Recombination in Avian Gamma-Coronavirus Infectious Bronchitis Virus

Abstract
:1. Introduction
2. Results and Discussion
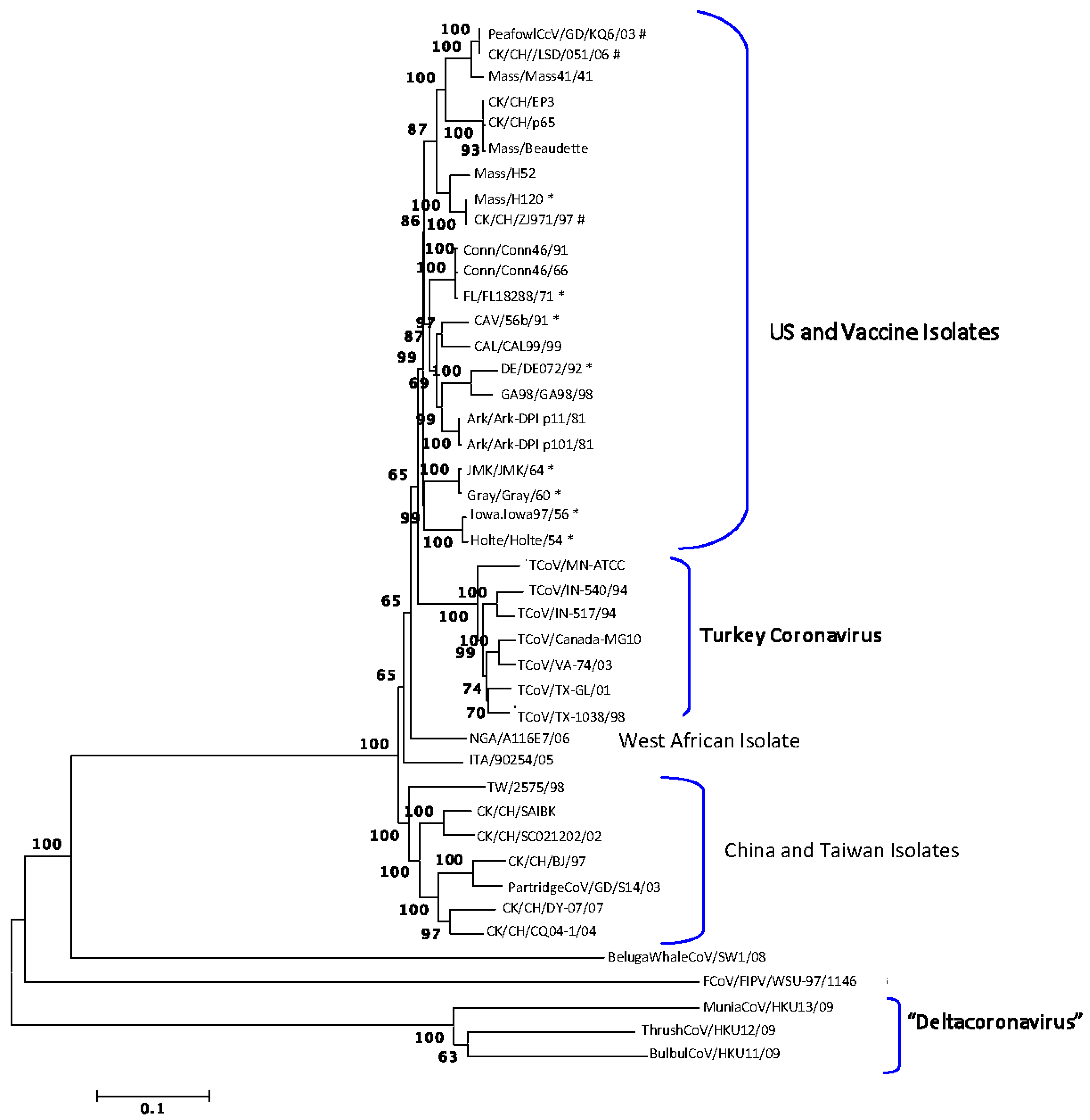
2.1. Sequence Analysis
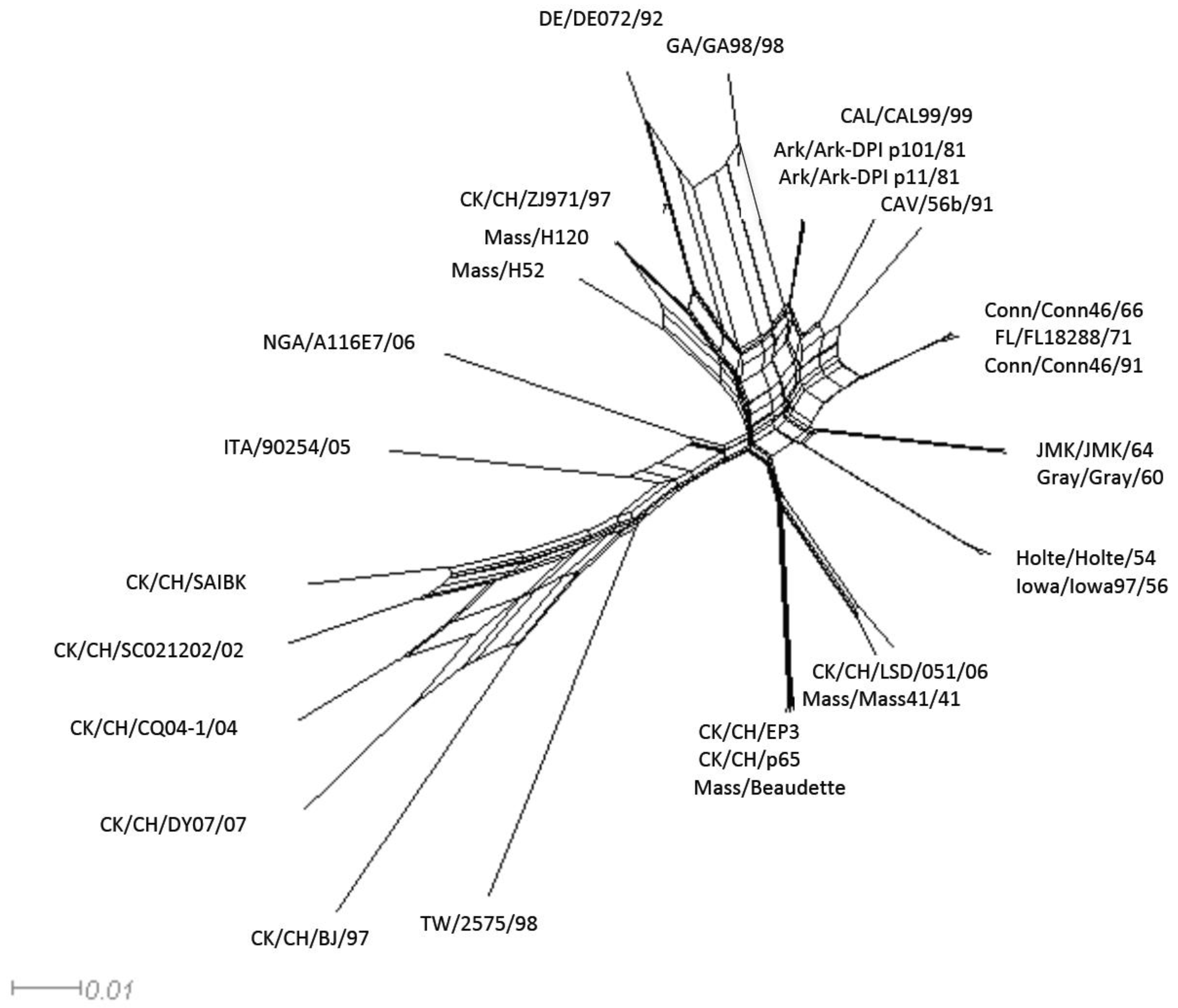
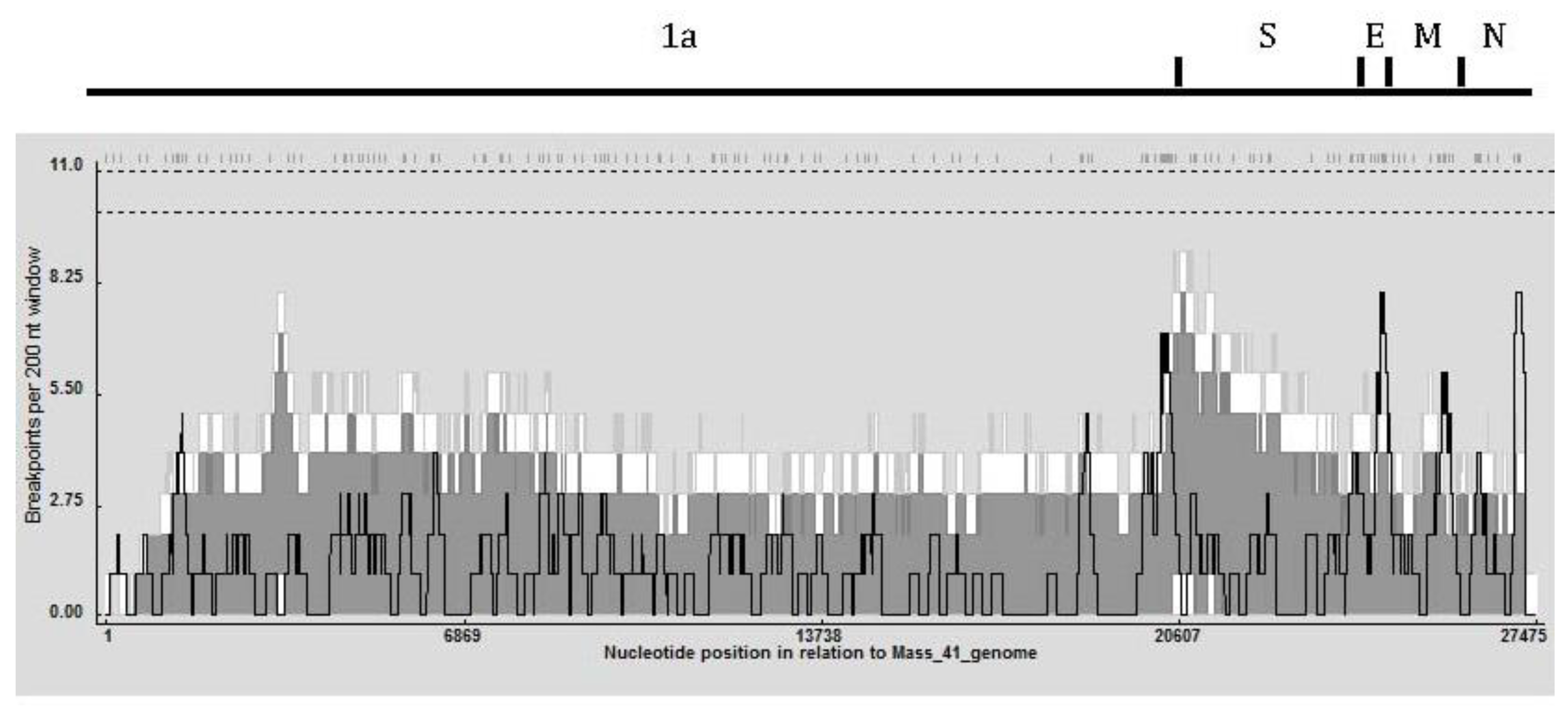
2.2. Recombination Analysis
3. Conclusions
4. Materials and Methods
4.1. Viruses and Viral RNA Extraction
4.2. RT-PCR Amplification and Sequencing
4.3. Genome Assembly and Analysis
4.4. GenBank Accession Numbers
4.5. Detection of Networked Relationships and Recombination Break Points
4.6. Phylogenic Analysis of Sequential Genome Fragments
4.7. Recombination Site Detection
Acknowledgements
References and Notes
- Lai, M.M.C.; Holmes, K.V. Coronaviridae: The viruses and their replication. In Fields Virology, 4th ed.; Knipe, D.M., Howley, P.M., Griffin, D.E., Lamb, R.A., Martin, M.A., Roizman, B., Straus, S.E., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2001; Volume 1, pp. 1163–1185. [Google Scholar]
- Cavanagh, D.; Mawditt, K.; Adzhar, A.; Gough, R.E.; Picault, J.P.; Naylor, C.J.; Haydon, D.; Shaw, K.; Britton, P. Does IBV change slowly despite the capacity of the spike protein to vary greatly? Adv. Exp. Med. Biol. 1998, 440, 729–734. [Google Scholar]
- Niesters, H.G.; Kusters, J.G.; Lenstra, J.A.; Spaan, W.J.; Horzined, M.C.; van der Zeijst, B.A. The neutralization epitopes on the spike protein of infectious bronchitis virus and their antigenic variation. Adv. Exp. Med. Biol. 1987, 218, 483–492. [Google Scholar]
- Holmes, E.C. The Evolution and Emergence of RNA Viruses, 1st ed.; Oxford University Press Inc.: New York, NY, USA, 2009. [Google Scholar]
- Lai, M. RNA Recombination in animal and plant viruses. Microbiol. Rev. 1992, 56, 61–79. [Google Scholar] [CrossRef] [PubMed]
- Kusters, J.G.; Jager, E.J.; Niesters, H.G.M.; van der Zeijst, B.A.M. Sequence evidence for RNA recombination in field isolates of avian coronavirus infectious bronchitis virus. Vaccine 1990, 8, 605–608. [Google Scholar] [CrossRef] [PubMed]
- Jia, W.; Karaca, K.; Parrish, C.R.; Naqi, S.A. A novel variant of avian infectious bronchitis virus resulting from recombination among three different strains. Arch. Virol. 1995, 140, 259–271. [Google Scholar] [CrossRef]
- Lee, C.W.; Jackwood, M.W. Origin and evolution of Georgia 98 (GA98), a new serotype of avian infectious bronchitis virus. Virus Res. 2001, 80, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.W.; Jackwood, M.W. Evidence of genetic diversity generated by recombination among avian coronavirus IBV. Arch. Virol. 2000, 145, 2135–48. [Google Scholar] [CrossRef]
- Estevez, C.; Villegas, P.; El-Attrache, J. A recombination event, induced in ovo, between a low passage infectious bronchitis virus field isolate and a highly embryo adaptedvaccine strain. Avian Dis. 2003, 47, 1282–1290. [Google Scholar] [CrossRef] [PubMed]
- Mardani, K.; Noormohammadi, A.H.; Ignjatovic, J.; Browning, G.F. Naturally occurring recombination between distant strains of infectious bronchitis virus. Arch. Virol. 2010, 155, 1581–1586. [Google Scholar] [CrossRef]
- Woo, P.C.; Lau, S.K.; Huang, Y.; Yuen, K.Y. Coronavirus diversity, phylogeny and interspecies jumping. Exp. Biol. Med. 2009, 234, 1117–1127. [Google Scholar] [CrossRef]
- Decaro, N.; Mari, V.; Campolo, M.; Lorusso, A.; Camero, M.; Elia, G.; Martella, V.; Cordioli, P.; Enjuanes, L.; Buonavoglia, C. Recombinant canine coronaviruses related to transmissible gastroenteritis virus of Swine are circulating in dogs. J. Virol. 2009, 83, 1532–1537. [Google Scholar] [CrossRef]
- Jackwood, M.W.; Boynton, T.O.; Hilt, D.A.; McKinley, E.T.; Kissinger, J.C.; Paterson, A.H.; Robertson, J.; Lemke, C.; McCall, A.W.; Williams, S.M.; Jackwood, J.W.; Byrd, L.A. Emergence of a group 3 coronavirus through recombination. Virology 2010, 398, 98–108. [Google Scholar] [CrossRef]
- Lee, C.W.; Hilt, D.A.; Jackwood, M.W. Identification and analysis of the Georgia 98 serotype, a new serotype of infectious bronchitis virus. Avian Dis. 2001, 45, 164–172. [Google Scholar] [CrossRef] [PubMed]
- National Center for Biotechnology Information. Available online: http://www.ncbi.nlm.nih.gov/ (accessed on 15 February 2011).
- Tamura, K.; Dudley, J.; Nei, M.; Kumar, S. MEGA4: Molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol. Biol. Evol. 2007, 24, 1596–1599. [Google Scholar] [CrossRef]
- Woo, P.C.Y.; Huang, Y.; Lau, S.K.; Yuen, K.Y. Coronavirus genomics and bioinformatics analysis. Viruses 2010, 2, 1804–1820. [Google Scholar] [CrossRef] [PubMed]
- Worobey, M.; Holmes, E.C. Evolutionary aspects of recombination in RNA viruses. J. Gen. Virol. 1999, 80, 2535–2543. [Google Scholar] [CrossRef] [PubMed]
- Bruen, T.C.; Philippe, H.; Bryant, D. A simple and robust statistical test for detecting the presence of recombination. Genetics 2006, 172, 2665–2681. [Google Scholar] [CrossRef]
- Heath, L.; van der Walt, E.; Varsani, A.; Martin, D.P. Recombination patterns in aphthoviruses mirror those found in other picornaviruses. J. Virol. 2006, 80, 11827–11832. [Google Scholar] [CrossRef]
- Martin, D.P. Recombination detection and analysis using RDP3. Methods Mol. Biol. 2009, 537, 185–205. [Google Scholar]
- Zhang, Y.; Wang, H.-N.; Wang, T.; Fan, W.-Q.; Zhang, A.-Y.; Wei, K.; Tian, G.-B.; Yang, X. Complete genome sequence and recombination analysis of infectious bronchitis virus attenuated vaccine strain H120. Virus Genes 2010, 41, 377–388. [Google Scholar] [CrossRef]
- Hein, R. Poultry Health Consultant, Georgetown, DE, USA. Personal Communication, 2010. [Google Scholar]
- Woo, P.C.Y.; Lau, S.K.P.; Yip, C.C.Y.; Huang, Y.; Tsoi, H.-W.; Yuen, K-Y. Comparative analysis of 22 coronavirus HKU1 genomes reveals a novel genotype and evidence of natural recombination in coronavirus HKU1. J. Virol. 2006, 80, 7136–7145. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P.; Welch, J. Frequency and dynamics of recombination within different species of human enteroviruses. J. Virol. 2006, 80, 483–493. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P.; Midgley, S. Recombination in the genesis and evolution of hepatitis B virus genotypes. J. Virol. 2005, 79, 15467–15476. [Google Scholar] [CrossRef]
- Armesto, M.; Cavanagh, D.; Britton, P. The replicase gene of avian coronavirus infectious bronchitis virus is a determinant of pathogenicity. PLoS One 2009, 4, e7384. [Google Scholar] [CrossRef]
- Hagemeijer, M.C.; Verheije, M.H.; Ulasli, M.; Shaltiel, I.A.; de Vries, L.A.; Reggiori, F.; Rottier, P.J.; de Haan, C.A. Dynamics of coronavirus replication-transcription complexes. J. Virol. 2010, 84, 2134–2149. [Google Scholar] [CrossRef]
- Lindner, H.A.; Fotouhi-Ardakani, N.; Lytvyn, V.; Lachance, P.; Sulea, T.; Menard, R. The papain-like protease from the severe acute respiratory syndrome coronavirus is a deubiquitinating enzyme. J. Virol. 2005, 79, 15199–15208. [Google Scholar] [CrossRef]
- Zheng, D.; Chen, G.; Guo, B.; Cheng, G.; Tang, H. PLP2, a potent deubiquitinase from murine hepatitis virus, strongly inhibits cellular type I interferon production. Cell Res. 2008, 18, 1105–1113. [Google Scholar] [CrossRef] [PubMed]
- Gorbalenya, A.E.; Koonin, E.V.; Donchenko, A.P.; Blinov, V.M. Coronavirus genome: Prediction of putative functional domains in the non-structural polyprotein by comparative amino acid sequence analysis. Nucleic. Acids Res. 1989, 17, 4847–4861. [Google Scholar] [CrossRef]
- Gorbalenya, A.E.; Koonin, E.V.; Lai, M.M. Putative papain-related thiol proteases of positive-strand RNA viruses. Identification of rubi- and aphthovirus proteases and delineation of a novel conserved domain associated with proteases of rubi-, alpha- and coronaviruses. FEBS Lett. 1991, 288, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, K.K.; Cervantes-Barragan, L.; Ludewig, B.; Thiel, V. Mouse hepatitis virus liver pathology is dependent on ADP-ribose-1’’-phosphatase, a viral function conserved in the alpha-like supergroup. J. Virol. 2008, 82, 12325–12334. [Google Scholar] [CrossRef]
- Zhang, X.W.; Yap, Y.L.; Danchin, A. Testing the hypothesis of a recombinant origin of the SARS-associated coronavirus. Arch. Virol. 2005, 150, 1–20. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, Q.; Li, J.; Cao, W.; Zhang, J.X.; Zhang, L.; Zhang, W.; Shao, Z.J.; Yan, Y. Analysis of recombination and natural selection in human enterovirus 71. Virology 2010, 398, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Gelb, J.J.; Jackwood, M.W. Infectious Bronchitis. In A Laboratory Manual for the Isolation, Identification, and Characterization of Avian Pathogens, 5th ed.; Dufour-Zavala, L., Swayne, D.E., Glisson, J.R., Pearson, J.E., Reed, W.M., Jackwood, M.W., Woolcock, P., Eds.; American Association of Avian Pathologists: Kennett Square, PA, USA, 2008; pp. 146–149. [Google Scholar]
- Huson, D.H.; Bryant, D. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 2006, 23, 254–267. [Google Scholar] [CrossRef] [PubMed]
- Bryant, D.; Moulton, V. Neighbor-net: An agglomerative method for the construction of phylogenetic networks. Mol. Biol. Evol. 2004, 21, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Posada, D.; Crandall, K.A.; Williamson, C. A modified bootscan algorithm for automated identification of recombinant sequences and recombination breakpoints. AIDS Res. Hum. Retroviruses 2005, 21, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Padidam, M.; Sawyer, S.; Fauquet, C.M. Possible emergence of new geminiviruses by frequent recombination. Virology 1999, 265, 218–225. [Google Scholar] [CrossRef]
- Smith, J.M. Analyzing the mosaic structure of genes. J. Mol. Evol. 1992, 34, 126–129. [Google Scholar] [CrossRef] [PubMed]
- Posada, D.; Crandall, K.A. Evaluation of methods for detecting recombination from DNA sequences: Computer simulations. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 13757–61372. [Google Scholar] [CrossRef]
- Gibbs, M.J.; Armstrong, J.S.; Gibbs, A.J. Sister-scanning: A Monte Carlo procedure for assessing signals in recombinant sequences. Bioinformatics 2000, 16, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Boni, M.F.; Posada, D.; Feldman, M.W. An exact nonparametric method for inferring mosaic structure in sequence triplets. Genetics 2007, 176, 1035–1047. [Google Scholar] [CrossRef]
- Posada, D. Evaluation of methods for detecting recombination from DNA sequences: Empirical data. Mol. Biol. Evol. 2002, 19, 708–717. [Google Scholar] [CrossRef] [PubMed]
- Salminen, M.A.M. Detecting and characterising individual recombination events: practice. In The Phylogenetic Handbook: A Practical Approach to Phylogenetic Analysis and Hypothesis Testing; Lemey, P., Salemi, M., Vandamme, A.M., Eds.; Cambridge University Press: Cambridge, UK, 2010; p. 723. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| ORFa | CAV/CAV56b/91 | DE/DE072/92 | FL/FL18288/71 | Gray/Gray/60 | Mass/H120 | Holte/Holte/54 | Iowa/Iowa97/56 | JMK/JMK/64 | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Location | ntb | aac | Location | nt | aa | Location | nt | aa | Location | nt | aa | Location | nt | aa | Location | nt | aa | Location | nt | aa | Location | nt | aa | |
| 5’UTR | 1–527 | 527 | – | 1–528 | 528 | – | 1–528 | 528 | – | 1–528 | 528 | – | 1–528 | 528 | – | 1–528 | 528 | – | 1–528 | 528 | – | 1–528 | 528 | – |
| 1a | 528–12389 | 1,862 | 3953 | 529–12309 | 11781 | 3926 | 529–12387 | 11859 | 3952 | 529–12387 | 11859 | 3952 | 529–12330 | 11802 | 3933 | 529–12384 | 11856 | 3951 | 529–12390 | 11802 | 3933 | 529–12387 | 11859 | 3952 |
| 1ab | 528–20422 | 19895 | 6631 | 529–20336 | 19808 | 6602 | 529–20420 | 19892 | 6630 | 529–20420 | 19892 | 6630 | 529–20363 | 19835 | 6611 | 529–20414 | 19886 | 6628 | 529–20423 | 19895 | 6631 | 529–20421 | 19893 | 6630 |
| Spike | 20373–23873 | 3501 | 1166 | 20287–23739 | 3453 | 1150 | 20371–23838 | 3468 | 1155 | 20371–23874 | 3504 | 1167 | 20314–23802 | 3489 | 1162 | 20365–23871 | 3507 | 1168 | 20374–23880 | 3507 | 1168 | 20371–23877 | 3507 | 1168 |
| 3a | 23873–24046 | 174 | 57 | 23785–23958 | 174 | 57 | 23838–24011 | 164 | 54 | 23874–24047 | 174 | 57 | 23802–23975 | 174 | 57 | 23871–24044 | 174 | 57 | 23880–24053 | 174 | 57 | 23877–24050 | 174 | 57 |
| 3b | 24046–24240 | 195 | 64 | 23958–24152 | 195 | 64 | 24011–24202 | 192 | 63 | 24047–24241 | 195 | 64 | 23975–24169 | 195 | 64 | 24044–24238 | 195 | 64 | 24053–24247 | 195 | 64 | 24050–24244 | 195 | 64 |
| Envelope | 24221–24502 | 282 | 93 | 24133–24462 | 330 | 109 | 24186–24488 | 303 | 100 | 24222–24545 | 324 | 107 | 24150–24479 | 330 | 109 | 24219–24542 | 324 | 107 | 24228–24551 | 324 | 107 | 24225–24548 | 324 | 107 |
| Membrane | 24651–25175 | 525 | 174 | 24434–25111 | 678 | 225 | 24488–25156 | 669 | 222 | 24523–25188 | 666 | 221 | 24451–25128 | 678 | 225 | 24520–25188 | 667 | 222 | 24529–25140 | 612 | 203 | 24526–25197 | 672 | 223 |
| 4b | 25176–25460 | 285 | 94 | 25112–25396 | 285 | 94 | 25157–25441 | 285 | 94 | 25189–25428 | 240 | 79 | 25129–25371 | 243 | 80 | 25189–25473 | 285 | 94 | 25194–25478 | 285 | 94 | 25198–25329 | 132 | 43 |
| 4c | 25381–25554 | 174 | 57 | 25317–25487 | 171 | 56 | 25362–25532 | 171 | 56 | 25340–25510 | 171 | 56 | 25334–25504 | 171 | 56 | 25394–25534 | 141 | 46 | 25399–25539 | 141 | 46 | 25374–25568 | 195 | 64 |
| 5a | 25538–25735 | 198 | 65 | 25471–25668 | 198 | 65 | 25516–25713 | 198 | 65 | 25494–25691 | 198 | 65 | 25488–25685 | 198 | 65 | 25547–25744 | 198 | 65 | 25552–25749 | 198 | 65 | 25552–25749 | 198 | 65 |
| 5b | 25732–25980 | 249 | 82 | 25665–25913 | 249 | 82 | 25710–25958 | 249 | 82 | 25688–25936 | 249 | 82 | 25682–25930 | 249 | 82 | 25741–25989 | 249 | 82 | 25746–25994 | 249 | 82 | 25746–25994 | 249 | 82 |
| Nucleocapsid | 25923–27152 | 1230 | 409 | 25856–27085 | 1230 | 409 | 25901–27130 | 1230 | 409 | 25879–27111 | 1233 | 410 | 25873–27102 | 1230 | 409 | 25932–27161 | 1230 | 409 | 25937–27166 | 1230 | 409 | 25937–27166 | 1230 | 409 |
| 6b | 27161–27385 | 225 | 74 | 27094–27318 | 225 | 74 | 27139–27363 | 225 | 74 | – | – | – | 27126–27356 | 231 | 76 | – | – | – | 27175–27399 | 225 | 74 | 27175–27399 | 225 | 74 |
| 3’UTR | 27386–27663 | 248 | – | 27319–27591 | 273 | – | 27364–27616 | 253 | – | 27112–27568 | 455 | – | 27357–27632 | 276 | – | 27162–27246 | 85 | – | 27340–27662 | 323 | – | 27400–27793 | 393 | – |
| Recombinant | Breakpoints | Genes b | Major Sequence c | Minor Sequence d | Detection Method | ||
|---|---|---|---|---|---|---|---|
| Begin | End | ||||||
| Ark/Ark-DPI-11/81 | 3,498 | 8,667 | 1ab (nsp 3, 4, and 5) | Conn/Conn46/66 | DE/DE072/92 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | |
| 4,312 | 10,590 | 1ab (nsp 3, 4, 5, and 6) | CK/CH/LSD/051 | DE/DE072/92 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 13,072 | 20,186 | 1ab (nsp 11/12, 13, 14, 15, and 16) | Unknowne (JMK/JMK/64) | CAL/CAL99/99 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 20,292 | 23,909 | 1ab (nsp16), Spike, 3a | Conn/Conn46/66 | Unknown (Mass/Mass41) | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 21,613 | 23,856* | Spike, 3a | CAL/CAL99/99 | JMK/JMK/64 | RDP, Maxchi, Chimaera, SiSscan, 3Seq | ||
| Ark/Ark-DPI-101/81 | 3,498 | 8,667 | 1ab (nsp 3, 4, and 5) | Conn/Conn46/66 | DE/DE072/92 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | |
| 4,312 | 10,590 | 1ab (nsp 3, 4, 5, and 6) | CK/CH/LSD/051 | DE/DE072/92 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 13,072 | 20,186 | 1ab (nsp 11/12, 13, 14, 15, and 16) | Unknown (JMK/JMK/64) | CAL/CAL99/99 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 20,292 | 23,909 | 1ab (nsp16), Spike, 3a | Conn/Conn46/66 | Unknown (Mass/Mass41) | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 21,613 | 23,856* | Spike, 3a | CAL/CAL99/99 | JMK/JMK/64 | RDP, Maxchi, Chimaera, SiSscan, 3Seq | ||
| CAL/CAL99/99 | 0* | 4,368* | 5’UTR,1ab (nsp 2 and 3) | Ark/Ark-DPI/81 | Unknown (DE/DE072/92) | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | |
| 2,382 | 4,255* | 1ab (nsp2,nsp3) | DE/DE072/92 | Conn/Conn46/66 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 4,312 | 10,590 | 1ab (nsp 3, 4, 5, and 6) | CK/CH/LSD/051 | DE/DE072/92 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 8,104 | 10,649* | 1ab (nsp 4, 5, and 6) | DE/DE072/92 | Conn/Conn46/66 | RDP, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 24,587* | 25,773 | Envelope, Membrane, 4b, 4c, 5a, 5b | Unknown (GA/GA98/98) | Ark/Ark-DPI/81 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| CAV/56b/91 | 0* | 1,512 | 1ab (nsp 2) | ITA/90254/2005 | DE/DE072/92 | RDP, GENECONV, Maxchi, Chimaera, 3Seq | |
| 0* | 4,368* | 5’UTR,1ab (nsp 2 and 3) | Ark/Ark-DPI/81 | Unknown (DE/DE072/92) | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 4,312 | 10,590 | 1ab (nsp 3, 4, 5, and 6) | CK/CH/LSD/051 | DE/DE072/92 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 4,392* | 4,558 | 1ab (nsp3) | Ark/Ark-DPI/81 | Conn/Conn46/91 | GENECONV, Maxchi, Chimaera, 3Seq | ||
| 8,104 | 10,649* | 1ab (nsp 4, 5, and 6) | DE/DE072/92 | Conn/Conn46/66 | RDP, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 13,072 | 20,186 | 1ab (nsp 11/12, 13, 14, 15, and 16) | Unknown (JMK/JMK/64) | CAL/CAL99/99 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 20,292 | 23,909 | 1ab (nsp16), Spike, 3a | Conn/Conn46/66 | Unknown (Mass/Mass41) | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 24,556 | 25,748 | Envelope, Membrane, 4b, 4c, 5a, 5b | Ark/Ark-DPI/81 | Unknown (CAL/CAL99/99) | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| CK/CH/BJ/97 | 31* | 5,600 | 5’UTR, 1ab (nsp 2 and 3) | CK/CH/SAIBK | Unknown (CK/CH/CQ041/04) | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | |
| CK/CH/CQ04-1/04 | 60* | 4,711 | 5’UTR, 1ab (nsp 2 and 3) | CK/CH/SC021202/02 | CK/CH/DY-07/07 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | |
| 8,751 | 9,018 | 1 ab (nsp 5) | CK/CH/SC021202/02 | CK/CH/DY-07/07 | RDP, GENECONV, Maxchi, Chimaera | ||
| 9,626 | 18,737 | 1ab (nsp 5, 6, 7, 8, 9, 10, 11/12, 13, 14, 15) | CK/CH/SAIBK | CK/CH/DY-07/07 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 18,738* | 20,350 | 1ab (nsp 15 and 16) | CK/CH/SAIBK | ITA/90254/2005 | RDP, GENECONV, Maxchi, Chimaera | ||
| 20,160 | 21,138 | 1ab (nsp 16), Spike | JMK/JMK/64 | CK/CH/BJ/97 | RDP, GENECONV, Maxchi, Chimaera, SiSscan | ||
| 27,120 | 27,354 | Nucleocapsid, 6b | JMK/JMK/64 | CK/CH/DY-07/07 | GENECONV, Maxchi, Chimaera, SiSscan | ||
| CK/CH/DY-07/07 | 1,170 | 5,017 | 1ab (nsp 2 and 3) | DE/DE072/92 | CK/CH/SAIBK | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | |
| 22,216 | 23,963 | Spike, 3a | CK/CH/BJ/97 | CK/CH/CQ04-1/04 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 25,455 | 25,662 | 4c, 5a | CK/CH/BJ/97 | CK/CH/CQ04-1/04 | RDP, GENECONV, Maxchi, Chimaera, SiSscan | ||
| CK/CH/LSD/051/06 | 306 | 3,628* | 5’UTR, 1ab (nsp 2 and 3) | Mass/Mass41 | Ark/Ark-DPI/81 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | |
| 1,453 | 2,743 | 1ab (nsp 2 and 3) | Mass/H52 | Mass/Mass41/41 | GENECONV, Maxchi, Chimaera, 3Seq | ||
| 13,668 | 14,734 | 1ab, (nsp 11/12) | Mass/Mass41/41 | DE/DE072/92 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 15,447 | 15,821 | 1ab (nsp 13) | Mass/Mass41/41 | DE/DE072/92 | RDP, GENECONV, Maxchi, Chimaera, SiSscan | ||
| 20,203 | 24,772 | 1ab (nsp 16), Spike, 3a, 3b, Envelope, Membrane | NGA/A116E7/06 | Mass/Mass41 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 25,063 | 25,776 | Membrane, 4b, 4c, 5a, 5b | Unknown (Mass/Mass41/41) | Mass/H120 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 25,774* | 26,341 | 5b, Nucleocapsid | Mass/Mass41/41 | Mass/H120 | RDP, GENECONV, SiSscan, 3Seq | ||
| CK/CH/SAIBK | 7,241 | 9,126 | 1ab (nsp 3, 4,5) | CK/CH/SC0212/02 | DE/DE072/92 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | |
| 20,160 | 21,138 | 1ab (nsp 16), Spike | JMK/JMK/64 | CK/CH/BJ/97 | RDP, GENECONV, Maxchi, Chimaera, SiSscan | ||
| CK/CH/SC021202/02 | 13,342 | 14,784 | 1ab (nsp 11/12) | CK/CH/SAIBK | CK/CH/DY-07/07 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | |
| 20,160 | 21,138 | 1ab (nsp 16), Spike | JMK/JMK/64 | CK/CH/BJ/97 | RDP, GENECONV, Maxchi, Chimaera, SiSscan | ||
| 27,120 | 27,354 | Nucleocapsid, 6b | JMK/JMK/64 | CK/CH/DY-07/07 | GENECONV, Maxchi, Chimaera, SiSscan | ||
| CK/CH/ZJ971/97 | 0* | 11,115 | 5’UTR, 1ab (nsp 2, 3, 4, 5, 6, 7, and 8) | NGA/A116E7/06 | Ark/Ark-DPI/81 | RDP, GENECONV, Maxchi, Chimaera, SiSscan | |
| 306 | 3,628* | 5’UTR, 1ab (nsp 2 and 3) | Mass/Mass41 | Ark/Ark-DPI/81 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 4,312 | 10,590 | 1ab (nsp 3, 4, 5, and 6) | CK/CH/LSD/051 | DE/DE072/92 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 20,203 | 24,772 | 1ab (nsp 16), Spike, 3a, 3b, Envelope, Membrane | NGA/A116E7/06 | Mass/Mass41 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 26,286 | 27,027 | Nucleocapsid, 6b, 3’UTR | Iowa/Iowa97/56 | CAL/CAL99/99 | RDP, GENECONV, Maxchi, Chimaera, 3Seq | ||
| 27,094 | 27,244 | Nucleocapsid, 6b | Iowa/Iowa97/56 | Unknown (TW/2575/98) | RDP, GENECONV, Maxchi, Chimaera, SiSscan | ||
| Conn/Conn46/66 | 0* | 1,512 | 1ab (nsp 2) | ITA/90254/2005 | DE/DE072/92 | RDP, GENECONV, Maxchi, Chimaera, 3Seq | |
| 0* | 4,368* | 5’UTR,1ab (nsp 2 and 3) | Ark/Ark-DPI/81 | Unknown (DE/DE072/92) | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 13,072 | 20,186 | 1ab (nsp 11/12, 13, 14, 15, and 16) | Unknown (JMK/JMK/64) | CAL/CAL99/99 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 20,361 | 21,981 | Spike | CAL/CAL99/99 | Mass/Mass41 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| Conn/Conn46/91 | 0* | 1,512 | 1ab (nsp 2) | ITA/90254/2005 | DE/DE072/92 | RDP, GENECONV, Maxchi, Chimaera, 3Seq | |
| 0* | 4,368* | 5’UTR,1ab (nsp 2 and 3) | Ark/Ark-DPI/81 | Unknown (DE/DE072/92) | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 13,072 | 20,186 | 1ab (nsp 11/12, 13, 14, 15, and 16) | Unknown (JMK/JMK/64) | CAL/CAL99/99 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 20,361 | 21,981 | Spike | CAL/CAL99/99 | Mass/Mass41 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| DE/DE072/92 | 0* | 11,115 | 5’UTR, 1ab (nsp 2, 3, 4, 5, 6, 7, and 8) | NGA/A116E7/06 | Ark/Ark-DPI/81 | RDP, GENECONV, Maxchi, Chimaera, SiSscan | |
| 18,776 | 19,911* | 1ab (nsp 15 and 16) | Mass/H120 | Ark/Ark-DPI/81 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 19,934 | 24,431 | 1ab (nsp16), Spike, 3a, 3b, Envelope | Mass/H120 | Unknown (Mass/Mass41) | RDP, GENECOV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 20,203 | 24,772 | 1ab (nsp 16), Spike, 3a, 3b, Envelope, Membrane | NGA/A116E7/06 | Mass/Mass41 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 23,504 | 24,431* | Spike, 3a, 3b, Envelope | CK/CH/CQ04-1/04 | CALCAL99/99 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 25,575 | 27,482* | 5a, 5b, Nucleocapsid, 6b, 3’UTR | CK/CH/ZJ971/97 | JMK/JMK/64 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| FL/FL18288/71 | 0* | 1,512 | 1ab (nsp 2) | ITA/90254/2005 | DE/DE072/92 | RDP, GENECONV, Maxchi, Chimaera, 3Seq | |
| 0* | 4,368* | 5’UTR,1ab (nsp 2 and 3) | Ark/Ark-DPI/81 | Unknown (DE/DE072/92) | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 13,072 | 20,186 | 1ab (nsp 11/12, 13, 14, 15, and 16) | Unknown (JMK/JMK/64) | CAL/CAL99/99 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 20,361 | 21,981 | Spike | CAL/CAL99/99 | Mass/Mass41 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| GA98/0470/98 | 0* | 4,368* | 5’UTR,1ab (nsp 2 and 3) | Ark/Ark-DPI/81 | Unknown (DE/DE072/92) | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | |
| 2,382 | 4,255* | 1ab, (nsp2 and 3) | DE/DE072/92 | Conn/Conn46/66 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 3,498 | 8,667 | 1ab (nsp 3, 4, and 5) | Conn/Conn46/66 | DE/DE072/92 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 9,569 | 9,770 | 1ab (nsp 5) | Gray/Gray/60 | Unknown (NGA/A116E7/06) | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 13,072 | 20,186 | 1ab (nsp 11/12, 13, 14, 15, and 16) | Unknown (JMK/JMK/64) | CAL/CAL99/99 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 23,504 | 24,431* | Spike, 3a, 3b, Envelope | CK/CH/CQ04-1/04 | CALCAL99/99 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 24,500* | 25,438 | Membrane, 4b | Conn/Conn46/66 | Mass/Mass41/41 | RDP, GENECONV, Chimaera, SiSscan, 3Seq | ||
| Gray/Gray/60 | 0* | 4,368* | 5’UTR,1ab (nsp 2 and 3) | Ark/Ark-DPI/81 | Unknown (DE/DE072/92) | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | |
| 8,488 | 12,055 | 1ab (nsp 4, 5, 6, 7, 8, 9, and 10) | Unknown (CK/CH/LSD/051/06) | Conn/Conn46/91 | RDP, GENECONV, Maxchi, Chimaera | ||
| 13,070* | 14,216 | 1ab (nsp 11/12) | Unknown (CAV/56b/91) | Ark/Ark-DPI/81 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 24,131 | 27,145 | 3b, Envelope, Membrane, 4b, 4c, 5a, 5b, Nucleocapsid, 3’UTR | Ark/Ark-DPI/81 | Unknown (Conn/Conn46/91) | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| Holte/Holte/54 | 0* | 4,368* | 5’UTR,1ab (nsp 2 and 3) | Ark/Ark-DPI/81 | Unknown (DE/DE072/92) | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | |
| Iowa/Iowa97/56 | 0* | 4,368 | 5’UTR,1ab (nsp 2 and 3) | Ark/Ark-DPI/81 | Unknown (DE/DE072/92) | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | |
| 4,368 | 5,144 | 1ab (nsp 3) | Holte/Holte/54 | DE/DE072/92 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| ITA/90254/05 | 16,367 | 25,699 | 1ab (nsp 13, 14, 15, 16) Spike, 3a, 3b, Envelope, Membrane, 4b, 4c, 5a | GA98/0470/98 | CK/CH/BJ/97 | RDP, GENCOV, Maxchi, SiSscan | |
| 22,216 | 23,963 | Spike, 3a | CK/CH/BJ/97 | CK/CH/CQ04-1/04 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 24,423 | 25,632* | Envelope, Membrane, 4b, 4c, 5a | CK/CH/DY-07/07 | NGA/A116E7/06 | RDP, GENECONV, Maxchi, Chimaera, 3Seq | ||
| JMK/JMK/64 | 0* | 1,512 | 1ab (nsp 2) | ITA/90254/2005 | DE/DE072/92 | RDP, GENECONV, Maxchi, Chimaera, 3Seq | |
| 0* | 4,368* | 5’UTR,1ab (nsp 2 and 3) | Ark/Ark-DPI/81 | Unknown (DE/DE072/92) | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 8,488 | 12,055 | 1ab (nsp 4, 5, 6, 7, 8, 9, and 10) | Unknown (CK/CH/LSD/051/06) | Conn/Conn46/91 | RDP, GENECONV, Maxchi, Chimaera | ||
| 13,070* | 14,216 | 1ab (nsp 11/12) | Unknown (CAV/56b/91) | Ark/Ark-DPI/81 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 24,131 | 27,145 | 3b, Envelope, Membrane, 4b, 4c, 5a, 5b, Nucleocapsid, 3’UTR | Ark/Ark-DPI/81 | Unknown (Conn/Conn46/91) | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| Mass/H52 | 306 | 3,628* | 5’UTR, 1ab (nsp 2 and 3) | Mass/Mass41 | Ark/Ark-DPI/81 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | |
| 4,312 | 10,590 | 1ab (nsp 3, 4, 5, and 6) | CK/CH/LSD/051 | DE/DE072/92 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 19,925 | 20,168* | 1ab (nsp 16) | Mass/Mass41/41 | Mass/H120 | GENECONV, Maxchi, Chimaera, SiSscan | ||
| 20,203 | 24,772 | 1ab (nsp 16), Spike, 3a, 3b, Envelope, Membrane | NGA/A116E7/06 | Mass/Mass41/41 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 25,063 | 25,776 | Membrane, 4b, 4c, 5a, 5b | Unknown (Mass/Mass41/41) | Mass/H120 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 26,286 | 27,027 | Nucleocapsid, 6b, 3’UTR | Iowa/Iowa97/56 | CAL/CAL99/99 | RDP, GENECONV, Maxchi, Chimaera, 3Seq | ||
| 26,372 | 27,526* | Nucleocapsid, 6b, 3’UTR | Unknown (DE/DE072/92) | Mass/H120 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 27,094 | 27,244 | Nucleocapsid, 6b | Iowa/Iowa97/56 | Unknown (TW/2575/98) | RDP, GENECONV, Maxchi, Chimaera, SiSscan | ||
| Mass/H120 | 0* | 11,115 | 5’UTR, 1ab (nsp 2, 3, 4, 5, 6, 7, and 8) | NGA/A116E7/06 | Ark/Ark-DPI/81 | RDP, GENECONV, Maxchi, Chimaera, SiSscan | |
| 306 | 3,628* | 5’UTR, 1ab (nsp 2 and 3) | Mass/Mass41 | Ark/Ark-DPI/81 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 4,312 | 10,590 | 1ab (nsp 3, 4, 5, and 6) | CK/CH/LSD/051 | DE/DE072/92 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 20,203 | 24,772 | 1ab (nsp 16), Spike, 3a, 3b, Envelope, Membrane | NGA/A116E7/06 | Mass/Mass41 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 26,286 | 27,027 | Nucleocapsid, 6b, 3’UTR | Iowa/Iowa97/56 | CAL/CAL99/99 | RDP, GENECONV, Maxchi, Chimaera, 3Seq | ||
| 27,094 | 27,244 | Nucleocapsid, 6b | Iowa/Iowa97/56 | Unknown (TW/2575/98) | RDP, GENECONV, Maxchi, Chimaera, SiSscan | ||
| NGA/A116E7/06 | 7,035 | 8,271 | 1ab (nsp 3 and 4) | Holte/Holte/54 | DE/DE072/92 | RDP, GENECONV, Maxchi, Chimaera, 3Seq | |
| TW/2575/98 | 20,160 | 21,138 | 1ab (nsp 16), Spike | JMK/JMK/64 | CK/CH/BJ/97 | RDP, GENECONV, Maxchi, Chimaera, SiSscan | |
| 27,120 | 27,354 | Nucleocapsid, 6b | JMK/JMK/64 | CK/CH/DY-07/07 | GENECONV, Maxchi, Chimaera, SiSscan | ||
| Genomic Region | Number of Fragments a | % of Total |
|---|---|---|
| 5’UTR b | 8 | 4.2 |
| Nsp c 2 | 20 | 10.5 |
| nsp 3 | 33 | 17.3 |
| nsp 4 | 17 | 8.9 |
| nsp 5 | 15 | 7.9 |
| nsp6 | 10 | 8.3 |
| nsp 7 | 6 | 3.2 |
| nsp 8 | 6 | 3.2 |
| nsp9 | 4 | 2.1 |
| nsp 10 | 4 | 2.1 |
| nsp 11/12 | 13 | 6.8 |
| nsp 13 | 12 | 6.3 |
| nsp 14 | 10 | 5.3 |
| nsp 15 | 10 | 5.3 |
| nsp 16 | 19 | 10.0 |
| Spike | 30 | 15.8 |
| 3a | 14 | 7.4 |
| 3b | 13 | 6.8 |
| Envelope | 17 | 8.9 |
| Membrane | 17 | 8.9 |
| 4b | 12 | 6.3 |
| 4c | 12 | 6.3 |
| 5a | 15 | 7.9 |
| 5b | 11 | 5.8 |
| Nucleocapsid | 14 | 7.4 |
| 3’UTR | 13 | 6.8 |
| Strain | Serotype | Origin | Source |
|---|---|---|---|
| CAV/CAV56b/91 | CAV | California, USA | P. Woolcock a |
| DE/DE072/92 | DE | Delmarva, USA | J. Gelb Jr b |
| FL/FL18288/71 | FL | Florida, USA | P. Villegas c |
| Gray/Gray/60 | Gray | Delmarva, USA | J. Gelb Jr. |
| Holte/Holte/54 | Holte | Wisconsin, USA | J. King d |
| Iowa/Iowa97/56 | Iowa | Iowa, USA | J. King |
| JMK/JMK/64 | JMK | Delmarva, USA | J. Gelb, Jr. |
| Mass/H120 | Mass | The Netherlands | J. King |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2011 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thor, S.W.; Hilt, D.A.; Kissinger, J.C.; Paterson, A.H.; Jackwood, M.W. Recombination in Avian Gamma-Coronavirus Infectious Bronchitis Virus. Viruses 2011, 3, 1777-1799. https://doi.org/10.3390/v3091777
Thor SW, Hilt DA, Kissinger JC, Paterson AH, Jackwood MW. Recombination in Avian Gamma-Coronavirus Infectious Bronchitis Virus. Viruses. 2011; 3(9):1777-1799. https://doi.org/10.3390/v3091777
Chicago/Turabian StyleThor, Sharmi W., Deborah A. Hilt, Jessica C. Kissinger, Andrew H. Paterson, and Mark W. Jackwood. 2011. "Recombination in Avian Gamma-Coronavirus Infectious Bronchitis Virus" Viruses 3, no. 9: 1777-1799. https://doi.org/10.3390/v3091777
APA StyleThor, S. W., Hilt, D. A., Kissinger, J. C., Paterson, A. H., & Jackwood, M. W. (2011). Recombination in Avian Gamma-Coronavirus Infectious Bronchitis Virus. Viruses, 3(9), 1777-1799. https://doi.org/10.3390/v3091777