Warburton, L.; Perrin, M.; Horsfield, W.; Kingsley, C.; Rybicki, E.P.; Williamson, A.L. Evidence of unique genotypes of beak and feather disease virus in southern Africa. J. Virol. 2004, 78, 9277–9284. [Google Scholar] [CrossRef]
- Rosario, K.; Marinov, M.; Stainton, D.; Kraberger, S.; Wiltshire, E.J.; Collings, D.A.; Walters, M.; Martin, D.P.; Breitbart, M.; Varsani, A. Dragonfly cyclovirus, a novel single-stranded DNA virus discovered in dragonflies (odonata: Anisoptera). J. Gen. Virol. 2011, 92, 1302–1308. [Google Scholar] [CrossRef]
- Lefeuvre, P.; Lett, J.M.; Varsani, A.; Martin, D.P. Widely conserved recombination patterns among single-stranded DNA viruses. J. Virol. 2009, 83, 2697–2707. [Google Scholar] [CrossRef]
- Hesse, R.; Kerrigan, M.; Rowland, R.R. Evidence for recombination between PCV2a and PCV2b in the field. Virus Res. 2008, 132, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Cheung, A.K. Homologous recombination within the capsid gene of porcine circovirus type 2 subgroup viruses via natural co-infection. Arch. Virol. 2009, 154, 531–534. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.M.; Hon, C.C.; Lam, T.Y.; Li, V.Y.; Wong, C.K.; de Oliveira, T.; Leung, F.C. Evidence for recombination in natural populations of porcine circovirus type 2 in hong kong and mainland china. J. Gen. Virol. 2007, 88, 1733–1737. [Google Scholar] [CrossRef] [PubMed]
- He, C.Q.; Ding, N.Z.; Fan, W.; Wu, Y.H.; Li, J.P.; Li, Y.L. Identification of chicken anemia virus putative intergenotype recombinants. Virology 2007, 366, 1–7. [Google Scholar] [CrossRef]
- Lefebvre, D.J.; Van Doorsselaere, J.; Delputte, P.L.; Nauwynck, H.J. Recombination of two porcine circovirus type 2 strains. Arch. Virol. 2009, 154, 875–879. [Google Scholar] [CrossRef]
- Hughes, A.L. Birth-and-death evolution of protein-coding regions and concerted evolution of non-coding regions in the multi-component genomes of nanoviruses. Mol. Phylogenet. Evol. 2004, 30, 287–294. [Google Scholar] [CrossRef]
- Hu, J.M.; Fu, H.C.; Lin, C.H.; Su, H.J.; Yeh, H.H. Reassortment and concerted evolution in banana bunchy top virus genomes. J. Virol. 2007, 81, 1746–1761. [Google Scholar] [CrossRef]
- Manni, F.; Rotola, A.; Caselli, E.; Bertorelle, G.; Di Luca, D. Detecting recombination in tt virus: A phylogenetic approach. J. Mol. Evol. 2002, 55, 563–572. [Google Scholar] [CrossRef]
- Biagini, P.; Gallian, P.; Attoui, H.; Touinssi, M.; Cantaloube, J.; de Micco, P.; de Lamballerie, X. Genetic analysis of full-length genomes and subgenomic sequences of tt virus-like mini virus human isolates. J. Gen. Virol. 2001, 82, 379–383. [Google Scholar] [CrossRef]
- Worobey, M. Extensive homologous recombination among widely divergent tt viruses. J. Virol. 2000, 74, 7666–7670. [Google Scholar] [CrossRef]
- Lukashov, V.V.; Goudsmit, J. Evolutionary relationships among parvoviruses: Virus-host coevolution among autonomous primate parvoviruses and links between adeno-associated and avian parvoviruses. J. Virol. 2001, 75, 2729–2740. [Google Scholar] [CrossRef]
- Kapoor, A.; Simmonds, P.; Slikas, E.; Li, L.L.; Bodhidatta, L.; Sethabutr, O.; Triki, H.; Bahri, O.; Oderinde, B.S.; Baba, M.M.; et al. Human bocaviruses are highly diverse, dispersed, recombination prone, and prevalent in enteric infections. J. Infect. Dis. 2010, 201, 1633–1643. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Wang, X.; Ni, B.; Shen, H.; Wang, H.; Zhang, X.; Chen, S.; Shao, S.; Zhang, W. Recombination analysis based on the complete genome of bocavirus. Virol. J. 2011, 8, 182. [Google Scholar] [CrossRef] [PubMed]
- Shackelton, L.A.; Hoelzer, K.; Parrish, C.R.; Holmes, E.C. Comparative analysis reveals frequent recombination in the parvoviruses. J. Gen. Virol. 2007, 88, 3294–3301. [Google Scholar] [CrossRef]
- Kapoor, A.; Simmonds, P.; Slikas, E.; Li, L.; Bodhidatta, L.; Sethabutr, O.; Triki, H.; Bahri, O.; Oderinde, B.S.; Baba, M.M.; et al. Human bocaviruses are highly diverse, dispersed, recombination prone, and prevalent in enteric infections. J. Infect. Dis. 2010, 201, 1633–1643. [Google Scholar] [CrossRef] [PubMed]
- Grigoras, I.; Timchenko, T.; Katul, L.; Grande-Perez, A.; Vetten, H.J.; Gronenborn, B. Reconstitution of authentic nanovirus from multiple cloned dnas. J. Virol. 2009, 83, 10778–10787. [Google Scholar] [CrossRef]
- Desai, M.; Pal, R.; Deshmukh, R.; Banker, D. Replication of tt virus in hepatocyte and leucocyte cell lines. J. Med. Virol. 2005, 77, 136–143. [Google Scholar] [CrossRef]
- Kakkola, L.; Tommiska, J.; Boele, L.C.; Miettinen, S.; Blom, T.; Kekarainen, T.; Qiu, J.; Pintel, D.; Hoeben, R.C.; Hedman, K.; et al. Construction and biological activity of a full-length molecular clone of human torque teno virus (ttv) genotype 6. FEBS J. 2007, 274, 4719–4730. [Google Scholar] [CrossRef]
- Leppik, L.; Gunst, K.; Lehtinen, M.; Dillner, J.; Streker, K.; de Villiers, E.M. In vivo and in vitro intragenomic rearrangement of tt viruses. J. Virol. 2007, 81, 9346–9356. [Google Scholar] [CrossRef]
- Cheung, A.K. Palindrome regeneration by template strand-switching mechanism at the origin of DNA replication of porcine circovirus via the rolling-circle melting-pot replication model. J. Virol. 2004, 78, 9016–9029. [Google Scholar] [CrossRef]
- Timchenko, T.; de Kouchkovsky, F.; Katul, L.; David, C.; Vetten, H.J.; Gronenborn, B. A single rep protein initiates replication of multiple genome components of faba bean necrotic yellows virus, a single-stranded DNA virus of plants. J. Virol. 1999, 73, 10173–10182. [Google Scholar] [CrossRef] [PubMed]
- Saunders, K.; Lucy, A.; Stanley, J. DNA forms of the geminivirus african cassava mosaic-virus consistent with a rolling circle mechanism of replication. Nucleic Acids Res. 1991, 19, 2325–2330. [Google Scholar] [CrossRef] [PubMed]
- Cheung, A.K. Identification of an octanucleotide motif sequence essential for viral protein, DNA, and progeny virus biosynthesis at the origin of DNA replication of porcine circovirus type 2. Virology 2004, 324, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Cheung, A.K. Identification of the essential and non-essential transcription units for protein synthesis, DNA replication and infectious virus production of porcine circovirus type 1. Arch. Virol. 2004, 149, 975–988. [Google Scholar] [CrossRef]
- Gutierrez, C.; Ramirez-Parra, E.; Castellano, M.M.; Sanz-Burgos, A.P.; Luque, A.; Missich, R. Geminivirus DNA replication and cell cycle interactions. Vet. Microbiol. 2004, 98, 111–119. [Google Scholar] [CrossRef]
- Tattersall, P.; Ward, D.C. Rolling hairpin model for replication of parvovirus and linear chromosomal DNA. Nature 1976, 263, 106–109. [Google Scholar] [CrossRef]
- Cotmore, S.F.; Tattersall, P. Resolution of parvovirus dimer junctions proceeds through a novel heterocruciform intermediate. J. Virol. 2003, 77, 6245–6254. [Google Scholar] [CrossRef]
- Cotmore, S.F.; Tattersall, P. Parvovirus DNA Replication. In DNA Replication in Eukaryotic Cells; DePamphilis, M.L., Ed.; Cold Spring Harbour Laboratory press: Cold Spring Harbor, NY, USA, 1996; pp. 199–813. [Google Scholar]
- Abouzid, A.M.; Frischmuth, T.; Jeske, H. A putative replicative form of the abutilon mosaic-virus (gemini group) in a chromatin-like structure. Mol. Gen. Genet. 1988, 212, 252–258. [Google Scholar] [CrossRef]
- Pilartz, M.; Jeske, H. Abutilon mosaic geminivirus double-stranded DNA is packed into minichromosomes. Virology 1992, 189, 800–802. [Google Scholar] [CrossRef]
- Pilartz, M.; Jeske, H. Mapping of abutilon mosaic geminivirus minichromosomes. J. Virol. 2003, 77, 10808–10818. [Google Scholar]
- Heyraud-Nitschke, F.; Schumacher, S.; Laufs, J.; Schaefer, S.; Schell, J.; Gronenborn, B. Determination of the origin cleavage and joining domain of geminivirus rep proteins. Nucleic Acids Res. 1995, 23, 910–916. [Google Scholar] [CrossRef] [PubMed]
- Cheung, A.K. Detection of template strand switching during initiation and termination of DNA replication of porcine circovirus. J. Virol. 2004, 78, 4268–4277. [Google Scholar] [CrossRef] [PubMed]
- Laufs, J.; Traut, W.; Heyraud, F.; Matzeit, V.; Rogers, S.G.; Schell, J.; Gronenborn, B. In-vitro cleavage and joining at the viral origin of replication by the replication initiator protein of tomato yellow leaf curl virus. Proc. Natl. Acad. Sci. U. S. A. 1995, 92, 3879–3883. [Google Scholar] [CrossRef] [PubMed]
- Orozco, B.M.; Hanley-Bowdoin, L. Conserved sequence and structural motifs contribute to the DNA binding and cleavage activities of a geminivirus replication protein. J. Biol. Chem. 1998, 273, 24448–24456. [Google Scholar] [CrossRef]
- Castellano, M.M.; Sanz-Burgos, A.P.; Gutierrez, C. Initiation of DNA replication in a eukaryotic rolling-circle replicon: Identification of multiple rna-protein complexes at the geminivirus origin. J. Mol. Biol. 1999, 290, 639–652. [Google Scholar] [CrossRef]
- Hafner, G.J.; Stafford, M.R.; Wolter, L.C.; Harding, R.M.; Dale, J.L. Nicking and joining activity of banana bunchy top virus replication protein in vitro. J. Gen. Virol. 1997, 78, 1795–1799. [Google Scholar] [CrossRef]
- Jeske, H.; Lutgemeier, M.; Preiss, W. DNA forms indicate rolling circle and recombination dependent replication of abutilon mosaic virus. EMBO J. 2001, 20, 6158–6167. [Google Scholar] [CrossRef]
- Okamoto, H.; Ukita, M.; Nishizawa, T.; Kishimoto, J.; Hoshi, Y.; Mizuo, H.; Tanaka, T.; Miyakawa, Y.; Mayumi, M. Circular double-stranded forms of tt virus DNA in the liver. J. Virol. 2000, 74, 5161–5167. [Google Scholar] [CrossRef]
- Okamoto, H.; Takahashi, M.; Nishizawa, T.; Tawara, A.; Sugai, Y.; Sai, T.; Tanaka, T.; Tsuda, F. Replicative forms of tt virus DNA in bone marrow cells. Biochem. Biophys. Res. Commun. 2000, 270, 657–662. [Google Scholar] [CrossRef]
- de Villiers, E.M.; Borkosky, S.S.; Kimmel, R.; Gunst, K.; Fei, J.W. The diversity of torque teno viruses: In vitro replication leads to the formation of additional replication-competent subviral molecules. J. Virol. 2011, 85, 7284–7295. [Google Scholar] [CrossRef]
- Heyraud, F.; Matzeit, V.; Schaefer, S.; Schell, J.; Gronenborn, B. The conserved nonanucleotide motif of the geminivirus stem-loop sequence promotes replicational release of virus molecules from redundant copies. Biochimie 1993, 75, 605–615. [Google Scholar] [CrossRef]
- Owor, B.E.; Martin, D.P.; Shepherd, D.N.; Edema, R.; Monjane, A.L.; Rybicki, E.P.; Thomson, J.A.; Varsani, A. Genetic analysis of maize streak virus isolates from uganda reveals widespread distribution of a recombinant variant. J. Gen. Virol. 2007, 88, 3154–3165. [Google Scholar] [CrossRef]
- Xu, Y.; Price, B.D. Chromatin dynamics and the repair of DNA double strand breaks. Cell Cycle 2011, 10, 261–267. [Google Scholar] [CrossRef]
- Alberter, B.; Rezaian, M.A.; Jeske, H. Replicative intermediates of tomato leaf curl virus and its satellite dnas. Virology 2005, 331, 441–448. [Google Scholar] [CrossRef]
- Jovel, J.; Preiss, W.; Jeske, H. Characterization of DNA intermediates of an arising geminivirus. Virus Res. 2007, 130, 63–70. [Google Scholar] [CrossRef]
- Erdmann, J.B.; Shepherd, D.N.; Martin, D.P.; Varsani, A.; Rybicki, E.P.; Jeske, H. Replicative intermediates of maize streak virus found during leaf development. J. Gen. Virol. 2010, 91, 1077–1081. [Google Scholar] [CrossRef]
- Preiss, W.; Jeske, H. Multitasking in replication is common among geminiviruses. J. Virol. 2003, 77, 2972–2980. [Google Scholar] [CrossRef]
- Varsani, A.; Shepherd, D.N.; Monjane, A.L.; Owor, B.E.; Erdmann, J.B.; Rybicki, E.P.; Peterschmitt, M.; Briddon, R.W.; Markham, P.G.; Oluwafemi, S.; et al. Recombination, decreased host specificity and increased mobility may have driven the emergence of maize streak virus as an agricultural pathogen. J. Gen. Virol. 2008, 89, 2063–2074. [Google Scholar] [CrossRef]
- Stenger, D.C.; Revington, G.N.; Stevenson, M.C.; Bisaro, D.M. Replicational release of geminivirus genomes from tandemly repeated copies-Evidence for rolling-circle replication of a plant viral-DNA. Proc. Natl. Acad. Sci. U. S. A. 1991, 88, 8029–8033. [Google Scholar] [CrossRef]
- Fenaux, M.; Halbur, P.G.; Haqshenas, G.; Royer, R.; Thomas, P.; Nawagitgul, P.; Gill, M.; Toth, T.E.; Meng, X.J. Cloned genomic DNA of type 2 porcine circovirus is infectious when injected directly into the liver and lymph nodes of pigs: Characterization of clinical disease, virus distribution, and pathologic lesions. J. Virol. 2002, 76, 541–551. [Google Scholar] [CrossRef]
- Burns, T.M.; Harding, R.M.; Dale, J.L. The genome organization of banana bunchy top virus—analysis of 6 ssdna components. J. Gen. Virol. 1995, 76, 1471–1482. [Google Scholar]
- Katul, L.; Timchenko, T.; Gronenborn, B.; Vetten, H.J. Ten distinct circular ssdna components, four of which encode putative replication-associated proteins, are associated with the faba bean necrotic yellows virus genome. J. Gen. Virol. 1998, 79, 3101–3109. [Google Scholar] [CrossRef]
- Boevink, P.; Chu, P.W.; Keese, P. Sequence of subterranean clover stunt virus DNA: Affinities with the geminiviruses. Virology 1995, 207, 354–361. [Google Scholar] [CrossRef]
- Sano, Y.; Wada, M.; Hashimoto, Y.; Matsumoto, T.; Kojima, M. Sequences of ten circular ssdna components associated with the milk vetch dwarf virus genome. J. Gen. Virol. 1998, 79, 3111–3118. [Google Scholar] [CrossRef]
- Hill, J.E.; Strandberg, J.O.; Hiebert, E.; Lazarowitz, S.G. Asymmetric infectivity of pseudorecombinants of cabbage leaf curl virus and squash leaf curl virus: Implications for bipartite geminivirus evolution and movement. Virology 1998, 250, 283–292. [Google Scholar] [CrossRef]
- Chakraborty, S.; Vanitharani, R.; Chattopadhyay, B.; Fauquet, C.M. Supervirulent pseudorecombination and asymmetric synergism between genomic components of two distinct species of begomovirus associated with severe tomato leaf curl disease in india. J. Gen. Virol. 2008, 89, 818–828. [Google Scholar] [CrossRef]
- Gilbertson, R.L.; Hidayat, S.H.; Paplomatas, E.J.; Rojas, M.R.; Hou, Y.M.; Maxwell, D.P. Pseudorecombination between infectious cloned DNA-components of tomato mottle and bean dwarf mosaic geminiviruses. J. Gen. Virol. 1993, 74, 23–31. [Google Scholar] [CrossRef]
- von Arnim, A.; Stanley, J. Determinants of tomato golden mosaic-virus symptom development located on DNA-b. Virology 1992, 186, 286–293. [Google Scholar] [CrossRef]
- Pita, J.S.; Fondong, V.N.; Sangare, A.; Otim-Nape, G.W.; Ogwal, S.; Fauquet, C.M. Recombination, pseudorecombination and synergism of geminiviruses are determinant keys to the epidemic of severe cassava mosaic disease in uganda. J. Gen. Virol. 2001, 82, 655–665. [Google Scholar] [CrossRef]
- Unseld, S.; Ringel, M.; Hofer, P.; Hohnle, M.; Jeske, H.; Bedford, I.D.; Markham, P.G.; Frischmuth, T. Host range and symptom variation of pseudorecombinant virus produced by two distinct bipartite geminiviruses. Arch. Virol. 2000, 145, 1449–1454. [Google Scholar] [CrossRef]
- Unseld, S.; Ringel, M.; Konrad, A.; Lauster, S.; Frischmuth, T. Virus-specific adaptations for the production of a pseudorecombinant virus formed by two distinct bipartite geminiviruses from central america. Virology 2000, 274, 179–188. [Google Scholar] [CrossRef]
- Briddon, R.W.; Patil, B.L.; Bagewadi, B.; Nawaz-ul-Rehman, M.S.; Fauquet, C.M. Distinct evolutionary histories of the DNA-A and DNA-B components of bipartite begomoviruses. BMC Evol. Biol. 2010, 10, 97. [Google Scholar] [CrossRef]
- Saunders, K.; Salim, N.; Mali, V.R.; Malathi, V.G.; Briddon, R.; Markham, P.G.; Stanley, J. Characterisation of sri lankan cassava mosaic virus and indian cassava mosaic virus: Evidence for acquisition of a DNA b component by a monopartite begomovirus. Virology 2002, 293, 63–74. [Google Scholar] [CrossRef]
- Saunders, K.; Stanley, J. A nanovirus-like DNA component associated with yellow vein disease of ageratum conyzoides: Evidence for interfamilial recombination between plant DNA viruses. Virology 1999, 264, 142–152. [Google Scholar] [CrossRef]
- Chen, L.F.; Rojas, M.; Kon, T.; Gamby, K.; Xoconostle-Cazares, B.; Gilbertson, R.L. A severe symptom phenotype in tomato in mali is caused by a reassortant between a novel recombinant begomovirus (tomato yellow leaf curl mali virus) and a betasatellite. Mol. Plant. Pathol. 2009, 10, 415–430. [Google Scholar] [CrossRef]
- Lefeuvre, P.; Martin, D.P.; Hoareau, M.; Naze, F.; Delatte, H.; Thierry, M.; Varsani, A.; Becker, N.; Reynaud, B.; Lett, J.M. Begomovirus ’melting pot’ in the south-west indian ocean islands: Molecular diversity and evolution through recombination. J. Gen. Virol. 2007, 88, 3458–3468. [Google Scholar] [CrossRef]
- Bell, K.E.; Dale, J.L.; Ha, C.V.; Vu, M.T.; Revill, P.A. Characterisation of rep-encoding components associated with banana bunchy top nanovirus in vietnam. Arch. Virol. 2002, 147, 695–707. [Google Scholar] [CrossRef]
- Horser, C.L.; Karan, M.; Harding, R.M.; Dale, J.L. Additional rep-encoding dnas associated with banana bunchy top virus. Arch. Virol. 2001, 146, 71–86. [Google Scholar] [CrossRef]
- Sung, Y.K.; Coutts, R.H.A. Pseudorecombination and complementation between potato yellow mosaic geminivirus and tomato golden mosaic geminivirus. J. Gen. Virol. 1995, 76, 2809–2815. [Google Scholar] [CrossRef]
- Lazarowitz, S.G.; Wu, L.C.; Rogers, S.G.; Elmer, J.S. Sequence-specific interaction with the viral all protein identifies a geminivirus DNA-replication origin. Plant Cell 1992, 4, 799–809. [Google Scholar]
- Fontes, E.P.B.; Gladfelter, H.J.; Schaffer, R.L.; Petty, I.T.D.; Hanleybowdoin, L. Geminivirus replication origins have a modular organization. Plant Cell 1994, 6, 405–416. [Google Scholar] [PubMed]
- Hou, Y.M.; Gilbertson, R.L. Increased pathogenicity in a pseudorecombinant bipartite geminivirus correlates with intermolecular recombination. J. Virol. 1996, 70, 5430–5436. [Google Scholar] [CrossRef] [PubMed]
- Stenger, D.C. Strain-specific mobilization and amplification of a transgenic defective-interfering DNA of the geminivirus beet curly top virus. Virology 1994, 203, 397–402. [Google Scholar] [CrossRef] [PubMed]
- Arguello-Astorga, G.R.; Ruiz-Medrano, R. An iteron-related domain is associated to motif 1 in the replication proteins of geminiviruses: Identification of potential interacting amino acid-base pairs by a comparative approach. Arch. Virol. 2001, 146, 1465–1485. [Google Scholar] [CrossRef]
- Timchenko, T.; Katul, L.; Sano, Y.; de Kouchkovsky, F.; Vetten, H.J.; Gronenborn, B. The master rep concept in nanovirus replication: Identification of missing genome components and potential for natural genetic reassortment. Virology 2000, 274, 189–195. [Google Scholar] [CrossRef]
- Dry, I.B.; Krake, L.R.; Rigden, J.E.; Rezaian, M.A. A novel subviral agent associated with a geminivirus: The first report of a DNA satellite. Proc. Natl. Acad. Sci. U. S. A. 1997, 94, 7088–7093. [Google Scholar] [CrossRef]
- Lin, B.C.; Behjatnia, S.A.A.; Dry, I.B.; Randles, J.W.; Ali Rezaian, M. High-affinity rep-binding is not required for the replication of a geminivirus DNA and its satellite. Virology 2003, 305, 353–363. [Google Scholar] [CrossRef]
- Briddon, R.W.; Bull, S.E.; Amin, I.; Idris, A.M.; Mansoor, S.; Bedford, I.D.; Dhawan, P.; Rishi, N.; Siwatch, S.S.; Abdel-Salam, A.M.; et al. Diversity of DNA beta, a satellite molecule associated with some monopartite begomoviruses. Virology 2003, 312, 106–121. [Google Scholar] [CrossRef]
- Roberts, S.; Stanley, J. Lethal mutations within the conserved stem-loop of african cassava mosaic-virus DNA are rapidly corrected by genomic recombination. J. Gen. Virol. 1994, 75, 3203–3209. [Google Scholar] [CrossRef]
- Hyder, M.Z.; Shah, S.H.; Hameed, S.; Naqvi, S.M. Evidence of recombination in the banana bunchy top virus genome. Infect Genet Evol. 2011, 11, 1293–1300. [Google Scholar] [CrossRef]
- Jovel, J.; Reski, G.; Rothenstein, D.; Ringel, M.; Frischmuth, T.; Jeske, H. Sida micrantha mosaic is associated with a complex infection of begomoviruses different from abutilon mosaic virus. Arch. Virol. 2004, 149, 829–841. [Google Scholar] [CrossRef] [PubMed]
- Stanley, J.; Saunders, K.; Pinner, M.S.; Wong, S.M. Novel defective interfering dnas associated with ageratum yellow vein geminivirus infection of ageratum conyzoides. Virology 1997, 239, 87–96. [Google Scholar] [CrossRef]
- Tao, X.R.; Zhou, X.P. Pathogenicity of a naturally occurring recombinant DNA satellite associated with tomato yellow leaf curl china virus. J. Gen. Virol. 2008, 89, 306–311. [Google Scholar] [CrossRef] [PubMed]
- Gafni, Y.; Epel, B.L. The role of host and viral proteins in intra- and inter-cellular trafficking of geminiviruses. Physiol. Mol. Plant Pathol. 2002, 60, 231–241. [Google Scholar] [CrossRef]
- Saunders, K.; Norman, A.; Gucciardo, S.; Stanley, J. The DNA beta satellite component associated with ageratum yellow vein disease encodes an essential pathogenicity protein (beta c1). Virology 2004, 324, 37–47. [Google Scholar] [CrossRef]
- Duan, Y.P.; Powell, C.A.; Webb, S.E.; Purcifull, D.E.; Hiebert, E. Geminivirus resistance in transgenic tobacco expressing mutated bc1 protein. Mol. Plant Microbe Interact. 1997, 10, 617–623. [Google Scholar] [CrossRef]
- Briddon, R.W.; Mansoor, S.; Bedford, I.D.; Pinner, M.S.; Saunders, K.; Stanley, J.; Zafar, Y.; Malik, K.A.; Markham, P.G. Identification of DNA components required for induction of cotton leaf curl disease. Virology 2001, 285, 234–243. [Google Scholar] [CrossRef]
- Cui, X.F.; Tao, X.R.; Xie, Y.; Fauquet, C.M.; Zhou, X.P. A DNA beta associated with tomato yellow leaf curl china virus is required for symptom induction. J. Virol. 2004, 78, 13966–13974. [Google Scholar] [CrossRef]
- Briddon, R.W.; Stanley, J. Subviral agents associated with plant single-stranded DNA viruses. Virology 2006, 344, 198–210. [Google Scholar] [CrossRef]
- Saeed, M.; Zafar, Y.; Randles, J.W.; Rezaian, M.A. A monopartite begomovirus-associated DNA beta satellite substitutes for the DNA b of a bipartite begomovirus to permit systemic infection. J. Gen. Virol. 2007, 88, 2881–2889. [Google Scholar] [CrossRef]
- Kikuno, R.; Toh, H.; Hayashida, H.; Miyata, T. Sequence similarity between putative gene-products of geminiviral dnas. Nature 1984, 308, 562. [Google Scholar] [CrossRef] [PubMed]
- de Villiers, E.M.; Kimmel, R.; Leppik, L.; Gunst, K. Intragenomic rearrangement in tt viruses: A possible role in the pathogenesis of disease. In TT Viruses. The Still Elusive Human Pathogens; de Villiers, E.M., zur Hausen, H., Eds.; Springer: Berlin, Germany, 2009; Volume 331, pp. 91–107. [Google Scholar]
- Roberts, E.J.F.; Buck, K.W.; Coutts, R.H.A. Characterization of potato yellow mosaic-virus as a geminivirus with a bipartite genome. Intervirology 1988, 29, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Faust, E.A.; Ward, D.C. Incomplete genomes of the parvovirus minute virus of mice-Selective conservation of genome termini, including the origin for DNA-replication. J. Virol. 1979, 32, 276–292. [Google Scholar] [CrossRef] [PubMed]
- Hogan, A.; Faust, E.A. Nonhomologous recombination in the parvovirus chromosome: Role for a ctatttct motif. Mol. Cell. Biol. 1986, 6, 3005–3009. [Google Scholar]
- Clement, N.; Avalosse, B.; El Bakkouri, K.; Velu, T.; Brandenburger, A. Cloning and sequencing of defective particles derived from the autonomous parvovirus minute virus of mice for the construction of vectors with minimal cis-acting sequences. J. Virol. 2001, 75, 1284–1293. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.M.; Debelak, D.J.; Reynolds, T.C.; Miller, A.D. Identification and elimination of replication-competent adeno-associated virus (aav) that can arise by nonhomologous recombination during aav vector production. J. Virol. 1997, 71, 6816–6822. [Google Scholar] [CrossRef]
- Rhode, S.L., 3rd. Defective interfering particles of parvovirus h-1. J. Virol. 1978, 27, 347–356. [Google Scholar] [CrossRef]
- Hoelzer, K.; Shackelton, L.A.; Holmes, E.C.; Parrish, C.R. Within-host genetic diversity of endemic and emerging parvoviruses of dogs and cats. J. Virol. 2008, 82, 11096–11105. [Google Scholar] [CrossRef]
- MacDowell, S.W.; Coutts, R.H.; Buck, K.W. Molecular characterisation of subgenomic single stranded and double-stranded DNA forms isolated from plants infected with tomato golden mosaic virus. Nucleic Acids Res. 1986, 14, 7967–7984. [Google Scholar] [CrossRef]
- Liu, Y.L.; Robinson, D.J.; Harrison, B.D. Defective forms of cotton leaf curl virus DNA-a that have different combinations of sequence deletion, duplication, inversion and rearrangement. J. Gen. Virol. 1998, 79, 1501–1508. [Google Scholar] [CrossRef]
- Stanley, J.; Townsend, R. Characterisation of DNA forms associated with cassava latent virus infection. Nucleic Acids Res. 1985, 13, 2189–2206. [Google Scholar] [CrossRef]
- Macdonald, H.; Coutts, R.H.A.; Buck, K.W. Characterization of a subgenomic DNA isolated from triticum-aestivum plants infected with wheat dwarf virus. J. Gen. Virol. 1988, 69, 1339–1344. [Google Scholar] [CrossRef]
- Casado, C.G.; Javier Ortiz, G.; Padron, E.; Bean, S.J.; McKenna, R.; Agbandje-McKenna, M.; Boulton, M.I. Isolation and characterization of subgenomic dnas encapsidated in "single" t = 1 isometric particles of maize streak virus. Virology 2004, 323, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Ndunguru, J.; Legg, J.P.; Fofana, I.B.F.; Aveling, T.A.S.; Thompson, G.; Fauquet, C.M. Identification of a defective molecule derived from DNA-a of the bipartite begomovirus of east african cassava mosaic virus. Plant Pathol. 2006, 55, 2–10. [Google Scholar] [CrossRef]
- Patil, B.L.; Dutt, N.; Briddon, R.W.; Bull, S.E.; Rothenstein, D.; Borah, B.K.; Dasgupta, I.; Stanley, J.; Jeske, H. Deletion and recombination events between the DNA-a and DNA-b components of indian cassava-infecting geminiviruses generate defective molecules in nicotiana benthamiana. Virus Res. 2007, 124, 59–67. [Google Scholar] [CrossRef]
- Frischmuth, T.; Stanley, J. Strategies for the control of geminivirus diseases. Semin. Virol. 1993, 4, 329–337. [Google Scholar] [CrossRef]
- Horn, J.; Lauster, S.; Krenz, B.; Kraus, J.; Frischmuth, T.; Jeske, H. Ambivalent effects of defective DNA in beet curly top virus-infected transgenic sugarbeet plants. Virus Res. 2011, 158, 169–178. [Google Scholar]
- Stanley, J.; Frischmuth, T.; Ellwood, S. Defective viral-DNA ameliorates symptoms of geminivirus infection in transgenic plants. Proc. Natl. Acad. Sci. U. S. A. 1990, 87, 6291–6295. [Google Scholar] [CrossRef] [PubMed]
- Frischmuth, T.; Stanley, J. Beet curly top virus symptom amelioration in nicotiana benthamiana transformed with a naturally occurring viral subgenomic DNA. Virology 1994, 200, 826–830. [Google Scholar] [CrossRef]
- Frischmuth, T.; Engel, M.; Jeske, H. Beet curly top virus di DNA-mediated resistance is linked to its size. Mol. Breed. 1997, 3, 213–217. [Google Scholar] [CrossRef]
- Etessami, P.; Watts, J.; Stanley, J. Size reversion of african cassava mosaic-virus coat protein gene deletion mutants during infection of nicotiana-benthamiana. J. Gen. Virol. 1989, 70, 277–289. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.H.; Hohn, B. Mutational analysis of the small intergenic region of maize streak virus. Virology 1991, 183, 721–730. [Google Scholar] [CrossRef] [PubMed]
- Bisaro, D.M. Recombination in the geminiviruses: Mechanisms for maintaining genome size and generating genomic diversity. In Homologous Recombination and Gene Silencing in Plants; Paszkowski, J., Ed.; Kluwer Academic Publishers: Dordrecht, The Netherlands, 1994; pp. 39–60. [Google Scholar]
- Frischmuth, T.; Ringel, M.; Kocher, C. The size of encapsidated single-stranded DNA determines the multiplicity of african cassava mosaic virus particles. J. Gen. Virol. 2001, 82, 673–676. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.J.; Tan, Z.; Liu, Y.; Briddon, R.W.; Zhou, X.P. Size reversion of a truncated DNA beta associated with tobacco curly shoot virus. Virus Res. 2008, 131, 288–292. [Google Scholar] [CrossRef]
- Cheung, A.K. Mutational analysis of the direct tandem repeat sequences at the origin of DNA replication of porcine circovirus type 1. Virology 2005, 339, 192–199. [Google Scholar] [CrossRef]
- Frischmuth, T.; Stanley, J. Recombination between viral DNA and the transgenic coat protein gene of african cassava mosaic geminivirus. J. Gen. Virol. 1998, 79, 1265–1271. [Google Scholar] [CrossRef]
- Belyi, V.A.; Levine, A.J.; Skalka, A.M. Sequences from ancestral single-stranded DNA viruses in vertebrate genomes: The parvoviridae and circoviridae are more than 40 to 50 million years old. J. Virol. 2010, 84, 12458–12462. [Google Scholar] [CrossRef]
- Krupovic, M.; Forterre, P. Microviridae goes temperate: Microvirus-related proviruses reside in the genomes of bacteroidetes. PLoS ONE 2011, 6, e19893. [Google Scholar] [CrossRef]
- Liu, H.; Fu, Y.; Xie, J.; Cheng, J.; Ghabrial, S.A.; Li, G.; Peng, Y.; Yi, X.; Jiang, D. Widespread endogenization of densoviruses and parvoviruses in animal and human genomes. J. Virol. 2011. [Google Scholar] [CrossRef]
- Dutheil, N.; Shi, F.; Dupressoir, T.; Linden, R.M. Adeno-associated virus site-specifically integrates into a muscle-specific DNA region. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 4862–4866. [Google Scholar] [CrossRef]
- Ashby, M.K.; Warry, A.; Bejarano, E.R.; Khashoggi, A.; Burrell, M.; Lichtenstein, C.P. Analysis of multiple copies of geminiviral DNA in the genome of four closely related nicotiana species suggest a unique integration event. Plant Mol. Biol. 1997, 35, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Bejarano, E.R.; Khashoggi, A.; Witty, M.; Lichtenstein, C. Integration of multiple repeats of geminiviral DNA into the nuclear genome of tobacco during evolution. Proc. Natl. Acad. Sci. U. S.A. 1996, 93, 759–764. [Google Scholar] [CrossRef] [PubMed]
- Murad, L.; Bielawski, J.P.; Matyasek, R.; Kovarik, A.; Nichols, R.A.; Leitch, A.R.; Lichtenstein, C.P. The origin and evolution of geminivirus-related DNA sequences in nicotiana. Heredity 2004, 92, 352–358. [Google Scholar] [CrossRef] [PubMed]
- Kenton, A.; Khashoggi, A.; Parokonny, A.; Bennett, M.D.; Lichtenstein, C. Chromosomal location of endogenous geminivirus-related DNA-sequences in nicotiana-tabacum-l. Chromosome Res. 1995, 3, 346–350. [Google Scholar] [CrossRef]
- Gibbs, A.J.; Fargette, D.; Garcia-Arenal, F.; Gibbs, M.J. Time-The emerging dimension of plant virus studies. J. Gen. Virol. 2010, 91, 13–22. [Google Scholar] [CrossRef]
- Lefeuvre, P.; Harkins, G.W.; Lett, J.-M.; Briddon, R.W.; Leitch, A.R.; Chase, M.W.; Moury, B.; Martin, D.P. Evolutionary time-scale of begomoviruses: Evidence from integrated sequences in nicotiana genome. PLoS ONE 2011, 6, e19193. [Google Scholar] [CrossRef] [PubMed]
- Rojas, M.R.; Hagen, C.; Lucas, W.J.; Gilbertson, R.L. Exploiting chinks in the plant’s armor: Evolution and emergence of geminiviruses. Ann. Rev. Phytopathol. 2005, 43, 361–394. [Google Scholar] [CrossRef]
- Varsani, A.; Monjane, A.L.; Donaldson, L.; Oluwafemi, S.; Zinga, I.; Komba, E.K.; Plakoutene, D.; Mandakombo, N.; Mboukoulida, J.; Semballa, S.; et al. Comparative analysis of panicum streak virus and maize streak virus diversity, recombination patterns and phylogeography. Virol. J. 2009, 6, 194. [Google Scholar] [CrossRef] [PubMed]
- Varsani, A.; Regnard, G.L.; Bragg, R.; Hitzeroth, I.I.; Rybicki, E.P. Global genetic diversity and geographical and host-species distribution of beak and feather disease virus isolates. J. Gen. Virol. 2011, 92, 752–767. [Google Scholar] [CrossRef]
- Karan, M.; Harding, R.M.; Dale, J.L. Evidence for two groups of banana bunchy top virus isolates. J. Gen. Virol. 1994, 75, 3541–3546. [Google Scholar] [CrossRef]
- Prasanna, H.C.; Sinha, D.P.; Verma, A.; Singh, M.; Singh, B.; Rai, M.; Martin, D.P. The population genomics of begomoviruses: Global scale population structure and gene flow. Virol. J. 2010, 7, 220. [Google Scholar] [CrossRef] [PubMed]
- Briddon, R.W.; Bull, S.E.; Amin, I.; Mansoor, S.; Bedford, I.D.; Rishi, N.; Siwatch, S.S.; Zafar, Y.; Abdel-Salam, A.M.; Markham, P.G. Diversity of DNA 1: A satellite-like molecule associated with monopartite begomovirus-DNA beta complexes. Virology 2004, 324, 462–474. [Google Scholar] [CrossRef] [PubMed]
- Hoelzer, K.; Shackelton, L.A.; Parrish, C.R.; Holmes, E.C. Phylogenetic analysis reveals the emergence, evolution and dispersal of carnivore parvoviruses. J. Gen. Virol. 2008, 89, 2280–2289. [Google Scholar] [CrossRef]
- Saback, F.L.; Gomes, S.A.; Niel, C. High frequency of mixed tt virus infections in healthy adults and children detected by a simplified heteroduplex mobility assay. J. Virol. Meth. 2002, 101, 117–125. [Google Scholar] [CrossRef]
- Bigarre, L.; Beven, V.; de Boisseson, C.; Grasland, B.; Rose, N.; Biagini, P.; Jestin, A. Pig anelloviruses are highly prevalent in swine herds in france. J. Gen. Virol. 2005, 86, 631–635. [Google Scholar] [CrossRef] [PubMed]
- Morales, F.J. History and current distribution of begomoviruses in latin america. Plant Virus Epidemiol. 2006, 67, 127–162. [Google Scholar]
- Ribeiro, S.G.; De Ávila, A.C.; Bezerra, I.C.; Fernandes, J.J.; Faria, J.C.; Lima, M.F.; Gilbertson, R.L.; Maciel-Zambolim, E.; Zerbini, F.M. Widespread occurrence of tomato geminiviruses in brazil, associated with the new biotype of the whitefly vector. Plant Dis. 1998, 82, 830. [Google Scholar] [CrossRef]
- França, F.H.; Villas Bôas, G.L.; Branco, M.C. Occurrence of bemisia argentifolii bellows & perring (homoptera: Aleyrodidae) in the federal district. Anais da Sociedade Entomológica do Brasil 1996, 25, 369–372. [Google Scholar]
- Albuquerque, L.C.; Martin, D.P.; Avila, A.C.; Inoue-Nagata, A.K. Characterization of tomato yellow vein streak virus, a begomovirus from brazil. Virus Genes 2010, 40, 140–147. [Google Scholar] [CrossRef]
- Calegario, R.F.; Ferreira, S.D.S.; de Andrade, E.C.; Zerbini, F.M. Characterization of tomato yellow spot virus, a novel tomato-infecting begomovirus in brazil. Pesquisa Agropecuaria Brasileira 2007, 42, 1335–1343. [Google Scholar] [CrossRef]
- Castillo-Urquiza, G.P.; Beserra, J.E.A.; Bruckner, F.P.; Lima, A.T.M.; Varsani, A.; Alfenas Zerbini, P.; Zerbini, F.M. Six novel begomoviruses infecting tomato and associated weeds in southeastern brazil. Arch. Virol. 2008, 153, 1985–1989. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, F.R.; de Albuquerque, L.C.; Giordano, L.D.B.; Boiteux, L.S.; de Avila, A.C.; Inoue Nagata, A.K. Diversity and prevalence of brazilian bipartite begomovirus species associated to tomatoes. Virus Genes 2008, 36, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, S.G.; Martin, D.P.; Lacorte, C.; Simoes, L.C.; Orlandini, D.R.S.; Inoue-Nagata, A.K. Molecular and biological characterization of tomato chlorotic mottle virus suggests that recombination underlies the evolution and diversity of brazilian tomato begomoviruses. Phytopathology 2007, 97, 702–711. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, S.G.; Ambrozevicius, L.P.; Avila, A.C.; Bezerra, I.C.; Calegario, R.F.; Fernandes, J.J.; Lima, M.F.; de Mello, R.N.; Rocha, H.; Zerbini, F.M. Distribution and genetic diversity of tomato-infecting begomoviruses in brazil. Arch. Virol. 2003, 148, 281–295. [Google Scholar] [CrossRef] [PubMed]
- Prasanna, H.C.; Rai, M. Detection and frequency of recombination in tomato-infecting begomoviruses of south and southeast asia. Virol. J. 2007, 4, 111. [Google Scholar] [CrossRef]
- Martin, D.P.; Lefeuvre, P.; Varsani, A.; Hoareau, M.; Semegni, J.Y.; Dijoux, B.; Vincent, C.; Lett, J.M. Complex recombination patterns arising during geminivirus coinfections both preserve and demarcate biologically important intra-genome interaction networks. PLoS Pathog. 2011; in press. [Google Scholar]
- Worobey, M. Extensive homologous recombination among widely divergent tt viruses. J. Virol. 2000, 74, 7666–7670. [Google Scholar] [CrossRef]
- Mankertz, A.; Persson, F.; Mankertz, J.; Blaess, G.; Buhk, H.J. Mapping and characterization of the origin of DNA replication of porcine circovirus. J. Virol. 1997, 71, 2562–2566. [Google Scholar] [CrossRef]
- Stenger, D.C.; Davis, K.R.; Bisaro, D.M. Recombinant beet curly top virus genomes exhibit both parental and novel pathogenic phenotypes. Virology 1994, 200, 677–685. [Google Scholar] [CrossRef]
- Cheung, A.K. Homologous recombination plays minor role in excision of unit-length viral genomes from head-to-tail direct tandem repeats of porcine circovirus during DNA replication in escherichia coli. Arch. Virol. 2007, 152, 1531–1539. [Google Scholar] [CrossRef]
- Garcia-Andres, S.; Tomas, D.M.; Sanchez-Campos, S.; Navas-Castillo, J.; Moriones, E. Frequent occurrence of recombinants in mixed infections of tomato yellow leaf curl disease-associated begomoviruses. Virology 2007, 365, 210–219. [Google Scholar] [CrossRef]
- Rowe, C.L.; Fleming, J.O.; Nathan, M.J.; Sgro, J.Y.; Palmenberg, A.C.; Baker, S.C. Generation of coronavirus spike deletion variants by high-frequency recombination at regions of predicted rna secondary structure. J. Virol. 1997, 71, 6183–6190. [Google Scholar] [CrossRef] [PubMed]
- Koev, G.; Mohan, B.R.; Miller, W.A. Primary and secondary structural elements required for synthesis of barley yellow dwarf virus subgenomic RNA1. J. Virol. 1999, 73, 2876–2885. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Gao, L.; Balakrishnan, M.; Bambara, R.A. A recombination hot spot in HIV-1 contains guanosine runs that can form a g-quartet structure and promote strand transfer in vitro. J. Biol. Chem. 2009, 284, 33883–33893. [Google Scholar] [CrossRef] [PubMed]
- Duch, M.; Carrasco, M.L.; Jespersen, T.; Aagaard, L.; Pedersen, F.S. An rna secondary structure bias for non-homologous reverse transcriptase-mediated deletions in vivo. Nucleic Acids Res. 2004, 32, 2039–2048. [Google Scholar] [CrossRef]
- Draghici, H.K.; Varrelmann, M. Evidence for similarity-assisted recombination and predicted stem-loop structure determinant in potato virus x RNA recombination. J. Gen. Virol. 2010, 91, 552–562. [Google Scholar] [CrossRef]
- Simon-Loriere, E.; Martin, D.P.; Weeks, K.M.; Negroni, M. Rna structures facilitate recombination-mediated gene swapping in HIV-1. J. Virol. 2010, 84, 12675–12682. [Google Scholar] [CrossRef]
- Shepherd, D.N.; Martin, D.P.; Varsani, A.; Thomson, J.A.; Rybicki, E.P.; Klump, H.H. Restoration of native folding of single-stranded DNA sequences through reverse mutations: An indication of a new epigenetic mechanism. Arch. Biochem. Biophys. 2006, 453, 108–122. [Google Scholar] [CrossRef]
- Brewer, B.J. When polymerases collide-Replication and the transcriptional organization of the escherichia-coli chromosome. Cell 1988, 53, 679–686. [Google Scholar] [CrossRef]
- Lett, J.M.; Lefeuvre, P.; Couston, L.; Hoareau, M.; Thierry, M.; Reynaud, B.; Martin, D.P.; Varsani, A. Complete genomic sequences of tomato yellow leaf curl mali virus isolates infecting tomato and pepper from the north province of cameroon. Arch. Virol. 2009, 154, 535–540. [Google Scholar] [CrossRef]
- Ben Asher, E.; Bratosin, S.; Aloni, Y. Intracellular DNA of the parvovirus minute virus of mice is organized in a minichromosome structure. J. Virol. 1982, 41, 1044–1054. [Google Scholar] [CrossRef]
- Felsenstein, J. The evolutionary advantage of recombination. Genetics 1974, 78, 737–756. [Google Scholar] [CrossRef] [PubMed]
- Keightley, P.D.; Otto, S.P. Interference among deleterious mutations favours sex and recombination in finite populations. Nature 2006, 443, 89–92. [Google Scholar] [CrossRef] [PubMed]
- Martin, G.; Otto, S.P.; Lenormand, T. Selection for recombination in structured populations. Genetics 2006, 172, 593–609. [Google Scholar] [CrossRef] [PubMed]
- Gagnon, C.A.; Music, N.; Fontaine, G.; Tremblay, D.; Harel, J. Emergence of a new type of porcine circovirus in swine (pcv): A type 1 and type 2 pcv recombinant. Vet. Microbiol. 2010, 144, 18–23. [Google Scholar] [CrossRef]
- Kim, H.K.; Luo, Y.; Moon, H.J.; Park, S.J.; Keum, H.O.; Rho, S.; Park, B.K. Phylogenetic and recombination analysis of genomic sequences of pcv2 isolated in korea. Virus Genes 2009, 39, 352–358. [Google Scholar] [CrossRef]
- Amin, I.; Mansoor, S.; Amrao, L.; Hussain, M.; Irum, S.; Zafar, Y.; Bull, S.E.; Briddon, R.W. Mobilisation into cotton and spread of a recombinant cotton leaf curl disease satellite-Brief report. Arch. Virol. 2006, 151, 2055–2065. [Google Scholar] [CrossRef]
- Martin, D.P.; Willment, J.A.; Billharz, R.; Velders, R.; Odhiambo, B.; Njuguna, J.; James, D.; Rybicki, E.P. Sequence diversity and virulence in zea mays of maize streak virus isolates. Virology 2001, 288, 247–255. [Google Scholar] [CrossRef]
- Harkins, G.W.; Martin, D.P.; Duffy, S.; Monjane, A.L.; Shepherd, D.N.; Windram, O.P.; Owor, B.E.; Donaldson, L.; van Antwerpen, T.; Sayed, R.A.; et al. Dating the origins of the maize adapted strain of maize streak virus, msv-a. J. Gen. Virol. 2009, 90, 3066–3074. [Google Scholar] [CrossRef]
- Zhou, X.P.; Robinson, D.J.; Harrison, B.D. Types of variation in DNA-a among isolates of east african cassava mosaic virus from kenya, malawi and tanzania. J. Gen. Virol. 1998, 79, 2835–2840. [Google Scholar] [CrossRef]
- Pita, J.S.; Fondong, V.N.; Sangare, A.; Kokora, R.N.N.; Fauquet, C.M. Genomic and biological diversity of the african cassava geminiviruses. Euphytica 2001, 120, 115–125. [Google Scholar] [CrossRef]
- Sanz, A.I.; Fraile, A.; Garcia-Arenal, F.; Zhou, X.P.; Robinson, D.J.; Khalid, S.; Butt, T.; Harrison, B.D. Multiple infection, recombination and genome relationships among begomovirus isolates found in cotton and other plants in pakistan. J. Gen. Virol. 2000, 81, 1839–1849. [Google Scholar] [CrossRef] [PubMed]
- Monci, F.; Sanchez-Campos, S.; Navas-Castillo, J.; Moriones, E. A natural recombinant between the geminiviruses tomato yellow leaf curl sardinia virus and tomato yellow leaf curl virus exhibits a novel pathogenic phenotype and is becoming prevalent in spanish populations. Virology 2002, 303, 317–326. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Andres, S.; Accotto, G.P.; Navas-Castillo, J.; Moriones, E. Founder effect, plant host, and recombination shape the emergent population of begomoviruses that cause the tomato yellow leaf curl disease in the mediterranean basin. Virology 2007, 359, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Lefeuvre, P.; Martin, D.P.; Harkins, G.; Lemey, P.; Gray, A.J.A.; Meredith, S.; Lakay, F.; Monjane, A.; Lett, J.M.; Varsani, A.; et al. The spread of tomato yellow leaf curl virus from the middle east to the world. PLoS Pathog. 2010, 6, e1001164. [Google Scholar] [CrossRef]
- Briddon, R.W.; Pinner, M.S.; Stanley, J.; Markham, P.G. Geminivirus coat protein gene replacement alters insect specificity. Virology 1990, 177, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Hofer, P.; Bedford, I.D.; Markham, P.G.; Jeske, H.; Frischmuth, T. Coat protein gene replacement results in whitefly transmission of an insect nontransmissible geminivirus isolate. Virology 1997, 236, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Petty, I.T.D. Genetic analysis of bipartite geminivirus tissue tropism. Virology 2001, 291, 311–323. [Google Scholar] [CrossRef]
- Martin, D.P.; Rybicki, E.P. Investigation of maize streak virus pathogenicity determinants using chimaeric genomes. Virology 2002, 300, 180–188. [Google Scholar] [CrossRef]
- Schnippenkoetter, W.H.; Martin, D.P.; Hughes, F.L.; Fyvie, M.; Willment, J.A.; James, D.; von Wechmar, M.B.; Rybicki, E.P. The relative infectivities and genomic characterisation of three distinct mastreviruses from south africa. Arch. Virol. 2001, 146, 1075–1088. [Google Scholar] [CrossRef]
- Koerber, J.T.; Jang, J.H.; Schaffer, D.V. DNA shuffling of adeno-associated virus yields functionally diverse viral progeny. Mol. Ther. 2008, 16, 1703–1709. [Google Scholar] [CrossRef]
- Maheshri, N.; Koerber, J.T.; Kaspar, B.K.; Schaffer, D.V. Directed evolution of adeno-associated virus yields enhanced gene delivery vectors. Nat. Biotechnol. 2006, 24, 198–204. [Google Scholar] [CrossRef] [PubMed]
- Grimm, D.; Lee, J.S.; Wang, L.; Desai, T.; Akache, B.; Storm, T.A.; Kay, M.A. In vitro and in vivo gene therapy vector evolution via multispecies interbreeding and retargeting of adeno associated viruses. J. Virol. 2008, 82, 5887–5911. [Google Scholar] [CrossRef] [PubMed]
- Morra, M.R.; Petty, I.T.D. Tissue specificity of geminivirus infection is genetically determined. Plant Cell 2000, 12, 2259–2270. [Google Scholar] [CrossRef]
- Li, W.; Asokan, A.; Wu, Z.; Van Dyke, T.; DiPrimio, N.; Johnson, J.S.; Govindaswamy, L.; Agbandje-McKenna, M.; Leichtle, S.; Redmond, D.E., Jr.; et al. Engineering and selection of shuffled aav genomes: A new strategy for producing targeted biological nanoparticles. Mol. Ther. 2008, 16, 1252–1260. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Jiang, J.; Drouin, L.M.; Agbandje-McKenna, M.; Chen, C.; Qiao, C.; Pu, D.; Hu, X.; Wang, D.Z.; Li, J.; et al. A myocardium tropic adeno-associated virus (aav) evolved by DNA shuffling and in vivo selection. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 3946–3951. [Google Scholar] [CrossRef]
- Koerber, J.T.; Schaffer, D.V. Transposon-based mutagenesis generates diverse adeno-associated viral libraries with novel gene delivery properties. Meth. Mol. Biol. 2008, 434, 161–170. [Google Scholar]
- Evans, D.; Jeske, H. DNA b facilitates, but is not essential for, the spread of abutilon mosaic-virus in agroinoculated nicotiana-benthamiana. Virology 1993, 194, 752–757. [Google Scholar] [CrossRef]
- Vuillaume, F.; Thebaud, G.; Urbino, C.; Forfert, N.; Granier, M.; Froissart, R.; Blanc, S.; Peterschmitt, M. Distribution of the phenotypic effects of random homologous recombination between two virus species. PLoS Pathog. 2011, 7, e1002028. [Google Scholar] [CrossRef]
- Davino, S.; Napoli, C.; Dellacroce, C.; Miozzi, L.; Noris, E.; Davino, M.; Accotto, G.P. Two new natural begomovirus recombinants associated with the tomato yellow leaf curl disease co-exist with parental viruses in tomato epidemics in italy. Virus Res. 2009, 143, 15–23. [Google Scholar] [CrossRef]
- van der Walt, E.; Palmer, K.E.; Martin, D.P.; Rybicki, E.P. Viable chimaeric viruses confirm the biological importance of sequence specific maize streak virus movement protein and coat protein interactions. Virol. J. 2008, 5, 61. [Google Scholar] [CrossRef]
- Garcia-Andres, S.; Monci, F.; Navas-Castillo, J.; Moriones, E. Begomovirus genetic diversity in the native plant reservoir solanum nigrum: Evidence for the presence of a new virus species of recombinant nature. Virology 2006, 350, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Andres, S.; Tomas, D.M.; Navas-Castillo, J.; Moriones, E. Resistance-driven selection of begomoviruses associated with the tomato yellow leaf curl disease. Virus Res. 2009, 146, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Accotto, G.-P. Istituto di Virologia Vegetale, C.N.R., Torino, Italy. 2010; Unpublished work. [Google Scholar]
- Rokyta, D.R.; Wichman, H.A. Genic incompatibilities in two hybrid bacteriophages. Mol. Biol. Evol. 2009, 26, 2831–2839. [Google Scholar] [CrossRef] [PubMed]
- Lefeuvre, P.; Lett, J.M.; Reynaud, B.; Martin, D.P. Avoidance of protein fold disruption in natural virus recombinants. PLoS Pathog. 2007, 3, 1782–1789. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; van der Walt, E.; Posada, D.; Rybicki, E.P. The evolutionary value of recombination is constrained by genome modularity. PLoS Genet. 2005, 1, 475–479. [Google Scholar] [CrossRef]
- Choi, L.R.; Stenger, D.C. Strain-specific determinants of beet curly top geminivirus DNA replication. Virology 1995, 206, 904–912. [Google Scholar] [CrossRef]
- Choi, I.R.; Stenger, D.C. The strain-specific cis-acting element of beet curly top geminivirus DNA replication maps to the directly repeated motif of the ori. Virology 1996, 226, 122–126. [Google Scholar] [CrossRef]
- Willment, J.A.; Martin, D.P.; Palmer, K.E.; Schnippenkoetter, W.H.; Shepherd, D.N.; Rybicki, E.P. Identification of long intergenic region sequences involved in maize streak virus replication. J. Gen. Virol. 2007, 88, 1831–1841. [Google Scholar] [CrossRef]
- Voigt, C.A.; Martinez, C.; Wang, Z.G.; Mayo, S.L.; Arnold, F.H. Protein building blocks preserved by recombination. Nat. Struct. Biol. 2002, 9, 553–558. [Google Scholar] [CrossRef]
- Hauck, B.; Xiao, W.D. Characterization of tissue tropism determinants of adeno-associated virus type 1. J. Virol. 2003, 77, 2768–2774. [Google Scholar] [CrossRef]
- Orozco, B.M.; HanleyBowdoin, L. A DNA structure is required for geminivirus replication origin function. J. Virol. 1996, 70, 148–158. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.C.; Tyson, J.J.; Lederman, M.; Stout, E.R.; Bates, R.C. A kinetic hairpin transfer model for parvoviral DNA replication. J. Mol. Biol. 1989, 208, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Cotmore, S.F.; Tattersall, P. High-mobility group 1/2 proteins are essential for initiating rolling circle-type DNA replication at a parvovirus hairpin origin. J. Virol. 1998, 72, 8477–8484. [Google Scholar] [CrossRef]
- Costello, E.; Sahli, R.; Hirt, B.; Beard, P. The mismatched nucleotides in the 5’-terminal hairpin of minute virus of mice are required for efficient viral-DNA replication. J. Virol. 1995, 69, 7489–7496. [Google Scholar] [CrossRef]
- Perros, M.; Spegelaere, P.; Dupont, F.; Vanacker, J.M.; Rommelaere, J. Cruciform structure of a DNA motif of parvovirus minute virus of mice (prototype strain) involved in the attenuation of gene-expression. J. Gen. Virol. 1994, 75, 2645–2653. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.E.; Korber, B.T. HIV-1 intra-subtype superinfection rates: Estimates using a structured coalescent with recombination. Infect. Genet. Evol. 2005, 5, 85–95. [Google Scholar] [CrossRef]
- Lemey, P.; Rambaut, A.; Drummond, A.J.; Suchard, M.A. Bayesian phylogeography finds its roots. PLoS Comput. Biol. 2009, 5, e1000520. [Google Scholar] [CrossRef] [PubMed]
- Lemey, P.; Rambaut, A.; Welch, J.J.; Suchard, M.A. Phylogeography takes a relaxed random walk in continuous space and time. Mol. Biol. Evol. 2010, 27, 1877–1885. [Google Scholar] [CrossRef]
- Currie, T.E.; Greenhill, S.J.; Gray, R.D.; Hasegawa, T.; Mace, R. Rise and fall of political complexity in island south-east asia and the pacific. Nature 2010, 467, 801–804. [Google Scholar] [CrossRef]
- van Bocxlaer, I.; Loader, S.P.; Roelants, K.; Biju, S.D.; Menegon, M.; Bossuyt, F. Gradual adaptation toward a range-expansion phenotype initiated the global radiation of toads. Science 2010, 327, 679–682. [Google Scholar] [CrossRef]
- Arenas, M.; Posada, D. The effect of recombination on the reconstruction of ancestral sequences. Genetics 2010, 184, 1133–1429. [Google Scholar] [CrossRef] [PubMed]
- Gullberg, M.; Tolf, C.; Jonsson, N.; Mulders, M.N.; Savolainen-Kopra, C.; Hovi, T.; Van Ranst, M.; Lemey, P.; Hafenstein, S.; Lindberg, A.M. Characterization of a putative ancestor of coxsackievirus b5. J. Virol. 2010, 84, 9695–9708. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2011 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (https://creativecommons.org/licenses/by/3.0/).
Share and Cite
Martin, D.P.; Biagini, P.; Lefeuvre, P.; Golden, M.; Roumagnac, P.; Varsani, A. Recombination in Eukaryotic Single Stranded DNA Viruses. Viruses 2011, 3, 1699-1738. https://doi.org/10.3390/v3091699
Martin DP, Biagini P, Lefeuvre P, Golden M, Roumagnac P, Varsani A. Recombination in Eukaryotic Single Stranded DNA Viruses. Viruses. 2011; 3(9):1699-1738. https://doi.org/10.3390/v3091699
Chicago/Turabian StyleMartin, Darren P., Philippe Biagini, Pierre Lefeuvre, Michael Golden, Philippe Roumagnac, and Arvind Varsani. 2011. "Recombination in Eukaryotic Single Stranded DNA Viruses" Viruses 3, no. 9: 1699-1738. https://doi.org/10.3390/v3091699
APA StyleMartin, D. P., Biagini, P., Lefeuvre, P., Golden, M., Roumagnac, P., & Varsani, A. (2011). Recombination in Eukaryotic Single Stranded DNA Viruses. Viruses, 3(9), 1699-1738. https://doi.org/10.3390/v3091699