
Single-Cell Genomics for Virology



Abstract
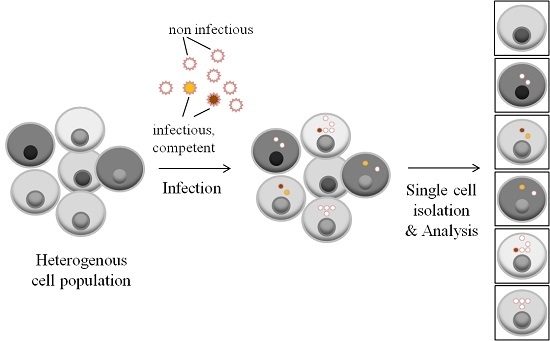
:
1. Introduction
2. The Origins of Single Cell Heterogeneity at the Transcriptome Level
3. Virus-Based Analyses
3.1. The Impact of Cellular Heterogeneity on Viral Replication (Expression and Yield)
3.1.1. Vesicular Stomatitis Virus (VSV)
3.1.2. Hepatitis C Virus (HCV)
3.1.3. Hepatitis B Virus (HBV)
3.1.4. Influenza A Virus (IAV)
4. Cell-Based Analyses
4.1. The Impact of Cellular Heterogeneity on Virus-Induced Cellular Immune Response
4.2. The Impact of Viral Infection on Cellular Transcriptome
4.3. The Impact of Cellular Heterogeneity on Viral Infection Outcome
5. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Wang, Y.; Navin, N.E. Advances and applications of single-cell sequencing technologies. Mol. Cell 2015, 58, 598–609. [Google Scholar] [CrossRef] [PubMed]
- Kolodziejczyk, A.A.; Kim, J.K.; Svensson, V.; Marioni, J.C.; Teichmann, S.A. The technology and biology of single-cell RNA sequencing. Mol. Cell 2015, 58, 610–620. [Google Scholar] [CrossRef] [PubMed]
- Grun, D.; van Oudenaarden, A. Design and Analysis of Single-Cell Sequencing Experiments. Cell 2015, 163, 799–810. [Google Scholar] [CrossRef] [PubMed]
- Navin, N.; Kendall, J.; Troge, J.; Andrews, P.; Rodgers, L.; McIndoo, J.; Cook, K.; Stepansky, A.; Levy, D.; Esposito, D.; et al. Tumour evolution inferred by single-cell sequencing. Nature 2011, 472, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Bendall, S.C.; Nolan, G.P. From single cells to deep phenotypes in cancer. Nat. Biotechnol. 2012, 30, 639–647. [Google Scholar] [CrossRef] [PubMed]
- Di Palma, S.; Bodenmiller, B. Unraveling cell populations in tumors by single-cell mass cytometry. Curr. Opin. Biotechnol. 2015, 31, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Hegde, P.S.; Clynes, R.; Foukas, P.G.; Harari, A.; Kleen, T.O.; Kvistborg, P.; Maccalli, C.; Maecker, H.T.; Page, D.B.; et al. Novel technologies and emerging biomarkers for personalized cancer immunotherapy. J. Immunother. Cancer 2016, 4, 1–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winter, D.R.; Ledergor, G.; Amit, I. From mass cytometry to cancer prognosis. Nat. Biotechnol. 2015, 33, 931–932. [Google Scholar] [CrossRef] [PubMed]
- Amir-El, A.D.; Davis, K.L.; Tadmor, M.D.; Simonds, E.F.; Levine, J.H.; Bendall, S.C.; Shenfeld, D.K.; Krishnaswamy, S.; Nolan, G.P.; Pe’er, D. viSNE enables visualization of high dimensional single-cell data and reveals phenotypic heterogeneity of leukemia. Nat. Biotechnol. 2013, 31, 545–552. [Google Scholar] [CrossRef] [PubMed]
- McConnell, M.J.; Lindberg, M.R.; Brennand, K.J.; Piper, J.C.; Voet, T.; Cowing-Zitron, C.; Shumilina, S.; Lasken, R.S.; Vermeesch, J.R.; Hall, I.M.; et al. Mosaic copy number variation in human neurons. Science 2013, 342, 632–637. [Google Scholar] [CrossRef] [PubMed]
- Snijder, B.; Sacher, R.; Ramo, P.; Damm, E.M.; Liberali, P.; Pelkmans, L. Population context determines cell-to-cell variability in endocytosis and virus infection. Nature 2009, 461, 520–523. [Google Scholar] [CrossRef] [PubMed]
- Mingueneau, M.; Kreslavsky, T.; Gray, D.; Heng, T.; Cruse, R.; Ericson, J.; Bendall, S.; Spitzer, M.H.; Nolan, G.P.; Kobayashi, K.; et al. The transcriptional landscape of alphabeta T cell differentiation. Nat. Immunol. 2013, 14, 619–632. [Google Scholar] [CrossRef] [PubMed]
- Rothenberg, E.V.; Champhekar, A.; Damle, S.; Del Real, M.M.; Kueh, H.Y.; Li, L.; Yui, M.A. Transcriptional establishment of cell-type identity: Dynamics and causal mechanisms of T-cell lineage commitment. Cold Spring Harb. Symp. Quant. Biol. 2013, 78, 31–41. [Google Scholar] [CrossRef] [PubMed]
- De Obaldia, M.E.; Bhandoola, A. Transcriptional regulation of innate and adaptive lymphocyte lineages. Annu. Rev. Immunol. 2015, 33, 607–642. [Google Scholar] [CrossRef] [PubMed]
- Rothenberg, E.V.; Ungerback, J.; Champhekar, A. Forging T-Lymphocyte Identity: Intersecting Networks of Transcriptional Control. Adv. Immunol. 2016, 129, 109–174. [Google Scholar] [PubMed]
- Buettner, F.; Natarajan, K.N.; Casale, F.P.; Proserpio, V.; Scialdone, A.; Theis, F.J.; Teichmann, S.A.; Marioni, J.C.; Stegle, O. Computational analysis of cell-to-cell heterogeneity in single-cell RNA-sequencing data reveals hidden subpopulations of cells. Nat. Biotechnol. 2015, 33, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Bahar Halpern, K.; Caspi, I.; Lemze, D.; Levy, M.; Landen, S.; Elinav, E.; Ulitsky, I.; Itzkovitz, S. Nuclear Retention of mRNA in Mammalian Tissues. Cell Rep. 2015, 13, 2653–2662. [Google Scholar] [CrossRef] [PubMed]
- Battich, N.; Stoeger, T.; Pelkmans, L. Control of Transcript Variability in Single Mammalian Cells. Cell 2015, 163, 1596–1610. [Google Scholar] [CrossRef] [PubMed]
- Janes, K.A. Cell-to-Cell Transcript Variability: Seeing Signal in the Noise. Cell 2015, 163, 1566–1568. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Dominissini, D.; Rechavi, G.; He, C. Gene expression regulation mediated through reversible m(6)A RNA methylation. Nat. Rev. Genet. 2014, 15, 293–306. [Google Scholar] [CrossRef] [PubMed]
- Razooky, B.S.; Gutierrez, E.; Terry, V.H.; Spina, C.A.; Groisman, A.; Weinberger, L.S. Microwell devices with finger-like channels for long-term imaging of HIV-1 expression kinetics in primary human lymphocytes. Lab Chip 2012, 12, 4305–4312. [Google Scholar] [CrossRef] [PubMed]
- Weinberger, L.S.; Dar, R.D.; Simpson, M.L. Transient-mediated fate determination in a transcriptional circuit of HIV. Nat. Genet. 2008, 40, 466–470. [Google Scholar] [CrossRef] [PubMed]
- Sen, N.; Mukherjee, G.; Arvin, A.M. Single cell mass cytometry reveals remodeling of human T cell phenotypes by varicella zoster virus. Methods 2015, 90, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Bendall, S.C.; Nolan, G.P.; Roederer, M.; Chattopadhyay, P.K. A deep profiler’s guide to cytometry. Trends Immunol. 2012, 33, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Cox, K.S.; Tang, A.; Chen, Z.; Horton, M.S.; Yan, H.; Wang, X.M.; Dubey, S.A.; DiStefano, D.J.; Ettenger, A.; Fong, R.H.; et al. Rapid isolation of dengue-neutralizing antibodies from single cell-sorted human antigen-specific memory B-cell cultures. MAbs 2016, 8, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Li, Y.; Zheng, C.Y. A novel single-cell quantitative real-time RT-PCR method for quantifying foot-and-mouth disease viral RNA. J. Virol. Methods 2009, 155, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.T.; Huang, S.Y.; Chen, L.; Liu, F.; Cai, X.H.; Guo, Y.F.; Wang, M.J.; Han, Y.; Yu, D.M.; Jiang, J.H.; et al. Characterization of Full-Length Genomes of Hepatitis B Virus Quasispecies in Sera of Patients at Different Phases of Infection. J. Clin. Microbiol. 2015, 53, 2203–2214. [Google Scholar] [CrossRef] [PubMed]
- Luna, J.M.; Scheel, T.K.; Danino, T.; Shaw, K.S.; Mele, A.; Fak, J.J.; Nishiuchi, E.; Takacs, C.N.; Catanese, M.T.; de Jong, Y.P.; et al. Hepatitis C virus RNA functionally sequesters miR-122. Cell 2015, 160, 1099–1110. [Google Scholar] [CrossRef] [PubMed]
- McWilliam Leitch, E.C.; McLauchlan, J. Determining the cellular diversity of hepatitis C virus quasispecies by single-cell viral sequencing. J. Virol. 2013, 87, 12648–12655. [Google Scholar] [CrossRef] [PubMed]
- Suspene, R.; Meyerhans, A. Quantification of unintegrated HIV-1 DNA at the single cell level in vivo. PLoS ONE 2012, 7, e36246. [Google Scholar] [CrossRef] [PubMed]
- Josefsson, L.; Palmer, S.; Faria, N.R.; Lemey, P.; Casazza, J.; Ambrozak, D.; Kearney, M.; Shao, W.; Kottilil, S.; Sneller, M.; et al. Single cell analysis of lymph node tissue from HIV-1 infected patients reveals that the majority of CD4+ T-cells contain one HIV-1 DNA molecule. PLoS Pathog. 2013, 9, e1003432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Margolis, D.; Karl Salzwedel, K.; Chomont, N.; Psomas, C.; Routy, J.-P.; Guido Poli, G.; Lafeuillade, A. Highlights from the Seventh International Workshop on HIV Persistence during Therapy, 8–11 December 2015, Miami, Florida, USA. J. Virus Erad. 2016, 2, 57–65. [Google Scholar]
- Ciuffi, A.; Quenneville, S.; Rausell, A.; Rato, S.; Muñoz, M.; Telenti, A. Single-cell analysis identifies biomarkers for HIV permissiveness (Abstract OP2.1). J. Virus Erad. 2015, 1 (Suppl. 1), 1–18. [Google Scholar]
- Wu, L.; Zhang, X.; Zhao, Z.; Wang, L.; Li, B.; Li, G.; Dean, M.; Yu, Q.; Wang, Y.; Lin, X.; et al. Full-length single-cell RNA-seq applied to a viral human cancer: Applications to HPV expression and splicing analysis in HeLa S3 cells. Gigascience 2015, 4, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Nakasone, H.; Yamazaki, R.; Sato, K.; Sato, M.; Terasako, K.; Kimura, S.; Okuda, S.; Kako, S.; Oshima, K.; et al. Single-cell analysis of T-cell receptor repertoire of HTLV-1 Tax-specific cytotoxic T cells in allogeneic transplant recipients with adult T-cell leukemia/lymphoma. Cancer Res. 2010, 70, 6181–6192. [Google Scholar] [CrossRef] [PubMed]
- Heldt, F.S.; Kupke, S.Y.; Dorl, S.; Reichl, U.; Frensing, T. Single-cell analysis and stochastic modelling unveil large cell-to-cell variability in influenza A virus infection. Nat. Commun. 2015, 6, 8938. [Google Scholar] [CrossRef] [PubMed]
- Laske, T.; Heldt, F.S.; Hoffmann, H.; Frensing, T.; Reichl, U. Modeling the intracellular replication of influenza A virus in the presence of defective interfering RNAs. Virus Res. 2016, 213, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Schulte, M.B.; Andino, R. Single-cell analysis uncovers extensive biological noise in poliovirus replication. J. Virol. 2014, 88, 6205–6212. [Google Scholar] [CrossRef] [PubMed]
- Schulte, M.B.; Draghi, J.A.; Plotkin, J.B.; Andino, R. Experimentally guided models reveal replication principles that shape the mutation distribution of RNA viruses. Elife 2015, 4, e03753. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Yongky, A.; Yin, J. Growth of an RNA virus in single cells reveals a broad fitness distribution. Virology 2009, 385, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Timm, A.; Yin, J. Kinetics of virus production from single cells. Virology 2012, 424, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Timm, C.; Akpinar, F.; Yin, J. Quantitative characterization of defective virus emergence by deep sequencing. J. Virol. 2014, 88, 2623–2632. [Google Scholar] [CrossRef] [PubMed]
- Akpinar, F.; Timm, A.; Yin, J. High-Throughput Single-Cell Kinetics of Virus Infections in the Presence of Defective Interfering Particles. J. Virol. 2015, 90, 1599–1612. [Google Scholar] [CrossRef] [PubMed]
- Combe, M.; Garijo, R.; Geller, R.; Cuevas, J.M.; Sanjuan, R. Single-Cell Analysis of RNA Virus Infection Identifies Multiple Genetically Diverse Viral Genomes within Single Infectious Units. Cell Host Microbe 2015, 18, 424–432. [Google Scholar] [CrossRef] [PubMed]
- Sen, N.; Mukherjee, G.; Sen, A.; Bendall, S.C.; Sung, P.; Nolan, G.P.; Arvin, A.M. Single-cell mass cytometry analysis of human tonsil T cell remodeling by varicella zoster virus. Cell Rep. 2014, 8, 633–645. [Google Scholar] [CrossRef] [PubMed]
- Tsioris, K.; Gupta, N.T.; Ogunniyi, A.O.; Zimnisky, R.M.; Qian, F.; Yao, Y.; Wang, X.; Stern, J.N.; Chari, R.; Briggs, A.W.; et al. Neutralizing antibodies against West Nile virus identified directly from human B cells by single-cell analysis and next generation sequencing. Integr. Biol. (Camb.) 2015, 7, 1587–1597. [Google Scholar] [CrossRef] [PubMed]
- Julia, M.; Telenti, A.; Rausell, A. Sincell: An R/Bioconductor package for statistical assessment of cell-state hierarchies from single-cell RNA-seq. Bioinformatics 2015. [Google Scholar] [CrossRef] [PubMed]
- Martin-Gayo, E.; Buzon, M.J.; Ouyang, Z.; Hickman, T.; Cronin, J.; Pimenova, D.; Walker, B.D.; Lichterfeld, M.; Yu, X.G. Potent Cell-Intrinsic Immune Responses in Dendritic Cells Facilitate HIV-1-Specific T Cell Immunity in HIV-1 Elite Controllers. PLoS Pathog. 2015, 11, e1004930. [Google Scholar] [CrossRef] [PubMed]
- Martin-Gayo, E.; Cole, M.; Kolb, K.E.; Ouyang, Z.; Kazer, S.W.; Walker, B.D.; Yosef, N.; Shalek, A.K.; Yu, X.G. Identification of a Highly Functional DC Subset in Controllers by Single-Cell RNA-Seq (Abstract 19). In Proceedings of the Conference on Retroviruses and Opportunistic Infections, Boston, MA, USA, 22–25 February 2016.
- Dey, S.S.; Kester, L.; Spanjaard, B.; Bienko, M.; van Oudenaarden, A. Integrated genome and transcriptome sequencing of the same cell. Nat. Biotechnol. 2015, 33, 285–289. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
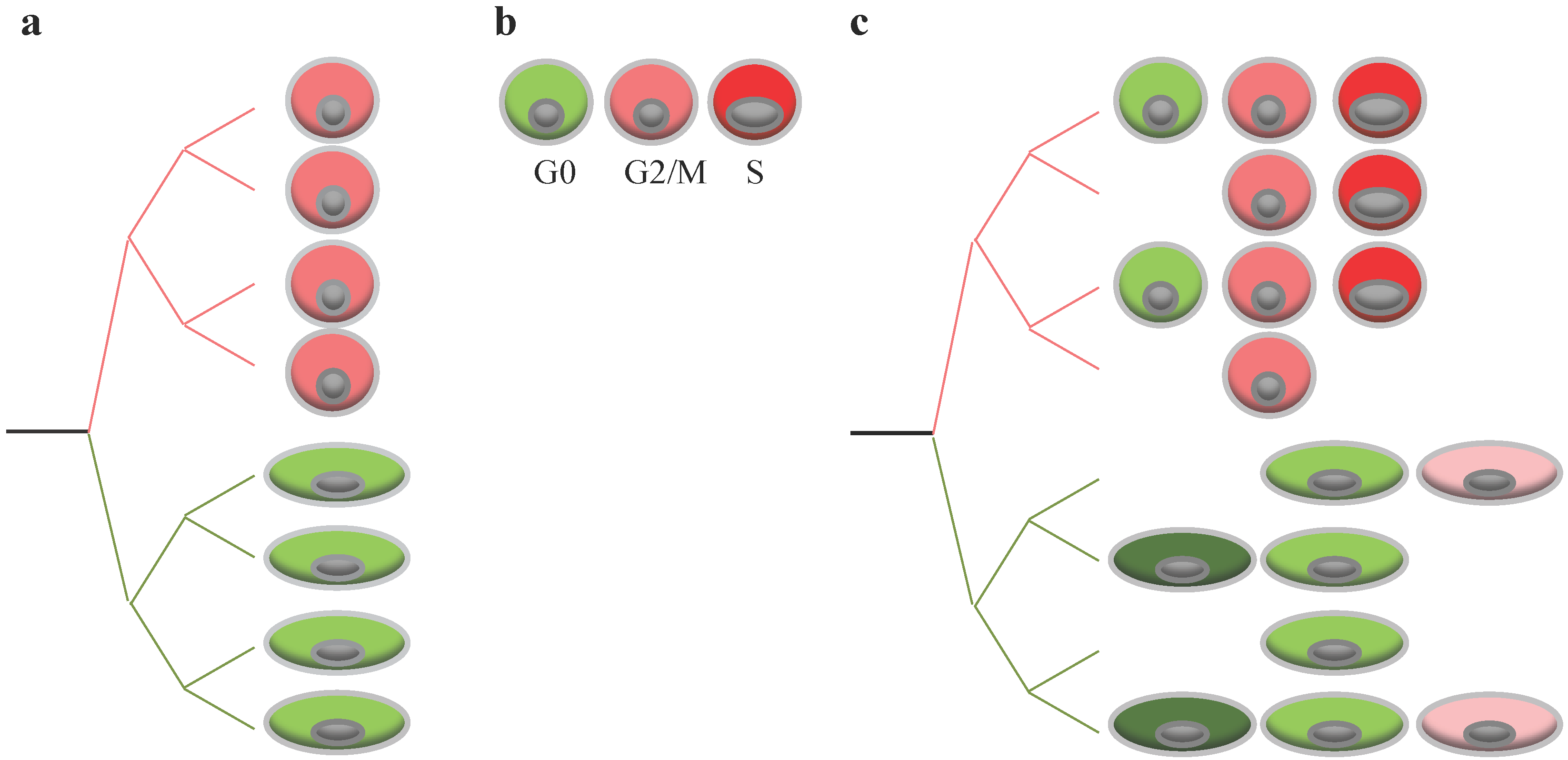{kind=link}
| Virus a | Cells b | Single Cell Isolation | Biological Sample Analyzed c | Method | Reference |
|---|---|---|---|---|---|
| SV40 (RV, MHV, DENV) | HeLa | - | Immunofluorescence | Microscopy and imaging | Snijder et al. [11] |
| DENV | B cells from DENV seropositive patients | FACS sorting | Cellular RNA | Deep-sequencing | Cox et al. [25] |
| FMDV | BHK-21 | Micromanipulation | vRNA | RT-PCR | Huang et al. [26] |
| HBV | Primary hepatocytes | Microscopy | vRNA, vDNA, cccDNA | In Situ Hybridization | Zhang et al. [27] |
| HCV | Huh-7.5 (hepatoma cell line) | FACS | Virus-encoded BFP and RFP reporters | FACS | Luna et al. [28] |
| HCV | Huh-7 (hepatoma cell line) | Micromanipulation | vRNA | Deep-sequencing, RT-qPCR | McWilliam Leitch and McLauchlan [29] |
| HIV | Jurkat (human T cell line) | Micromanipulation | Virus-encoded GFP | Time-lapse microscopy, FACS | Weinberger et al. [22] |
| HIV | Primary human CD4+ T cells (from blood) | Micromanipulation | Virus-encoded GFP | Time-lapse microscopy | Razooky et al. [21] |
| HIV | Primary human CD4+ T cells (from infected patients) | Microdissection | vDNA | PCR-Sequencing | Suspène and Meyerhans [30] |
| HIV | Primary human CD4+ T cells (from infected patients) | FACS sorting | vDNA, vRNA | Deep-sequencing | Josefsson et al. [31] |
| HIV | Primary human CD4+ T cells | Microfluidics (Fluidigm) | RNA | Deep-sequencing | Ciuffi et al. [32,33] |
| HPV | HeLaS3 | MIRALCS pipeline | Cellular RNA and vRNA | RNA-Seq | Wu et al. [34] |
| HTLV | Cytotoxic T lymphocytes from Adult-T-Leukemia (ATL) patients | FACS sorting | Cellular RNA (T-Cell Receptor repertoire) | Deep-sequencing | Tanaka et al. [35] |
| IAV | MDCK | Micromanipulation | vRNA, virion progeny | RT-qPCR, plaque assay | Heldt et al. [36]; Laske et al. [37] |
| Poliovirus | HeLaS3 | FACS sorting | Virion progeny, vRNA | Plaque assay, RT-qPCR | Schulte and Andino [38]; Schulte et al. [39] |
| VSV | BHK-21 | FACS sorting | Virion progeny | Plaque assay | Zhu et al. [40] |
| VSV | BHK-21 | Micromanipulation | Virion progeny | Plaque assay | Timm and Yin [41] |
| VSV | BHK-21 | Micromanipulation | Virion progeny (Virion-associated vRNA) | Deep-sequencing | Timm et al. [42] |
| VSV | BHK-21 | Micromanipulation | Virus-encoded GFP, virion progeny | Time-lapse microscopy, plaque assay | Akpinar et al. [43] |
| VSV | BHK-21 | Micromanipulation | Virion progeny (Virion-associated vRNA) | Deep-sequencing, plaque assay | Combe et al. [44] |
| VZV | Primary human T cells (from tonsils) | Mass Cytometry (CyTOF) | Set of 40 protein markers | Mass Cytometry (CyTOF) | Sen et al. [23,45] |
| WNV | Primary B cells (from infected patients) | Microengraving pipeline | Cellular RNA, Antibodies | Deep-sequencing, neutralization assay | Tsioris et al. [46] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ciuffi, A.; Rato, S.; Telenti, A. Single-Cell Genomics for Virology. Viruses 2016, 8, 123. https://doi.org/10.3390/v8050123
Ciuffi A, Rato S, Telenti A. Single-Cell Genomics for Virology. Viruses. 2016; 8(5):123. https://doi.org/10.3390/v8050123
Chicago/Turabian StyleCiuffi, Angela, Sylvie Rato, and Amalio Telenti. 2016. "Single-Cell Genomics for Virology" Viruses 8, no. 5: 123. https://doi.org/10.3390/v8050123
APA StyleCiuffi, A., Rato, S., & Telenti, A. (2016). Single-Cell Genomics for Virology. Viruses, 8(5), 123. https://doi.org/10.3390/v8050123