The Role of Brincidofovir in Preparation for a Potential Smallpox Outbreak


{kind=link}
{kind=link}
Abstract
:1. The Need for Smallpox Antivirals
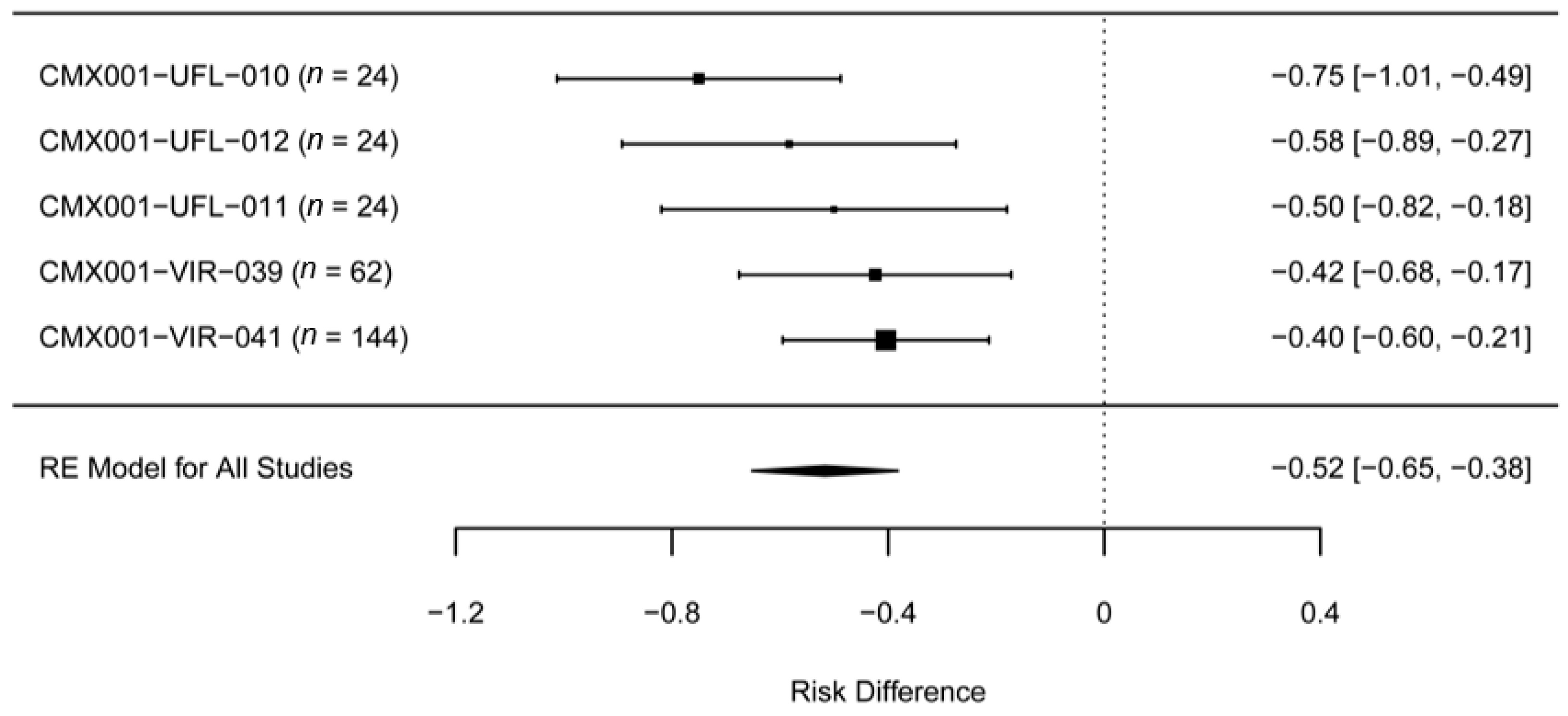
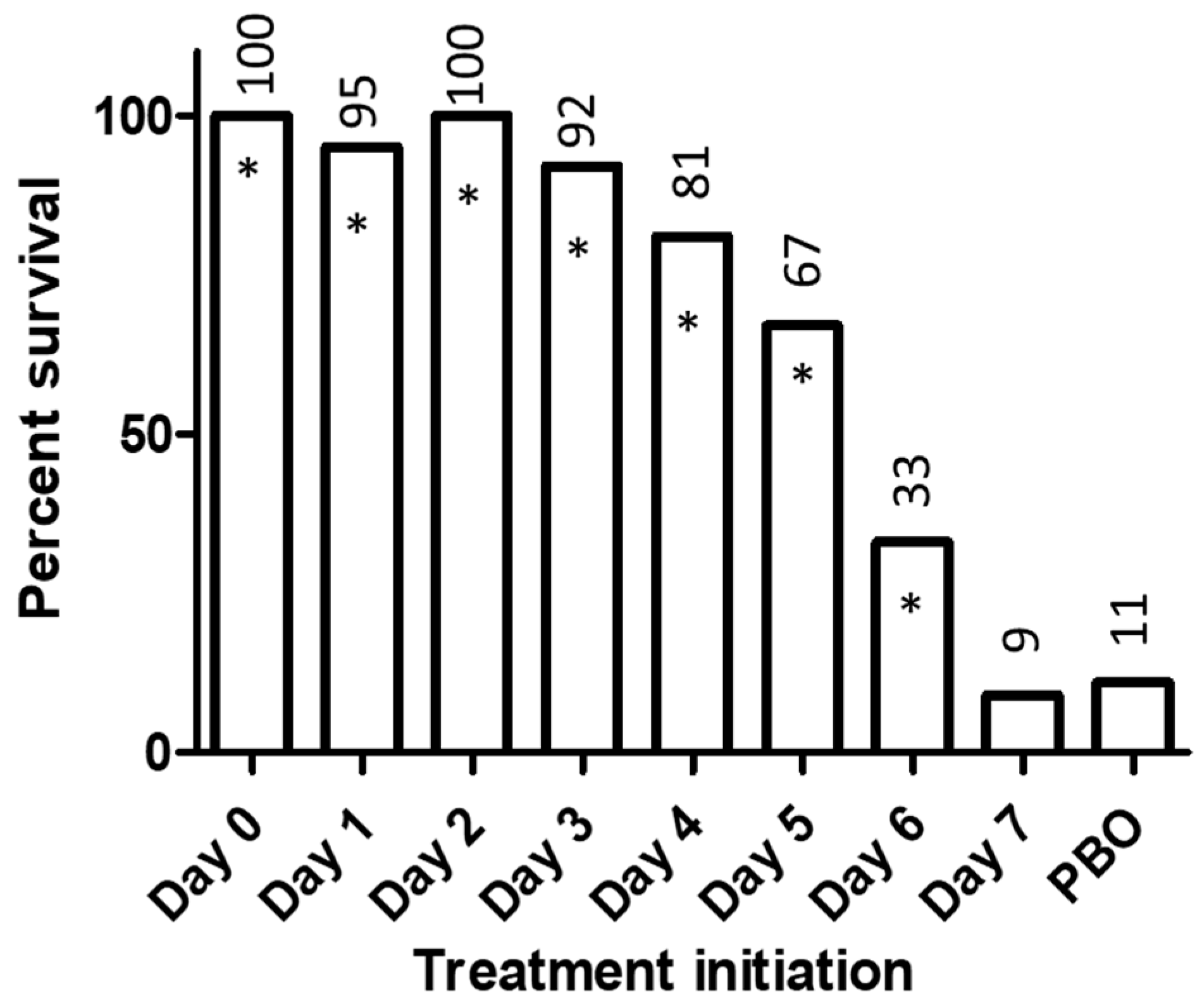
2. Experience with BCV
3. Drug-Resistant and Enhanced Virulence Viruses
4. Combination Therapy
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Henderson, D.A.; Inglesby, T.V.; Bartlett, J.G.; Ascher, M.S.; Eitzen, E.; Jahrling, P.B.; Hauer, J.; Layton, M.; McDade, J.; Osterholm, M.T.; et al. Smallpox as a biological weapon: Medical and public health management. JAMA 1999, 281, 2127–2137. [Google Scholar] [CrossRef] [PubMed]
- Gellman, B. 4 nations thought to possess smallpox: Iraq, N. Korea named, two officials say. Washington Post, 5 November 2001; A1. [Google Scholar]
- Alibek, K.; Handelman, S. Biohazard: The Chilling True Story of the Largest Biological Weapons Program in the World; Random House: New York, NY, USA, 1999; pp. 107–122. ISBN 978-0-385-33496-9. [Google Scholar]
- Kupferschmidt, K. Labmade smallpox is possible, study shows: Reconstitution of horsepox virus from mail-order DNA reignites synthetic biology debate. Science 2017, 357, 115–116. [Google Scholar] [CrossRef] [PubMed]
- Arvin, A.M.; Patel, D.M. Live Variola Virus: Considerations for Continuing Research; The National Academies Press: Washington, DC, USA, 2009; p. 133. ISBN 13978-0-309-13690-7. [Google Scholar]
- Hostetler, K.Y. Alkoxyalkyl prodrugs of acyclic nucleoside phosphonates enhance oral antiviral activity and reduce toxicity: Current state of the art. Antivir. Res. 2009, 82, A84–A98. [Google Scholar] [CrossRef] [PubMed]
- Lanier, R.; Trost, L.; Tippin, T.; Lampert, B.; Robertson, A.; Foster, S.; Rose, M.; Painter, W.; O’Mahony, R.; Almond, M.; et al. Development of CMX001 for the treatment of poxvirus infections. Viruses 2010, 2, 2740–2762. [Google Scholar] [CrossRef] [PubMed]
- Magee, W.C.; Hostetler, K.Y.; Evans, D.H. Mechanism of inhibition of vaccinia virus DNA polymerase by cidofovir diphosphate. Antimicrob. Agents Chemother. 2005, 49, 3153–3162. [Google Scholar] [CrossRef] [PubMed]
- Olson, V.A.; Smith, S.K.; Foster, S.; Li, Y.; Lanier, E.R.; Gates, I.; Trost, L.C.; Damon, I.K. In vitro efficacy of brincidofovir against variola virus. Antimicrob. Agents Chemother. 2014, 58, 5570–5571. [Google Scholar] [CrossRef] [PubMed]
- Kern, E.R.; Hartline, C.; Harden, E.; Keith, K.; Rodriguez, N.; Beadle, J.R.; Hostetler, K.Y. Enhanced inhibition of orthopoxvirus replication in vitro by alkoxyalkyl esters of cidofovir and cyclic cidofovir. Antimicrob. Agents Chemother. 2002, 46, 991–995. [Google Scholar] [CrossRef] [PubMed]
- Buller, R.M.; Owens, G.; Schriewer, J.; Melman, L.; Beadle, J.R.; Hostetler, K.Y. Efficacy of oral active ether lipid analogs of cidofovir in a lethal mousepox model. Virology 2004, 318, 474–481. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.M.; Rice, A.D.; Moyer, R.W. Rabbitpox virus and vaccinia virus infection of rabbits as a model for human smallpox. J. Virol. 2007, 81, 11084–11095. [Google Scholar] [CrossRef] [PubMed]
- Rice, A.D.; Adams, M.M.; Wallace, G.; Burrage, A.M.; Lindsey, S.F.; Smith, A.J.; Swetnam, D.; Manning, B.R.; Gray, S.A.; Lampert, B.; et al. Efficacy of CMX001 as a post exposure antiviral in New Zealand white rabbits infected with rabbitpox virus, a model for orthopoxvirus infections of humans. Viruses 2011, 3, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Rice, A.D.; Adams, M.M.; Lampert, B.; Foster, S.; Lanier, R.; Robertson, A.; Painter, G.R.; Moyer, R.W. Efficacy of CMX001 as a prophylactic and presymptomatic antiviral agent in New Zealand white rabbits infected with rabbitpox virus, a model for orthopoxvirus infections of humans. Viruses 2011, 3, 63–82. [Google Scholar] [CrossRef] [PubMed]
- Trost, L.C.; Rose, M.L.; Khouri, J.; Keilholz, L.; Long, J.; Godin, S.J.; Foster, S.A. The efficacy and pharmacokinetics of brincidofovir for the treatment of lethal rabbitpox virus infection: A model of smallpox disease. Antivir. Res. 2015, 117, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Grossi, I.M.; Foster, S.A.; Gainey, M.R.; Krile, R.T.; Dunn, J.A.; Brundage, T.; Khouri, J.M. Efficacy of delayed brincidofovir treatment against a lethal rabbitpox virus challenge in New Zealand White rabbits. Antivir. Res. 2017, 143, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Parker, S.; Schriewer, J.; Oberle, C.; Robertson, A.; Lanier, R.; Painter, G.; Buller, R.M. Using biomarkers to stage disease progression in a lethal mousepox model treated with CMX001. Antivir. Ther. 2008, 13, 863–873. [Google Scholar] [PubMed]
- Parker, S.; Siddiqui, A.M.; Oberle, C.; Hembrador, E.; Lanier, R.; Painter, G.; Robertson, A.; Buller, R.M. Mousepox in the C57BL/6 strain provides an improved model for evaluating anti-poxvirus therapies. Virology 2009, 385, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Parker, S.; Chen, N.G.; Foster, S.; Hartzler, H.; Hembrador, E.; Hruby, D.; Jordan, R.; Lanier, R.; Painter, G.; Painter, W.; et al. Evaluation of disease and viral biomarkers as triggers for therapeutic intervention in respiratory mousepox—An animal model of smallpox. Antivir. Res. 2012, 94, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Chittick, G.; Morrison, M.; Brundage, T.; Nichols, W.G. Short-term clinical safety profile of brincidofovir: A favorable benefit-risk proposition in the treatment of smallpox. Antivir. Res. 2017, 143, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Lederman, E.R.; Davidson, W.; Groff, H.L.; Smith, S.K.; Warkentien, T.; Li, Y.; Wilkins, K.A.; Karem, K.L.; Akondy, R.S.; Ahmed, R.; et al. Progressive vaccinia: Case description and laboratory-guided therapy with vaccinia immune globulin, ST-246, and CMX001. J. Infect. Dis. 2012, 206, 1372–1385. [Google Scholar] [CrossRef] [PubMed]
- Gazzani, P.; Gach, J.E.; Colmenero, I.; Martin, J.; Morton, H.; Brown, K.; Milford, D.V. Fatal disseminated cowpox virus infection in an adolescent renal transplant recipient. Pediatr. Nephrol. 2017, 32, 533–536. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.W.; Shafer, R.W. HIV-1 antiretroviral resistance: Scientific principles and clinical applications. Drugs 2012, 72, e1–e25. [Google Scholar] [CrossRef] [PubMed]
- Domingo, E.; Holland, J.J. RNA virus mutations and fitness for survival. Annu. Rev. Microbiol. 1997, 51, 151–178. [Google Scholar] [CrossRef] [PubMed]
- Duraffour, S.; Andrei, G.; Topalis, D.; Krečmerová, M.; Crance, J.M.; Garin, D.; Snoeck, R. Mutations conferring resistance to viral DNA polymerase inhibitors in camelpox virus give different drug-susceptibility profiles in vaccinia virus. J. Virol. 2012, 86, 7310–7325. [Google Scholar] [CrossRef] [PubMed]
- Andrei, G.; Gammon, D.B.; Fiten, P.; de Clercq, E.; Opdenakker, G.; Snoeck, R.; Evans, D.H. Cidofovir resistance in vaccinia virus is linked to diminished virulence in mice. J. Virol. 2006, 80, 9391–9401. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Pevear, D.C.; Davies, M.H.; Collett, M.S.; Bailey, T.; Rippen, S.; Barone, L.; Burns, C.; Rhodes, G.; Tohan, S.; et al. An orally bioavailable antipoxvirus compound (ST-246) inhibits extracellular virus formation and protects mice from lethal orthopoxvirus challenge. J. Virol. 2005, 79, 13139–13149. [Google Scholar] [CrossRef] [PubMed]
- Duraffour, S.; Lorenzo, M.M.; Zöller, G.; Topalis, D.; Grosenbach, D.; Hruby, D.E.; Andrei, G.; Blasco, R.; Meyer, H.; Snoeck, R. ST-246 is a key antiviral to inhibit the viral F13L phospholipase, one of the essential proteins for orthopoxvirus wrapping. J. Antimicrob. Chemother. 2015, 70, 1367–1380. [Google Scholar] [CrossRef] [PubMed]
- Jackson, R.J.; Ramsay, A.J.; Christensen, C.D.; Beaton, S.; Hall, D.F.; Ramshaw, I.A. Expression of mouse interleukin-4 by a recombinant ectromelia virus suppresses cytolytic lymphocyte responses and overcomes genetic resistance to mousepox. J. Virol. 2001, 75, 1205–1210. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Bellone, C.J.; Schriewer, J.; Owens, G.; Fredrickson, T.; Parker, S.; Buller, R.M. Poxvirus interleukin-4 expression overcomes inherent resistance and vaccine-induced immunity: Pathogenesis, prophylaxis, and antiviral therapy. Virology 2011, 409, 328–337. [Google Scholar] [CrossRef] [PubMed]
- Bean, B. Antiviral therapy: Current concepts and practices. Clin. Microbiol. Rev. 1992, 5, 146–182. [Google Scholar] [CrossRef] [PubMed]
- Hammer, S.M.; Squires, K.E.; Hughes, M.D.; Grimes, J.M.; Demeter, L.M.; Currier, J.S.; Eron, J.J., Jr.; Feinberg, J.E.; Balfour, H.H., Jr.; Deyton, L.R.; et al. A controlled trial of two nucleoside analogues plus indinavir in persons with human immunodeficiency virus infection and CD4 cell counts of 200 per cubic millimeter or less. N. Engl. J. Med. 1997, 337, 725–733. [Google Scholar]
- Gulick, R.M.; Mellors, J.W.; Havlir, D.; Eron, J.J.; Gonzalez, C.; McMahon, D.; Richman, D.D.; Valentine, F.T.; Jonas, L.; Meibohm, A.; et al. Treatment with indinavir, zidovudine, and lamivudine in adults with human immunodeficiency virus infection and prior antiretroviral therapy. N. Engl. J. Med. 1997, 337, 734–739. [Google Scholar] [CrossRef] [PubMed]
- Gulick, R.M.; Ribaudo, H.J.; Shikuma, C.M.; Lustgarten, S.; Squires, K.E.; Meyer, W.A., III; Acosta, E.P.; Schackman, B.R.; Pilcher, C.D.; Murphy, R.L.; et al. Triple-nucleoside regimens versus efavirenz-containing regimens for the initial treatment of HIV-1 infection. N. Engl. J. Med. 2004, 350, 1850–1861. [Google Scholar] [CrossRef] [PubMed]
- Guidelines for the Use of Antiretroviral Agents in HIV-1-Infected Adults and Adolescents. Available online: www.aidsinfo.nih.gov/guidelines (accessed on 11 September 2017).
- HCV Guidance: Recommendations for Testing, Managing, and Treating Hepatitis C. Available online: http://www.hcvguidelines.org/sites/default/files/full-guidance-pdf/HCVGuidance_April_12_2017_b.pdf (accessed on 11 September 2017).
- Quenelle, D.C.; Prichard, M.N.; Keith, K.A.; Hruby, D.E.; Jordan, R.; Painter, G.R.; Robertson, A.; Kern, E.R. Synergistic efficacy of the combination of ST-246 with CMX001 against orthopoxviruses. Antimicrob. Agents Chemother. 2007, 51, 4118–4124. [Google Scholar] [CrossRef] [PubMed]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Foster, S.A.; Parker, S.; Lanier, R. The Role of Brincidofovir in Preparation for a Potential Smallpox Outbreak. Viruses 2017, 9, 320. https://doi.org/10.3390/v9110320
Foster SA, Parker S, Lanier R. The Role of Brincidofovir in Preparation for a Potential Smallpox Outbreak. Viruses. 2017; 9(11):320. https://doi.org/10.3390/v9110320
Chicago/Turabian StyleFoster, Scott A., Scott Parker, and Randall Lanier. 2017. "The Role of Brincidofovir in Preparation for a Potential Smallpox Outbreak" Viruses 9, no. 11: 320. https://doi.org/10.3390/v9110320
APA StyleFoster, S. A., Parker, S., & Lanier, R. (2017). The Role of Brincidofovir in Preparation for a Potential Smallpox Outbreak. Viruses, 9(11), 320. https://doi.org/10.3390/v9110320