
A Role for the Host DNA Damage Response in Hepatitis B Virus cccDNA Formation—and Beyond?

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
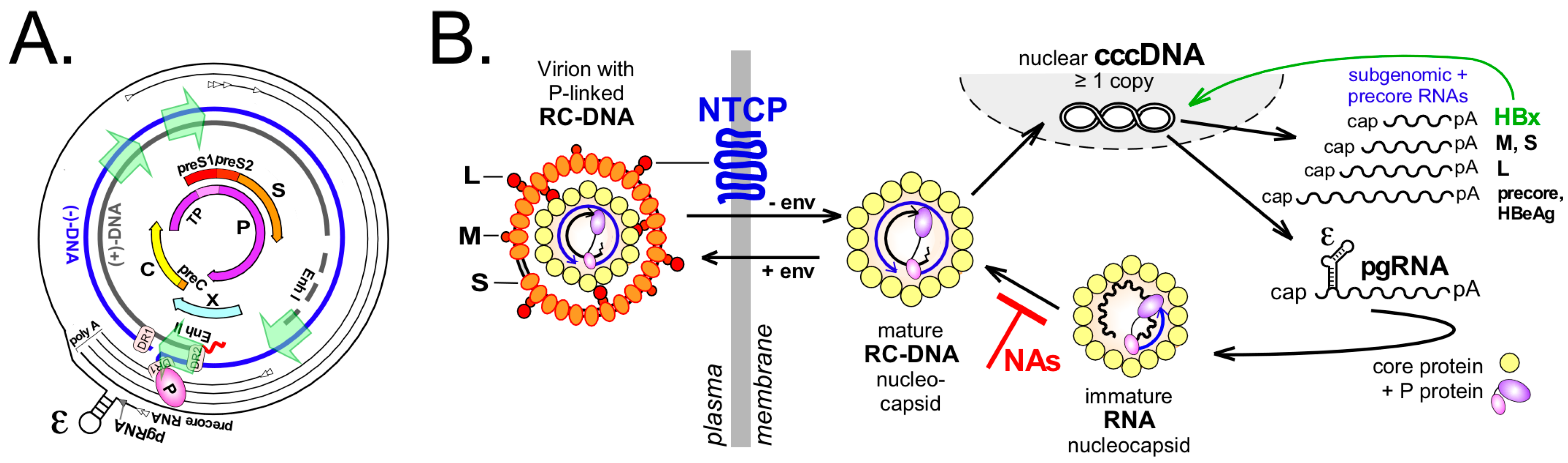
2. The Central Role of cccDNA in HBV Replication
3. The Actual Transcription Template: Poorly Understood HBV Minichromosome
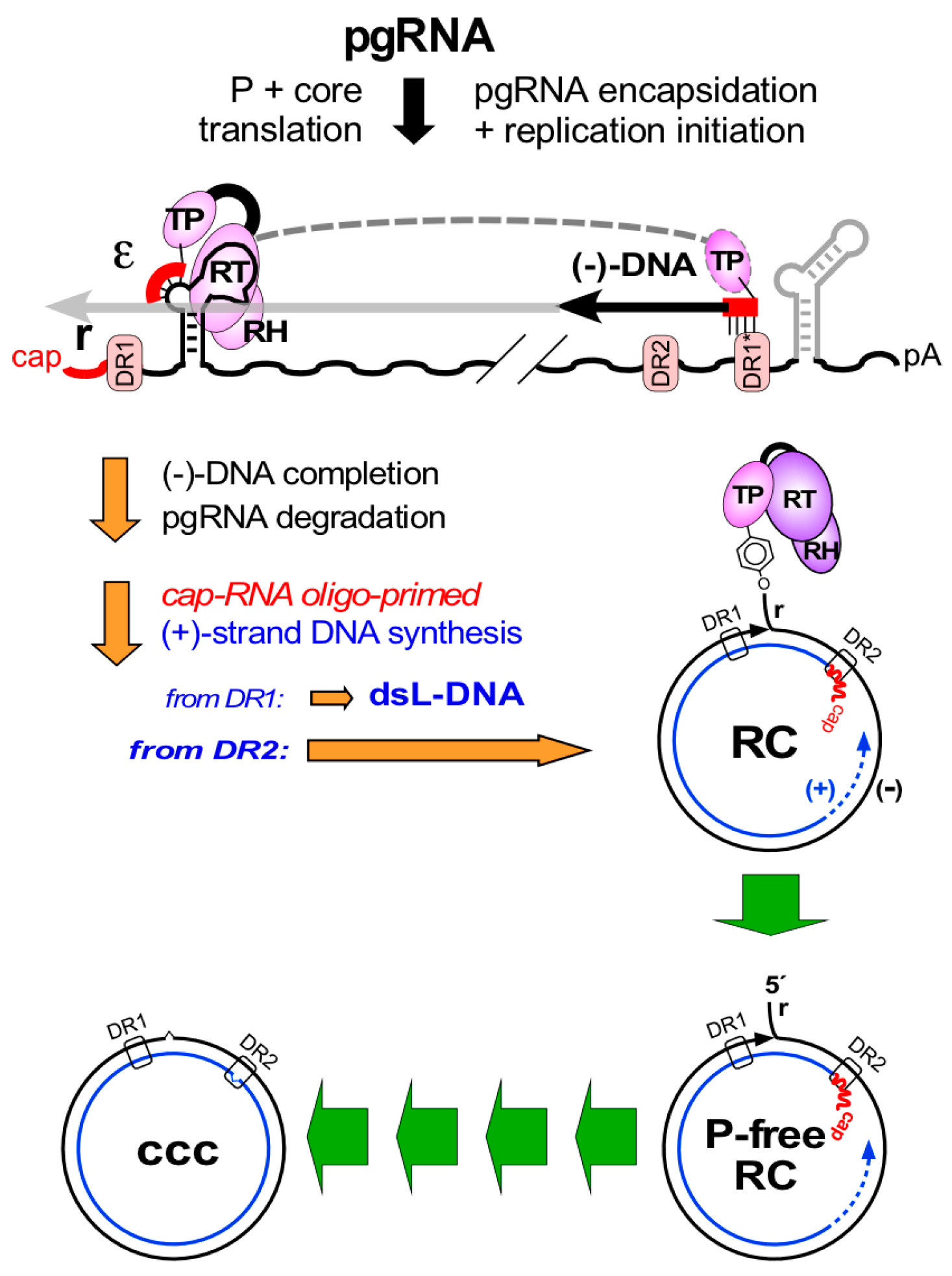
4. From P Protein-Linked RC-DNA to cccDNA in Multiple Steps—A Conceptual Overview
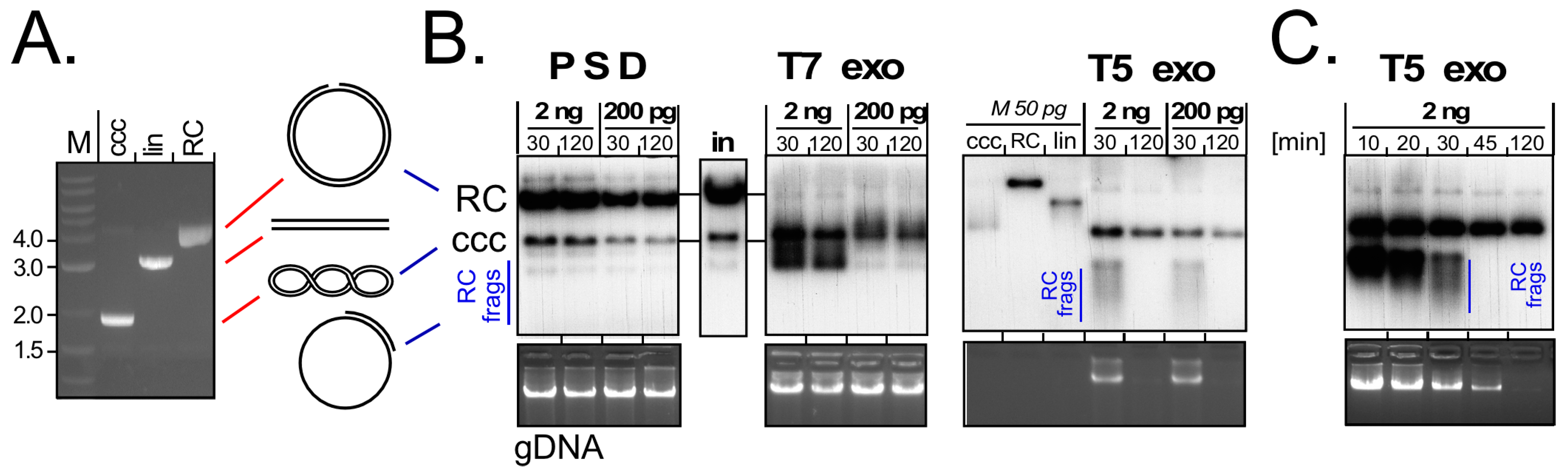
5. Human HBV cccDNA—Low Production Versus Difficult Specific Detection
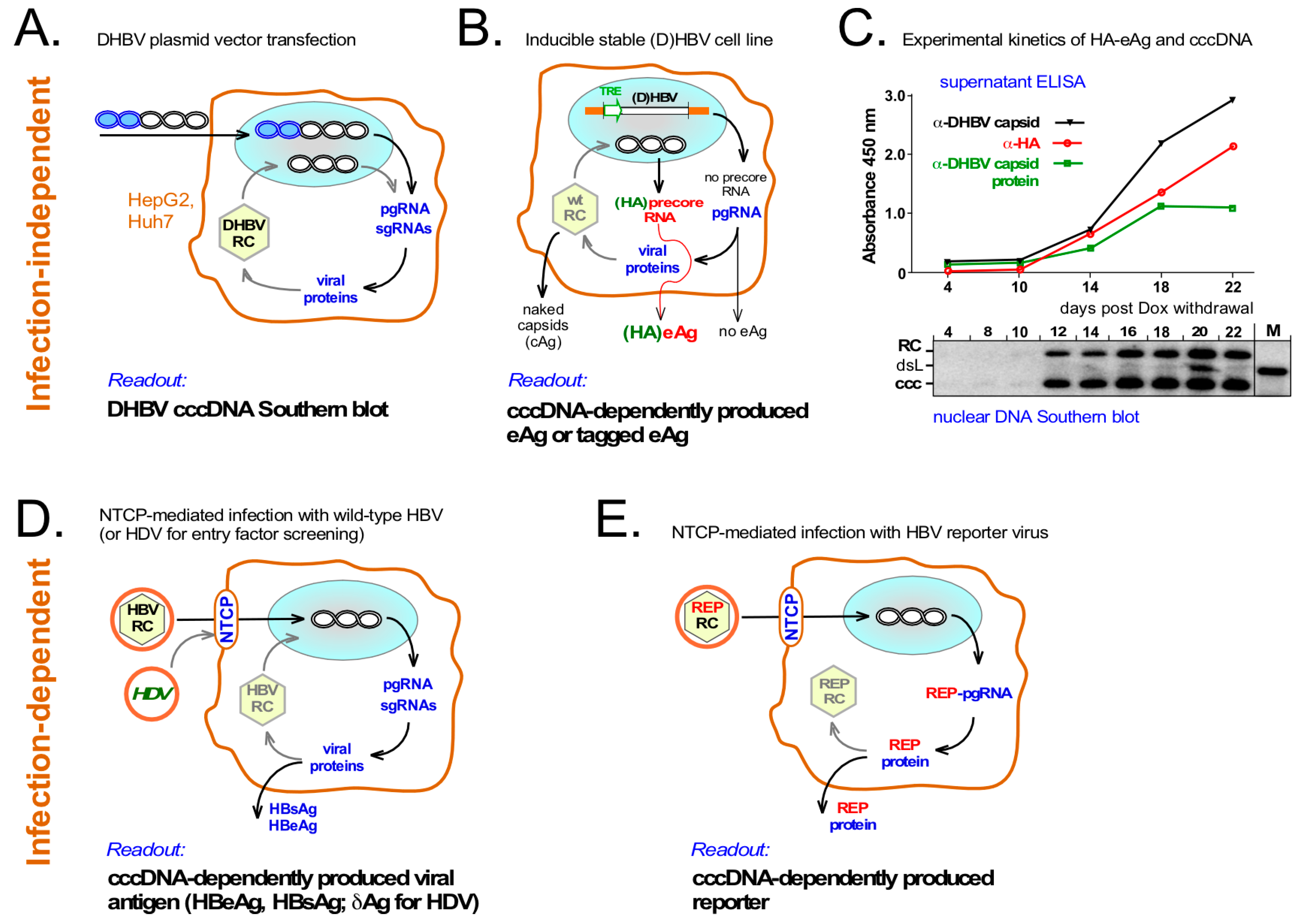
6. Surrogate Models for cccDNA Monitoring
6.1. Infection-Independent cccDNA Model Systems
6.2. Infection-Dependent Systems
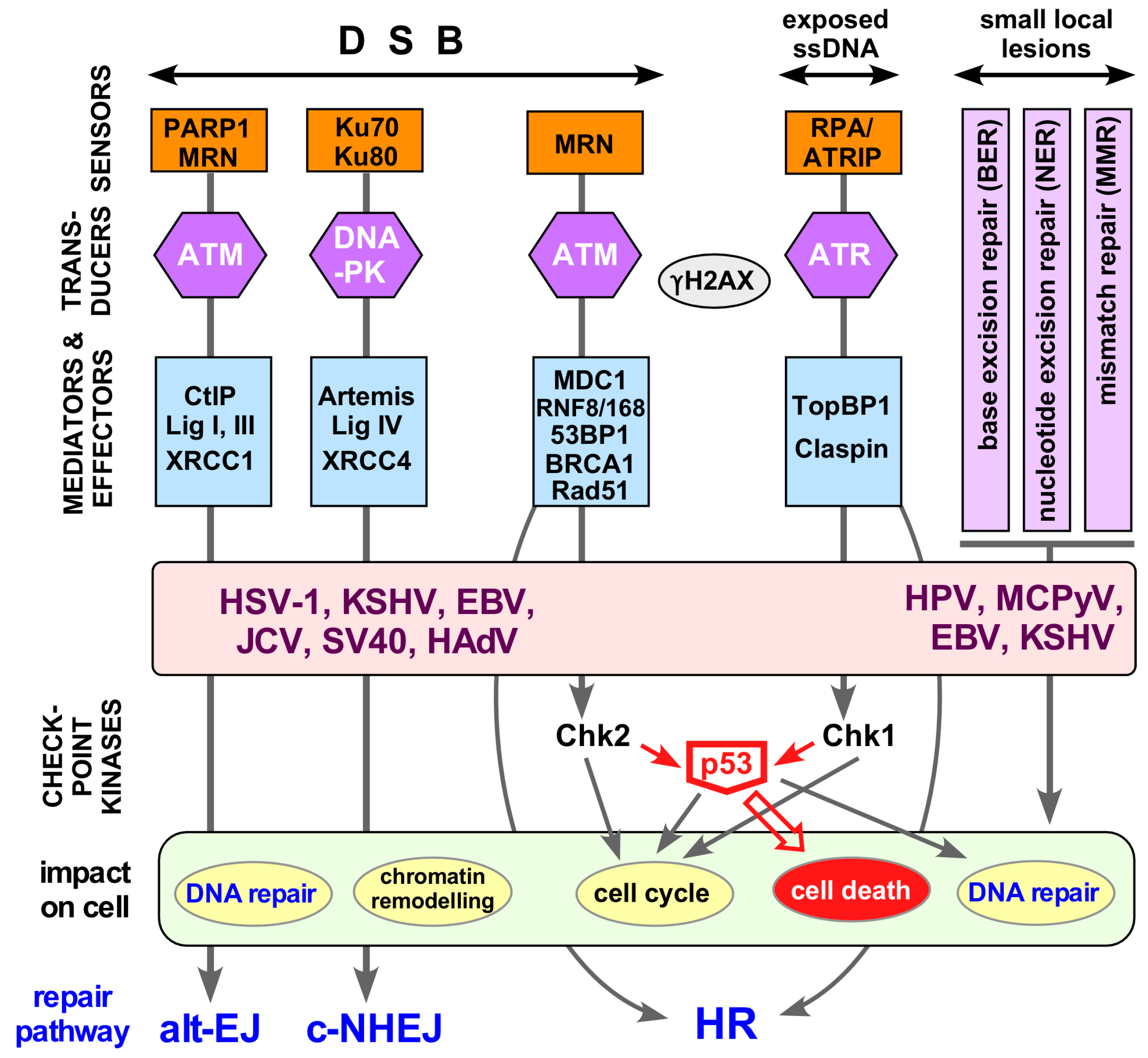
7. Evidence for a Connection between HBV and the Host DNA Damage Response
7.1. The Host DDR—A Simplified Overview
7.2. Other Viruses and the DDR
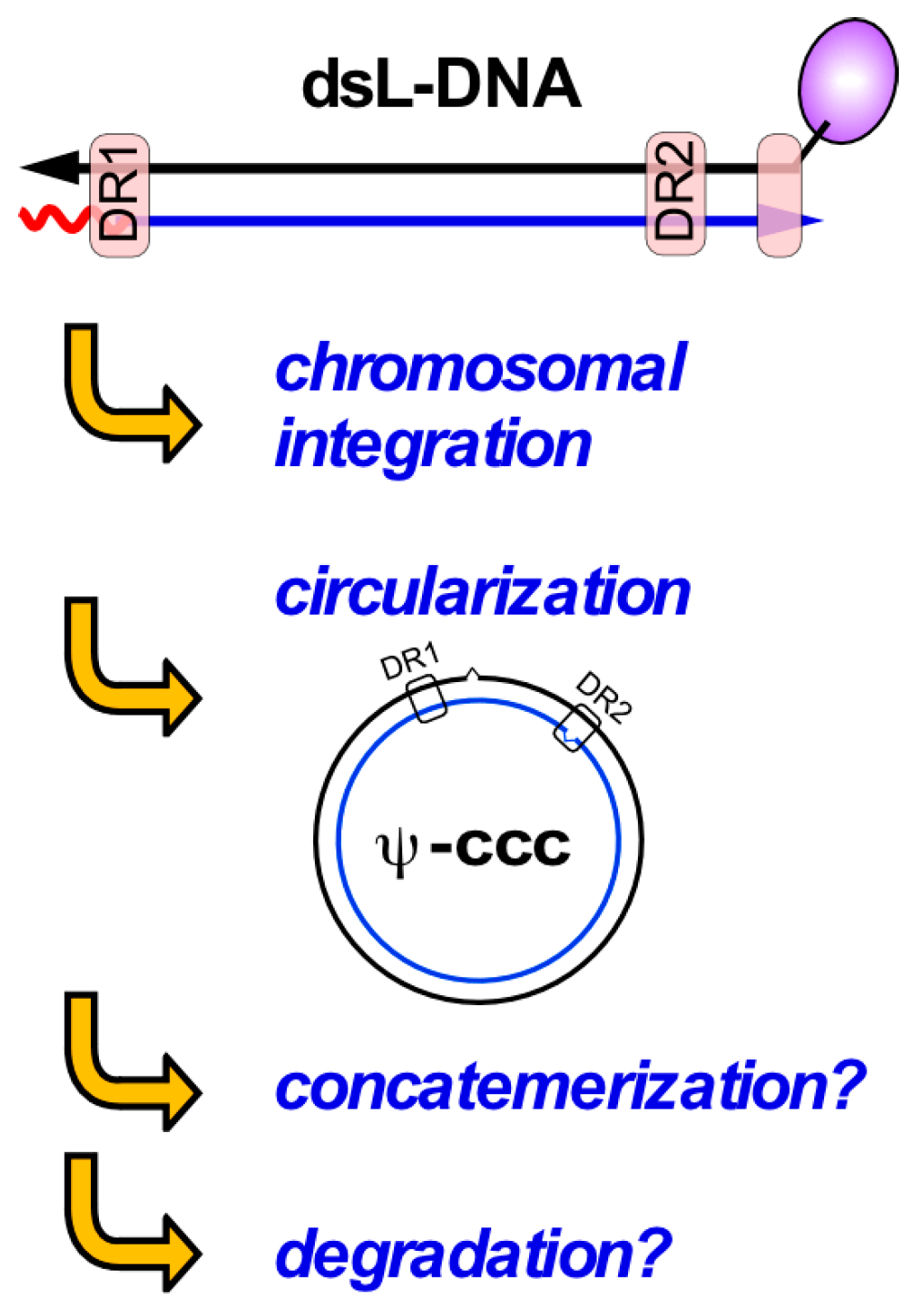
7.3. Crosstalk between HBV and DNA Repair—Integration and Viral dsL-DNA Circularization
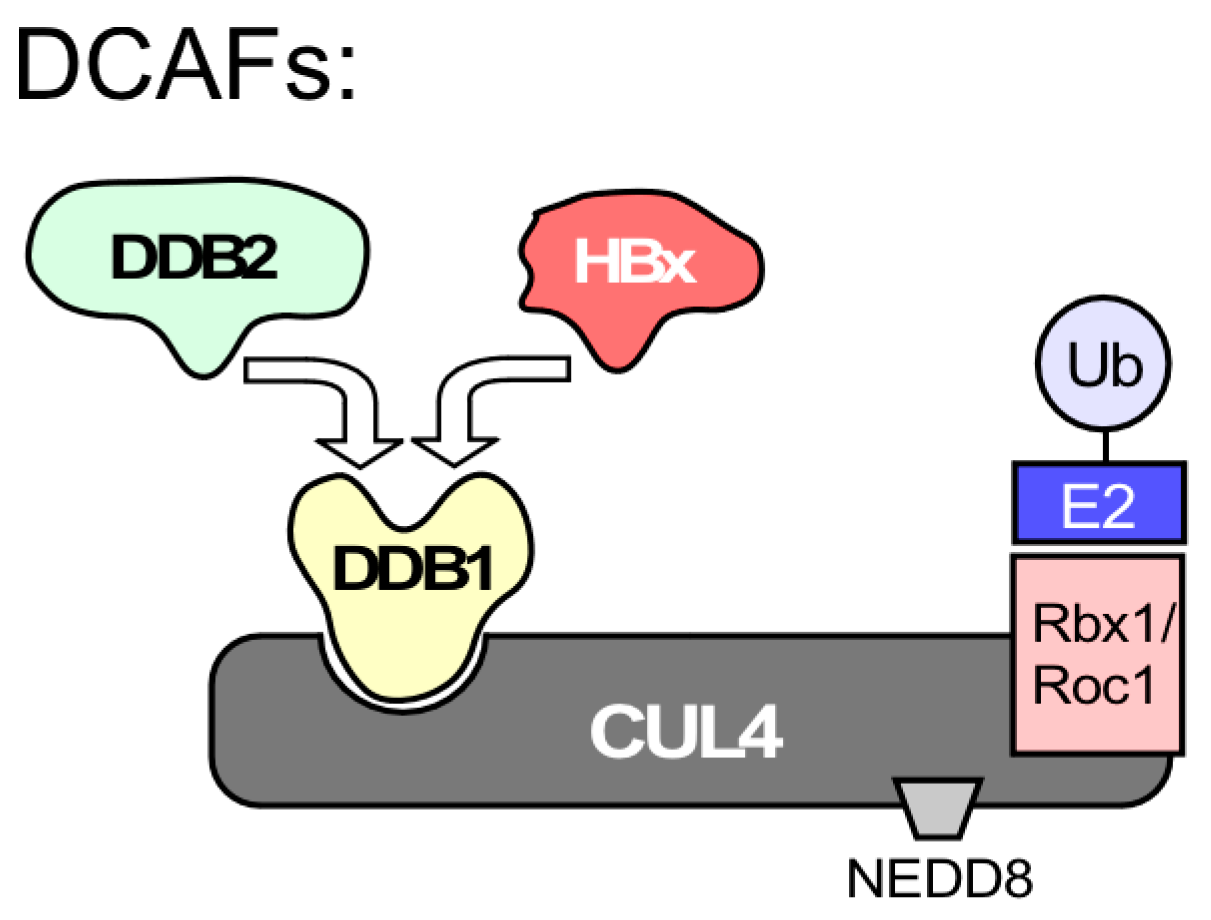
7.4. Does HBx Connect HBV to the Host DDR?
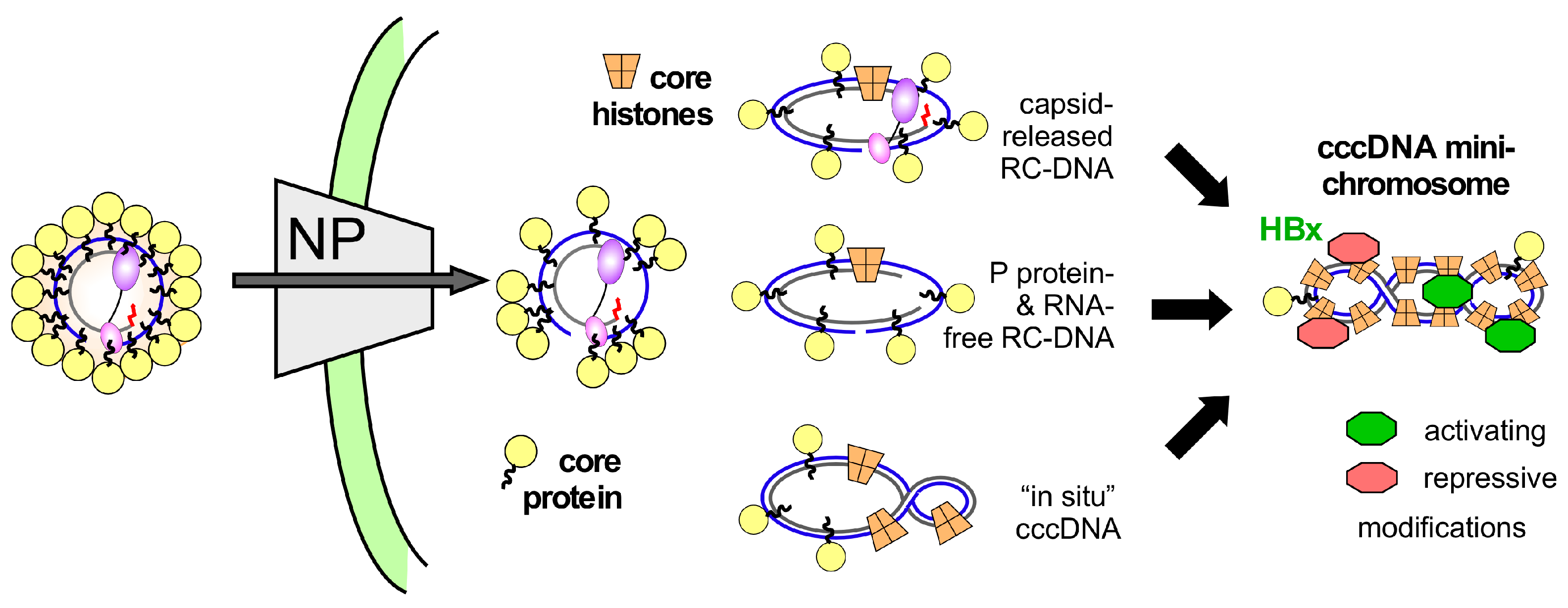
7.5. HBV RC-DNA to cccDNA Conversion—A Direct Case for Host DNA Repair Dependency
8. Conclusions and Open Questions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Trepo, C.; Chan, H.L.; Lok, A. Hepatitis B virus infection. Lancet 2014, 384, 2053–2063. [Google Scholar] [CrossRef]
- Stanaway, J.D.; Flaxman, A.D.; Naghavi, M.; Fitzmaurice, C.; Vos, T.; Abubakar, I.; Abu-Raddad, L.J.; Assadi, R.; Bhala, N.; Cowie, B.; et al. The global burden of viral hepatitis from 1990 to 2013: Findings from the Global Burden of Disease Study 2013. Lancet 2016, 388, 1081–1088. [Google Scholar] [CrossRef]
- WHO. Fact sheet 297 Cancer 2017. February 2017. [Google Scholar]
- Götte, M.; Feld, J.J. Direct-acting antiviral agents for hepatitis C: Structural and mechanistic insights. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 338–351. [Google Scholar] [CrossRef] [PubMed]
- Moradpour, D.; Grakoui, A.; Manns, M.P. Future landscape of hepatitis C research—Basic, translational and clinical perspectives. J. Hepatol. 2016, 65, S143–S155. [Google Scholar] [CrossRef] [PubMed]
- Beck, J.; Nassal, M. Hepatitis B virus replication. World J. Gastroenterol. 2007, 13, 48–64. [Google Scholar] [CrossRef]
- Nassal, M. Hepatitis B viruses: Reverse transcription a different way. Virus Res. 2008, 134, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Seeger, C.; Mason, W.S. Molecular biology of hepatitis B virus infection. Virology 2015, 479–480, 672–686. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, P.M. Epigenetics and Genetics of Viral Latency. Cell. Host Microbe 2016, 19, 619–628. [Google Scholar] [CrossRef] [PubMed]
- Tu, T.; Budzinska, M.A.; Shackel, N.A.; Urban, S. HBV DNA Integration: Molecular mechanisms and clinical implications. Viruses 2017, 9, 75. [Google Scholar] [CrossRef] [PubMed]
- Isorce, N.; Lucifora, J.; Zoulim, F.; Durantel, D. Immune-modulators to combat hepatitis B virus infection: From IFN-alpha to novel investigational immunotherapeutic strategies. Antiviral Res. 2015, 122, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Gish, R.; Jia, J.D.; Locarnini, S.; Zoulim, F. Selection of chronic hepatitis B therapy with high barrier to resistance. Lancet Infect. Dis. 2012, 12, 341–353. [Google Scholar] [CrossRef]
- Rehermann, B.; Ferrari, C.; Pasquinelli, C.; Chisari, F.V. The hepatitis B virus persists for decades after patients’ recovery from acute viral hepatitis despite active maintenance of a cytotoxic T-lymphocyte response. Nat. Med. 1996, 2, 1104–1108. [Google Scholar] [CrossRef] [PubMed]
- Seto, W.K.; Chan, T.S.; Hwang, Y.Y.; Wong, D.K.; Fung, J.; Liu, K.S.; Gill, H.; Lam, Y.F.; Lie, A.K.; Lai, C.L.; et al. Hepatitis B reactivation in patients with previous hepatitis B virus exposure undergoing rituximab-containing chemotherapy for lymphoma: A prospective study. J. Clin. Oncol. 2014, 32, 3736–3743. [Google Scholar] [CrossRef] [PubMed]
- Ni, Y.; Lempp, F.A.; Mehrle, S.; Nkongolo, S.; Kaufman, C.; Falth, M.; Stindt, J.; Königer, C.; Nassal, M.; Kubitz, R.; et al. Hepatitis B and D viruses exploit sodium taurocholate co-transporting polypeptide for species-specific entry into hepatocytes. Gastroenterology 2014, 146, 1070–1083. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Zhong, G.; Xu, G.; He, W.; Jing, Z.; Gao, Z.; Huang, Y.; Qi, Y.; Peng, B.; Wang, H.; et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. eLife 2012, 1, e00049. [Google Scholar] [CrossRef] [PubMed]
- Verrier, E.R.; Colpitts, C.C.; Bach, C.; Heydmann, L.; Weiss, A.; Renaud, M.; Durand, S.C.; Habersetzer, F.; Durantel, D.; Abou-Jaoude, G.; et al. A targeted functional RNA interference screen uncovers glypican 5 as an entry factor for hepatitis B and D viruses. Hepatology 2016, 63, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Gallucci, L.; Kann, M. Nuclear Import of Hepatitis B Virus Capsids and Genome. Viruses 2017, 9, 21. [Google Scholar] [CrossRef] [PubMed]
- Nassal, M. HBV cccDNA: Viral persistence reservoir and key obstacle for a cure of chronic hepatitis B. Gut 2015, 64, 1972–1984. [Google Scholar] [CrossRef] [PubMed]
- Chevaliez, S.; Hezode, C.; Bahrami, S.; Grare, M.; Pawlotsky, J.M. Long-term hepatitis B surface antigen (HBsAg) kinetics during nucleoside/nucleotide analogue therapy: Finite treatment duration unlikely. J. Hepatol. 2013, 58, 676–683. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.L.; Wong, D.; Ip, P.; Kopaniszen, M.; Seto, W.K.; Fung, J.; Huang, F.Y.; Lee, B.; Cullaro, G.; Chong, C.K.; et al. Reduction of covalently closed circular DNA with long-term nucleos(t)ide analogue treatment in chronic hepatitis B. J. Hepatol. 2017, 66, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Bock, C.T.; Schranz, P.; Schroder, C.H.; Zentgraf, H. Hepatitis B virus genome is organized into nucleosomes in the nucleus of the infected cell. Virus Genes 1994, 8, 215–229. [Google Scholar] [CrossRef] [PubMed]
- Bock, C.T.; Schwinn, S.; Locarnini, S.; Fyfe, J.; Manns, M.P.; Trautwein, C.; Zentgraf, H. Structural organization of the hepatitis B virus minichromosome. J. Mol. Biol. 2001, 307, 183–196. [Google Scholar] [CrossRef] [PubMed]
- Newbold, J.E.; Xin, H.; Tencza, M.; Sherman, G.; Dean, J.; Bowden, S.; Locarnini, S. The covalently closed duplex form of the hepadnavirus genome exists in situ as a heterogeneous population of viral minichromosomes. J. Virol. 1995, 69, 3350–3357. [Google Scholar] [PubMed]
- Lucifora, J.; Xia, Y.; Reisinger, F.; Zhang, K.; Stadler, D.; Cheng, X.; Sprinzl, M.F.; Koppensteiner, H.; Makowska, Z.; Volz, T.; et al. Specific and nonhepatotoxic degradation of nuclear hepatitis B virus cccDNA. Science 2014, 343, 1221–1228. [Google Scholar] [CrossRef] [PubMed]
- Belloni, L.; Pollicino, T.; De Nicola, F.; Guerrieri, F.; Raffa, G.; Fanciulli, M.; Raimondo, G.; Levrero, M. Nuclear HBx binds the HBV minichromosome and modifies the epigenetic regulation of cccDNA function. Proc. Natl. Acad. Sci. USA 2009, 106, 19975–19979. [Google Scholar] [CrossRef] [PubMed]
- Lucifora, J.; Arzberger, S.; Durantel, D.; Belloni, L.; Strubin, M.; Levrero, M.; Zoulim, F.; Hantz, O.; Protzer, U. Hepatitis B virus X protein is essential to initiate and maintain virus replication after infection. J. Hepatol. 2011, 55, 996–1003. [Google Scholar] [CrossRef] [PubMed]
- Minor, M.M.; Slagle, B.L. Hepatitis B virus HBx protein interactions with the ubiquitin proteasome system. Viruses 2014, 6, 4683–4702. [Google Scholar] [CrossRef]
- Slagle, B.L.; Bouchard, M.J. Hepatitis B Virus X and Regulation of Viral Gene Expression. Cold Spring Harb Perspect. Med. 2016, 6, a021402. [Google Scholar] [CrossRef] [PubMed]
- Tsuge, M.; Hiraga, N.; Akiyama, R.; Tanaka, S.; Matsushita, M.; Mitsui, F.; Abe, H.; Kitamura, S.; Hatakeyama, T.; Kimura, T.; et al. HBx protein is indispensable for development of viraemia in human hepatocyte chimeric mice. J. Gen. Virol. 2010, 91, 1854–1864. [Google Scholar] [CrossRef]
- Decorsiere, A.; Mueller, H.; van Breugel, P.C.; Abdul, F.; Gerossier, L.; Beran, R.K.; Livingston, C.M.; Niu, C.; Fletcher, S.P.; Hantz, O.; et al. Hepatitis B virus X protein identifies the Smc5/6 complex as a host restriction factor. Nature 2016, 531, 386–389. [Google Scholar] [CrossRef] [PubMed]
- Murphy, C.M.; Xu, Y.; Li, F.; Nio, K.; Reszka-Blanco, N.; Li, X.; Wu, Y.; Yu, Y.; Xiong, Y.; Su, L. Hepatitis B Virus X Protein Promotes Degradation of SMC5/6 to Enhance HBV Replication. Cell. Rep. 2016, 16, 2846–2854. [Google Scholar] [CrossRef] [PubMed]
- Niu, C.; Livingston, C.M.; Li, L.; Beran, R.K.; Daffis, S.; Ramakrishnan, D.; Burdette, D.; Peiser, L.; Salas, E.; Ramos, H.; et al. The Smc5/6 Complex Restricts HBV when Localized to ND10 without Inducing an Innate Immune Response and Is Counteracted by the HBV X Protein Shortly after Infection. PLoS ONE 2017, 12, e0169648. [Google Scholar] [CrossRef] [PubMed]
- Livingston, C.M.; Ramakrishnan, D.; Strubin, M.; Fletcher, S.P.; Beran, R.K. Identifying and Characterizing Interplay between Hepatitis B Virus X Protein and Smc5/6. Viruses 2017, 9, 69. [Google Scholar] [CrossRef] [PubMed]
- Saper, G.; Kler, S.; Asor, R.; Oppenheim, A.; Raviv, U.; Harries, D. Effect of capsid confinement on the chromatin organization of the SV40 minichromosome. Nucleic Acids Res. 2013, 41, 1569–1580. [Google Scholar] [CrossRef]
- Köck, J.; Rösler, C.; Zhang, J.J.; Blum, H.E.; Nassal, M.; Thoma, C. Generation of covalently closed circular DNA of hepatitis B viruses via intracellular recycling is regulated in a virus specific manner. PLoS Pathog. 2010, 6, e1001082. [Google Scholar] [CrossRef] [PubMed]
- Birnbaum, F.; Nassal, M. Hepatitis B virus nucleocapsid assembly: Primary structure requirements in the core protein. J. Virol. 1990, 64, 3319–3330. [Google Scholar]
- Nassal, M. The arginine-rich domain of the hepatitis B virus core protein is required for pregenome encapsidation and productive viral positive-strand DNA synthesis but not for virus assembly. J. Virol. 1992, 66, 4107–4116. [Google Scholar]
- Zlotnick, A.; Venkatakrishnan, B.; Tan, Z.; Lewellyn, E.; Turner, W.; Francis, S. Core protein: A pleiotropic keystone in the HBV lifecycle. Antiviral Res. 2015, 121, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Protzer, U. Hepatitis: Epigenetic control of HBV by HBx protein--releasing the break? Nat. Rev. Gastroenterol Hepatol. 2015, 12, 558–559. [Google Scholar] [CrossRef]
- Rothbart, S.B.; Strahl, B.D. Interpreting the language of histone and DNA modifications. Biochim. Biophys. Acta 2014, 1839, 627–643. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Cooper, S.; Brockdorff, N. The interplay of histone modifications—Writers that read. EMBO Rep. 2015, 16, 1467–1481. [Google Scholar] [CrossRef] [PubMed]
- Tropberger, P.; Mercier, A.; Robinson, M.; Zhong, W.; Ganem, D.E.; Holdorf, M. Mapping of histone modifications in episomal HBV cccDNA uncovers an unusual chromatin organization amenable to epigenetic manipulation. Proc. Natl. Acad. Sci. USA 2015, 112, E5715–E5724. [Google Scholar] [CrossRef] [PubMed]
- Porterfield, J.Z.; Dhason, M.S.; Loeb, D.D.; Nassal, M.; Stray, S.J.; Zlotnick, A. Full-length hepatitis B virus core protein packages viral and heterologous RNA with similarly high levels of cooperativity. J. Virol. 2010, 84, 7174–7184. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Kang, W.; Lei, X.; Li, Y.; Xiang, A.; Liu, Y.; Zhao, J.; Zhang, J.; Yan, Z. Hepatitis B viral core protein disrupts human host gene expression by binding to promoter regions. BMC Genom. 2012, 13, 563. [Google Scholar] [CrossRef]
- Qi, Y.; Gao, Z.; Xu, G.; Peng, B.; Liu, C.; Yan, H.; Yao, Q.; Sun, G.; Liu, Y.; Tang, D.; et al. DNA Polymerase kappa is a key cellular factor for the formation of covalently closed circular DNA of Hepatitis B Virus. PLoS Pathog. 2016, 12, e1005893. [Google Scholar] [CrossRef]
- Wang, G.Z.; Wang, Y.; Goff, S.P. Histones are rapidly loaded onto unintegrated retroviral DNAs soon after nuclear entry. Cell. Host Microbe 2016, 20, 798–809. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.S.; Kaneko, S.; Girones, R.; Anderson, R.W.; Hornbuckle, W.E.; Tennant, B.C.; Cote, P.J.; Gerin, J.L.; Purcell, R.H.; Miller, R.H. The woodchuck hepatitis virus X gene is important for establishment of virus infection in woodchucks. J. Virol. 1993, 67, 1218–1226. [Google Scholar]
- Königer, C.; Wingert, I.; Marsmann, M.; Rösler, C.; Beck, J.; Nassal, M. Involvement of the host DNA-repair enzyme TDP2 in formation of the covalently closed circular DNA persistence reservoir of hepatitis B viruses. Proc. Natl. Acad. Sci. USA 2014, 111, E4244–E4253. [Google Scholar] [CrossRef]
- Köck, J.; Schlicht, H.J. Analysis of the earliest steps of hepadnavirus replication: Genome repair after infectious entry into hepatocytes does not depend on viral polymerase activity. J. Virol. 1993, 67, 4867–4874. [Google Scholar] [PubMed]
- Köck, J.; Baumert, T.F.; Delaney, W.E.T.; Blum, H.E.; von Weizsacker, F. Inhibitory effect of adefovir and lamivudine on the initiation of hepatitis B virus infection in primary tupaia hepatocytes. Hepatology 2003, 38, 1410–1418. [Google Scholar] [PubMed]
- Delmas, J.; Schorr, O.; Jamard, C.; Gibbs, C.; Trepo, C.; Hantz, O.; Zoulim, F. Inhibitory effect of adefovir on viral DNA synthesis and covalently closed circular DNA formation in duck hepatitis B virus-infected hepatocytes in vivo and in vitro. Antimicrob. Agents Chemother. 2002, 46, 425–433. [Google Scholar] [CrossRef] [PubMed]
- Moraleda, G.; Saputelli, J.; Aldrich, C.E.; Averett, D.; Condreay, L.; Mason, W.S. Lack of effect of antiviral therapy in nondividing hepatocyte cultures on the closed circular DNA of woodchuck hepatitis virus. J. Virol. 1997, 71, 9392–9399. [Google Scholar] [PubMed]
- Hantz, O.; Parent, R.; Durantel, D.; Gripon, P.; Guguen-Guillouzo, C.; Zoulim, F. Persistence of the hepatitis B virus covalently closed circular DNA in HepaRG human hepatocyte-like cells. J. Gen. Virol. 2009, 90, 127–135. [Google Scholar] [CrossRef]
- Xia, Y.; Carpentier, A.; Cheng, X.; Block, P.D.; Zhao, Y.; Zhang, Z.; Protzer, U.; Liang, T.J. Human stem cell-derived hepatocytes as a model for hepatitis B virus infection, spreading and virus-host interactions. J. Hepatol. 2017, 66, 494–503. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y.; Sun, Y.; Huang, S.N.; Nitiss, J.L. Roles of eukaryotic topoisomerases in transcription, replication and genomic stability. Nat. Rev. Mol. Cell. Biol. 2016, 17, 703–721. [Google Scholar] [CrossRef]
- Zhang, Y.Y.; Zhang, B.H.; Theele, D.; Litwin, S.; Toll, E.; Summers, J. Single-cell analysis of covalently closed circular DNA copy numbers in a hepadnavirus-infected liver. Proc. Natl. Acad. Sci. USA 2003, 100, 12372–12377. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Yamamoto, T.; Cullen, J.; Saputelli, J.; Aldrich, C.E.; Miller, D.S.; Litwin, S.; Furman, P.A.; Jilbert, A.R.; Mason, W.S. Kinetics of hepadnavirus loss from the liver during inhibition of viral DNA synthesis. J. Virol. 2001, 75, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Summers, J.; Smith, P.M.; Horwich, A.L. Hepadnavirus envelope proteins regulate covalently closed circular DNA amplification. J. Virol. 1990, 64, 2819–2824. [Google Scholar]
- Summers, J.; Smith, P.M.; Huang, M.J.; Yu, M.S. Morphogenetic and regulatory effects of mutations in the envelope proteins of an avian hepadnavirus. J. Virol. 1991, 65, 1310–1317. [Google Scholar] [PubMed]
- Tuttleman, J.S.; Pourcel, C.; Summers, J. Formation of the pool of covalently closed circular viral DNA in hepadnavirus-infected cells. Cell 1986, 47, 451–460. [Google Scholar] [CrossRef]
- Werle-Lapostolle, B.; Bowden, S.; Locarnini, S.; Wursthorn, K.; Petersen, J.; Lau, G.; Trepo, C.; Marcellin, P.; Goodman, Z.; Delaney, W.E.T.; et al. Persistence of cccDNA during the natural history of chronic hepatitis B and decline during adefovir dipivoxil therapy. Gastroenterology 2004, 126, 1750–1758. [Google Scholar] [CrossRef]
- Volz, T.; Allweiss, L.; Ben, M.M.; Warlich, M.; Lohse, A.W.; Pollok, J.M.; Alexandrov, A.; Urban, S.; Petersen, J.; Lutgehetmann, M.; et al. The entry inhibitor Myrcludex-B efficiently blocks intrahepatic virus spreading in humanized mice previously infected with hepatitis B virus. J. Hepatol. 2013, 58, 861–867. [Google Scholar] [CrossRef]
- Sun, D.; Nassal, M. Stable HepG2- and Huh7-based human hepatoma cell lines for efficient regulated expression of infectious hepatitis B virus. J. Hepatol. 2006, 45, 636–645. [Google Scholar] [CrossRef] [PubMed]
- Lentz, T.B.; Loeb, D.D. Roles of the envelope proteins in the amplification of covalently closed circular DNA and completion of synthesis of the plus-strand DNA in hepatitis B virus. J. Virol. 2011, 85, 11916–11927. [Google Scholar] [CrossRef] [PubMed]
- Iannacone, M.; Guidotti, L.G. Mouse models of hepatitis B virus pathogenesis. Cold Spring Harb. Perspect. Med. 2015, 5, 5. [Google Scholar] [CrossRef]
- Cai, D.; Nie, H.; Yan, R.; Guo, J.T.; Block, T.M.; Guo, H. A southern blot assay for detection of hepatitis B virus covalently closed circular DNA from cell cultures. Methods Mol. Biol. 2013, 1030, 151–161. [Google Scholar]
- Chisari, F.V.; Mason, W.S.; Seeger, C. Virology. Comment on “Specific and nonhepatotoxic degradation of nuclear hepatitis B virus cccDNA”. Science 2014, 344, 1237. [Google Scholar] [CrossRef]
- Xia, Y.; Lucifora, J.; Reisinger, F.; Heikenwalder, M.; Protzer, U. Virology. Response to Comment on “Specific and nonhepatotoxic degradation of nuclear hepatitis B virus cccDNA”. Science 2014, 344, 1237. [Google Scholar] [CrossRef] [PubMed]
- Allweiss, L.; Volz, T.; Lutgehetmann, M.; Giersch, K.; Bornscheuer, T.; Lohse, A.W.; Petersen, J.; Ma, H.; Klumpp, K.; Fletcher, S.P.; et al. Immune cell responses are not required to induce substantial hepatitis B virus antigen decline during pegylated interferon-alpha administration. J. Hepatol. 2014, 60, 500–507. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Lu, W.; Zheng, Y.; Wang, W.; Bai, L.; Chen, L.; Feng, Y.; Zhang, Z.; Yuan, Z. In situ analysis of intrahepatic virological events in chronic hepatitis B virus infection. J. Clin. Investig. 2016, 126, 1079–1092. [Google Scholar] [CrossRef]
- Xia, Y.; Stadler, D.; Ko, C.; Protzer, U. Analyses of HBV cccDNA Quantification and Modification. Methods Mol. Biol. 2017, 1540, 59–72. [Google Scholar]
- Sayers, J.R.; Eckstein, F. A single-strand specific endonuclease activity copurifies with overexpressed T5 D15 exonuclease. Nucleic Acids Res. 1991, 19, 4127–4132. [Google Scholar] [CrossRef]
- Mu, D.; Yan, L.; Tang, H.; Liao, Y. A sensitive and accurate quantification method for the detection of hepatitis B virus covalently closed circular DNA by the application of a droplet digital polymerase chain reaction amplification system. Biotechnol. Lett. 2015, 37, 2063–2073. [Google Scholar] [CrossRef]
- Suspene, R.; Thiers, V.; Vartanian, J.P.; Wain-Hobson, S. PCR mediated recombination impacts the analysis of hepatitis B Virus covalently closed circular DNA. Retrovirology 2016, 13, 84. [Google Scholar] [CrossRef]
- Cui, X.; McAllister, R.; Boregowda, R.; Sohn, J.A.; Cortes Ledesma, F.; Caldecott, K.W.; Seeger, C.; Hu, J. Does tyrosyl DNA phosphodiesterase-2 play a role in hepatitis B virus genome repair? PLoS ONE 2015, 10, e0128401. [Google Scholar] [CrossRef] [PubMed]
- Ladner, S.K.; Otto, M.J.; Barker, C.S.; Zaifert, K.; Wang, G.H.; Guo, J.T.; Seeger, C.; King, R.W. Inducible expression of human hepatitis B virus (HBV) in stably transfected hepatoblastoma cells: A novel system for screening potential inhibitors of HBV replication. Antimicrob Agents Chemother 1997, 41, 1715–1720. [Google Scholar]
- Cai, D.; Mills, C.; Yu, W.; Yan, R.; Aldrich, C.E.; Saputelli, J.R.; Mason, W.S.; Xu, X.; Guo, J.T.; Block, T.M.; et al. Identification of disubstituted sulfonamide compounds as specific inhibitors of hepatitis B virus covalently closed circular DNA formation. Antimicrob. Agents Chemother. 2012, 56, 4277–4288. [Google Scholar] [CrossRef] [PubMed]
- Döring, T.; Prange, R. Rab33B and its autophagic Atg5/12/16L1 effector assist in hepatitis B virus naked capsid formation and release. Cell. Microbiol. 2015, 17, 747–764. [Google Scholar] [CrossRef]
- Cai, D.; Wang, X.; Yan, R.; Mao, R.; Liu, Y.; Ji, C.; Cuconati, A.; Guo, H. Establishment of an inducible HBV stable cell line that expresses cccDNA-dependent epitope-tagged HBeAg for screening of cccDNA modulators. Antiviral Res. 2016, 132, 26–37. [Google Scholar] [CrossRef]
- Guo, X.; Chen, P.; Hou, X.; Xu, W.; Wang, D.; Wang, T.Y.; Zhang, L.; Zheng, G.; Gao, Z.L.; He, C.Y.; et al. The recombined cccDNA produced using minicircle technology mimicked HBV genome in structure and function closely. Sci. Rep. 2016, 6, 25552. [Google Scholar] [CrossRef]
- Li, F.; Cheng, L.; Murphy, C.M.; Reszka-Blanco, N.J.; Wu, Y.; Chi, L.; Hu, J.; Su, L. Minicircle HBV cccDNA with a Gaussia luciferase reporter for investigating HBV cccDNA biology and developing cccDNA-targeting drugs. Sci. Rep. 2016, 6, 36483. [Google Scholar] [CrossRef] [PubMed]
- Gripon, P.; Diot, C.; Theze, N.; Fourel, I.; Loreal, O.; Brechot, C.; Guguen-Guillouzo, C. Hepatitis B virus infection of adult human hepatocytes cultured in the presence of dimethyl sulfoxide. J. Virol. 1988, 62, 4136–4143. [Google Scholar] [PubMed]
- Köck, J.; Nassal, M.; MacNelly, S.; Baumert, T.F.; Blum, H.E.; von Weizsäcker, F. Efficient infection of primary tupaia hepatocytes with purified human and woolly monkey hepatitis B virus. J. Virol. 2001, 75, 5084–5089. [Google Scholar] [CrossRef] [PubMed]
- Von Weizsäcker, F.; Köck, J.; MacNelly, S.; Ren, S.; Blum, H.E.; Nassal, M. The tupaia model for the study of hepatitis B virus: Direct infection and HBV genome transduction of primary tupaia hepatocytes. Methods Mol. Med. 2004, 96, 153–161. [Google Scholar] [PubMed]
- Allweiss, L.; Dandri, M. Experimental in vitro and in vivo models for the study of human hepatitis B virus infection. J. Hepatol. 2016, 64, S17–S31. [Google Scholar] [CrossRef] [PubMed]
- Gripon, P.; Rumin, S.; Urban, S.; Le Seyec, J.; Glaise, D.; Cannie, I.; Guyomard, C.; Lucas, J.; Trepo, C.; Guguen-Guillouzo, C. Infection of a human hepatoma cell line by hepatitis B virus. Proc. Natl. Acad. Sci. USA 2002, 99, 15655–15660. [Google Scholar] [CrossRef]
- Zhang, Z.; Torii, N.; Hu, Z.; Jacob, J.; Liang, T.J. X-deficient woodchuck hepatitis virus mutants behave like attenuated viruses and induce protective immunity in vivo. J. Clin. Investig. 2001, 108, 1523–1531. [Google Scholar] [CrossRef]
- Zoulim, F.; Saputelli, J.; Seeger, C. Woodchuck hepatitis virus X protein is required for viral infection in vivo. J. Virol. 1994, 68, 2026–2030. [Google Scholar] [PubMed]
- Marion, M.J.; Hantz, O.; Durantel, D. The HepaRG cell line: Biological properties and relevance as a tool for cell biology, drug metabolism, and virology studies. Methods Mol. Biol. 2010, 640, 261–272. [Google Scholar]
- Li, W.; Urban, S. Entry of hepatitis B and hepatitis D virus into hepatocytes: Basic insights and clinical implications. J. Hepatol. 2016, 64, S32–S40. [Google Scholar] [CrossRef]
- Verrier, E.R.; Colpitts, C.C.; Schuster, C.; Zeisel, M.B.; Baumert, T.F. Cell Culture Models for the Investigation of Hepatitis B and D Virus Infection. Viruses 2016, 8, 261. [Google Scholar] [CrossRef] [PubMed]
- Bartenschlager, R.; Pietschmann, T. Efficient hepatitis C virus cell culture system: What a difference the host cell makes. Proc. Natl. Acad. Sci. USA 2005, 102, 9739–9740. [Google Scholar] [CrossRef] [PubMed]
- Shlomai, A.; Schwartz, R.E.; Ramanan, V.; Bhatta, A.; de Jong, Y.P.; Bhatia, S.N.; Rice, C.M. Modeling host interactions with hepatitis B virus using primary and induced pluripotent stem cell-derived hepatocellular systems. Proc. Natl. Acad. Sci. USA 2014, 111, 12193–12198. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, L.F.; Yu, S.; Wang, X.; Zhang, L.; Yu, J.; Xie, L.; Li, W.; Ali, R.; Qiu, H.J. Applications of Replicating-Competent Reporter-Expressing Viruses in Diagnostic and Molecular Virology. Viruses 2016, 8, 127. [Google Scholar] [CrossRef] [PubMed]
- Protzer, U.; Nassal, M.; Chiang, P.W.; Kirschfink, M.; Schaller, H. Interferon gene transfer by a hepatitis B virus vector efficiently suppresses wild-type virus infection. Proc. Natl. Acad. Sci. USA 1999, 96, 10818–10823. [Google Scholar] [CrossRef]
- Li, B.; Sun, S.; Li, M.; Cheng, X.; Li, H.; Kang, F.; Kang, J.; Dornbrack, K.; Nassal, M.; Sun, D. Suppression of hepatitis B virus antigen production and replication by wild-type HBV dependently replicating HBV shRNA vectors in vitro and in vivo. Antiviral Res. 2016, 134, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wu, L.; Cheng, X.; Liu, S.; Li, B.; Li, H.; Kang, F.; Wang, J.; Xia, H.; Ping, C.; et al. Replication-competent infectious hepatitis B virus vectors carrying substantially sized transgenes by redesigned viral polymerase translation. PLoS ONE 2013, 8, e60306. [Google Scholar] [CrossRef] [PubMed]
- Ryan, E.L.; Hollingworth, R.; Grand, R.J. Activation of the DNA damage response by RNA viruses. Biomolecules 2016, 6, 2. [Google Scholar] [CrossRef] [PubMed]
- Weitzman, M.D.; Lilley, C.E.; Chaurushiya, M.S. Genomes in conflict: Maintaining genome integrity during virus infection. Annu. Rev. Microbiol. 2010, 64, 61–81. [Google Scholar] [CrossRef]
- Weitzman, M.D.; Weitzman, J.B. What’s the damage? The impact of pathogens on pathways that maintain host genome integrity. Cell Host Microbe 2014, 15, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Luftig, M.A. Viruses and the DNA damage response: Activation and antagonism. Annu. Rev. Virol. 2014, 1, 605–625. [Google Scholar] [CrossRef] [PubMed]
- Dempsey, A.; Bowie, A.G. Innate immune recognition of DNA: A recent history. Virology 2015, 479–480, 146–152. [Google Scholar]
- Lindahl, T.; Barnes, D.E. Repair of endogenous DNA damage. Cold Spring Harb. Symp. Quant. Biol. 2000, 65, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Pateras, I.S.; Havaki, S.; Nikitopoulou, X.; Vougas, K.; Townsend, P.A.; Panayiotidis, M.I.; Georgakilas, A.G.; Gorgoulis, V.G. The DNA damage response and immune signaling alliance: Is it good or bad? Nature decides when and where. Pharmacol. Ther. 2015, 154, 36–56. [Google Scholar] [CrossRef] [PubMed]
- Schreiner, S.; Wimmer, P.; Dobner, T. Adenovirus degradation of cellular proteins. Future Microbiol. 2012, 7, 211–225. [Google Scholar] [CrossRef] [PubMed]
- Ceccaldi, R.; Rondinelli, B.; D’Andrea, A.D. Repair pathway choices and consequences at the double-strand break. Trends Cell. Biol. 2016, 26, 52–64. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.J.; Chen, B.P.; Chen, D.J. DNA-PK: A dynamic enzyme in a versatile DSB repair pathway. DNA Repair (Amst) 2014, 17, 21–29. [Google Scholar] [CrossRef]
- Hustedt, N.; Durocher, D. The control of DNA repair by the cell cycle. Nat. Cell. Biol. 2016, 19, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Hollingworth, R.; Grand, R.J. Modulation of DNA damage and repair pathways by human tumour viruses. Viruses 2015, 7, 2542–2591. [Google Scholar] [CrossRef] [PubMed]
- Paull, T.T. Mechanisms of ATM Activation. Annu. Rev. Biochem. 2015, 84, 711–738. [Google Scholar] [CrossRef] [PubMed]
- Lavin, M.F.; Kozlov, S.; Gatei, M.; Kijas, A.W. ATM-Dependent Phosphorylation of All Three Members of the MRN Complex: From Sensor to Adaptor. Biomolecules 2015, 5, 2877–2902. [Google Scholar] [CrossRef] [PubMed]
- Bakkenist, C.J.; Kastan, M.B. Chromatin perturbations during the DNA damage response in higher eukaryotes. DNA Repair (Amst) 2015, 36, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Cimprich, K.A.; Cortez, D. ATR: An essential regulator of genome integrity. Nat. Rev. Mol. Cell. Biol. 2008, 9, 616–627. [Google Scholar] [CrossRef] [PubMed]
- Nam, E.A.; Cortez, D. ATR signalling: More than meeting at the fork. Biochem. J. 2011, 436, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Matt, S.; Hofmann, T.G. The DNA damage-induced cell death response: A roadmap to kill cancer cells. Cell. Mol. Life Sci. 2016, 73, 2829–2850. [Google Scholar] [CrossRef] [PubMed]
- Nakad, R.; Schumacher, B. DNA damage response and immune defense: Links and mechanisms. Front. Genet. 2016, 7, 147. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Xu, P.; Zou, W.; Shen, W.; Peng, J.; Liu, K.; Engelhardt, J.F.; Yan, Z.; Qiu, J. DNA damage signaling is required for replication of human bocavirus 1 DNA in dividing HEK293 cells. J. Virol. 2017, 91, e01831-16. [Google Scholar] [CrossRef] [PubMed]
- Shah, G.A.; O’Shea, C.C. Viral and cellular genomes activate distinct DNA damage responses. Cell 2015, 162, 987–1002. [Google Scholar] [CrossRef] [PubMed]
- Burgess, R.C.; Misteli, T. Not all DDRs are created equal: Non-canonical DNA damage responses. Cell 2015, 162, 944–947. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Qiu, J. Parvovirus infection-induced DNA damage response. Future Virol. 2013, 8, 245–257. [Google Scholar] [CrossRef] [PubMed]
- McFadden, K.; Luftig, M.A. Interplay between DNA tumor viruses and the host DNA damage response. Curr. Top. Microbiol. Immunol. 2013, 371, 229–257. [Google Scholar] [PubMed]
- Xiaofei, E.; Kowalik, T.F. The DNA damage response induced by infection with human cytomegalovirus and other viruses. Viruses 2014, 6, 2155–2185. [Google Scholar] [PubMed]
- Bregnard, C.; Benkirane, M.; Laguette, N. DNA damage repair machinery and HIV escape from innate immune sensing. Front. Microbiol. 2014, 5, 176. [Google Scholar] [PubMed]
- McKinney, C.C.; Hussmann, K.L.; McBride, A.A. The Role of the DNA Damage Response throughout the Papillomavirus Life Cycle. Viruses 2015, 7, 2450–2469. [Google Scholar] [CrossRef] [PubMed]
- Anacker, D.C.; Moody, C.A. Modulation of the DNA damage response during the life cycle of human papillomaviruses. Virus Res. 2016, 231, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Damania, B. The cGAS-STING Defense Pathway and Its Counteraction by Viruses. Cell Host Microbe 2016, 19, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Sung, W.K.; Zheng, H.; Li, S.; Chen, R.; Liu, X.; Li, Y.; Lee, N.P.; Lee, W.H.; Ariyaratne, P.N.; Tennakoon, C.; et al. Genome-wide survey of recurrent HBV integration in hepatocellular carcinoma. Nat. Genet. 2012, 44, 765–769. [Google Scholar] [CrossRef] [PubMed]
- Mason, W.S.; Gill, U.S.; Litwin, S.; Zhou, Y.; Peri, S.; Pop, O.; Hong, M.L.; Naik, S.; Quaglia, A.; Bertoletti, A.; et al. HBV DNA Integration and Clonal Hepatocyte Expansion in Chronic Hepatitis B Patients Considered Immune Tolerant. Gastroenterology 2016, 151, 986–998 e4. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, P.T.F.; Litwin, S.; Dolman, G.E.; Bertoletti, A.; Mason, W.S. Immune Tolerant Chronic Hepatitis B: The Unrecognized Risks. Viruses 2017, 9, 96. [Google Scholar] [CrossRef] [PubMed]
- Bill, C.A.; Summers, J. Genomic DNA double-strand breaks are targets for hepadnaviral DNA integration. Proc. Natl. Acad. Sci. USA 2004, 101, 11135–11140. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.H.; Liu, X.; Yan, H.X.; Li, W.Y.; Zeng, X.; Yang, Y.; Zhao, J.; Liu, S.P.; Zhuang, X.H.; Lin, C.; et al. Genomic and oncogenic preference of HBV integration in hepatocellular carcinoma. Nat. Commun. 2016, 7, 12992. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Summers, J. Illegitimate replication of linear hepadnavirus DNA through nonhomologous recombination. J. Virol. 1995, 69, 4029–4036. [Google Scholar] [PubMed]
- Yang, W.; Summers, J. Infection of ducklings with virus particles containing linear double-stranded duck hepatitis B virus DNA: Illegitimate replication and reversion. J. Virol. 1998, 72, 8710–8717. [Google Scholar] [PubMed]
- Sloan, R.D.; Wainberg, M.A. The role of unintegrated DNA in HIV infection. Retrovirology 2011, 8, 52. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Xu, C.; Zhou, T.; Block, T.M.; Guo, J.T. Characterization of the host factors required for hepadnavirus covalently closed circular (ccc) DNA formation. PLoS ONE 2012, 7, e43270. [Google Scholar] [CrossRef] [PubMed]
- Deriano, L.; Roth, D.B. Modernizing the nonhomologous end-joining repertoire: Alternative and classical NHEJ share the stage. Annu. Rev. Genet. 2013, 47, 433–455. [Google Scholar] [CrossRef] [PubMed]
- Stracker, T.H.; Carson, C.T.; Weitzman, M.D. Adenovirus oncoproteins inactivate the Mre11-Rad50-NBS1 DNA repair complex. Nature 2002, 418, 348–352. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.L.; Tsai, T.Y. Promyelocytic leukemia nuclear bodies link the DNA damage repair pathway with hepatitis B virus replication: Implications for hepatitis B virus exacerbation during chemotherapy and radiotherapy. Mol. Cancer Res. 2009, 7, 1672–1685. [Google Scholar] [CrossRef] [PubMed]
- Ko, H.L.; Ren, E.C. Novel poly (ADP-ribose) polymerase 1 binding motif in hepatitis B virus core promoter impairs DNA damage repair. Hepatology 2011, 54, 1190–1198. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.L. Defective DNA damage response and repair in liver cells expressing hepatitis B virus surface antigen. FASEB J. 2013, 27, 2316–2327. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, K.; Wang, Z.; Chowdhury, S.; Simadu, M.; Koura, M.; Muramatsu, M. Uracil DNA glycosylase counteracts APOBEC3G-induced hypermutation of hepatitis B viral genomes: Excision repair of covalently closed circular DNA. PLoS Pathog. 2013, 9, e1003361. [Google Scholar] [CrossRef] [PubMed]
- Higgs, M.R.; Chouteau, P.; Lerat, H. ‘Liver let die’: Oxidative DNA damage and hepatotropic viruses. J. Gen. Virol. 2014, 95, 991–1004. [Google Scholar] [CrossRef] [PubMed]
- Ricardo-Lax, I.; Ramanan, V.; Michailidis, E.; Shamia, T.; Reuven, N.; Rice, C.M.; Shlomai, A.; Shaul, Y. Hepatitis B virus induces RNR-R2 expression via DNA damage response activation. J. Hepatol. 2015, 63, 789–796. [Google Scholar] [CrossRef] [PubMed]
- Hollingworth, R.; Skalka, G.L.; Stewart, G.S.; Hislop, A.D.; Blackbourn, D.J.; Grand, R.J. Activation of DNA damage response pathways during lytic replication of KSHV. Viruses 2015, 7, 2908–2927. [Google Scholar] [CrossRef] [PubMed]
- Slagle, B.L.; Andrisani, O.M.; Bouchard, M.J.; Lee, C.G.; Ou, J.H.; Siddiqui, A. Technical standards for hepatitis B virus X protein (HBx) research. Hepatology 2015, 61, 1416–1424. [Google Scholar] [CrossRef] [PubMed]
- Levrero, M.; Zucman-Rossi, J. Mechanisms of HBV-induced hepatocellular carcinoma. J. Hepatol. 2016, 64, S84–S101. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Xie, N.; He, W.; Liu, R.; Lei, Y.; Chen, Y.; Tang, H.; Liu, B.; Huang, C.; Wei, Y. An integrated proteomics and bioinformatics analyses of hepatitis B virus X interacting proteins and identification of a novel interactor apoA-I. J. Proteom. 2013, 84, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Capovilla, A.; Arbuthnot, P. Hepatitis B virus X protein does not influence essential steps of nucleotide excision repair effected by human liver extracts. Biochem. Biophys. Res. Commun. 2003, 312, 806–810. [Google Scholar] [CrossRef] [PubMed]
- Capovilla, A.; Carmona, S.; Arbuthnot, P. Hepatitis B virus X-protein binds damaged DNA and sensitizes liver cells to ultraviolet irradiation. Biochem. Biophys. Res. Commun. 1997, 232, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Wang, X.W.; Harris, C.C. Hepatitis B virus X protein inhibits nucleotide excision repair. Int. J. Cancer 1999, 80, 875–879. [Google Scholar] [CrossRef]
- van de Klundert, M.A.; van Hemert, F.J.; Zaaijer, H.L.; Kootstra, N.A. The hepatitis B virus x protein inhibits thymine DNA glycosylase initiated base excision repair. PLoS ONE 2012, 7, e48940. [Google Scholar] [CrossRef] [PubMed]
- Truant, R.; Antunovic, J.; Greenblatt, J.; Prives, C.; Cromlish, J.A. Direct interaction of the hepatitis B virus HBx protein with p53 leads to inhibition by HBx of p53 response element-directed transactivation. J. Virol. 1995, 69, 1851–1859. [Google Scholar] [PubMed]
- Lee, T.H.; Elledge, S.J.; Butel, J.S. Hepatitis B virus X protein interacts with a probable cellular DNA repair protein. J. Virol. 1995, 69, 1107–1114. [Google Scholar] [PubMed]
- Van der Crabben, S.N.; Hennus, M.P.; McGregor, G.A.; Ritter, D.I.; Nagamani, S.C.; Wells, O.S.; Harakalova, M.; Chinn, I.K.; Alt, A.; Vondrova, L.; et al. Destabilized SMC5/6 complex leads to chromosome breakage syndrome with severe lung disease. J. Clin. Investig. 2016, 126, 2881–2892. [Google Scholar] [CrossRef] [PubMed]
- Scrima, A.; Konickova, R.; Czyzewski, B.K.; Kawasaki, Y.; Jeffrey, P.D.; Groisman, R.; Nakatani, Y.; Iwai, S.; Pavletich, N.P.; Thoma, N.H. Structural basis of UV DNA-damage recognition by the DDB1-DDB2 complex. Cell 2008, 135, 1213–1223. [Google Scholar] [CrossRef] [PubMed]
- Stoyanova, T.; Roy, N.; Kopanja, D.; Raychaudhuri, P.; Bagchi, S. DDB2 (damaged-DNA binding protein 2) in nucleotide excision repair and DNA damage response. Cell. Cycle 2009, 8, 4067–4071. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.S.; Jackson, S.P. Ubiquitylation, neddylation and the DNA damage response. Open Biol. 2015, 5, 150018. [Google Scholar] [CrossRef] [PubMed]
- Hrecka, K.; Hao, C.; Shun, M.C.; Kaur, S.; Swanson, S.K.; Florens, L.; Washburn, M.P.; Skowronski, J. HIV-1 and HIV-2 exhibit divergent interactions with HLTF and UNG2 DNA repair proteins. Proc. Natl. Acad. Sci. USA 2016, 113, E3921–E3930. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhou, X.; Barnes, C.O.; DeLucia, M.; Cohen, A.E.; Gronenborn, A.M.; Ahn, J.; Calero, G. The DDB1-DCAF1-Vpr-UNG2 crystal structure reveals how HIV-1 Vpr steers human UNG2 toward destruction. Nat. Struct. Mol. Biol. 2016, 23, 933–940. [Google Scholar] [CrossRef] [PubMed]
- Ballana, E.; Este, J.A. SAMHD1: At the crossroads of cell proliferation, immune responses, and virus restriction. Trends Microbiol. 2015, 23, 680–692. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Robert, E.I.; van Breugel, P.C.; Strubin, M.; Zheng, N. A promiscuous alpha-helical motif anchors viral hijackers and substrate receptors to the CUL4-DDB1 ubiquitin ligase machinery. Nat. Struct. Mol. Biol. 2010, 17, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Guerrieri, F.; Belloni, L.; D’Andrea, D.; Pediconi, N.; Le Pera, L.; Testoni, B.; Scisciani, C.; Floriot, O.; Zoulim, F.; Tramontano, A.; et al. Genome-wide identification of direct HBx genomic targets. BMC Genom. 2017, 18, 184. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y.; Huang, S.Y.; Gao, R.; Das, B.B.; Murai, J.; Marchand, C. Tyrosyl-DNA-phosphodiesterases (TDP1 and TDP2). DNA Repair (Amst) 2014, 19, 114–129. [Google Scholar] [CrossRef] [PubMed]
- Menon, V.; Povirk, L.F. End-processing nucleases and phosphodiesterases: An elite supporting cast for the non-homologous end joining pathway of DNA double-strand break repair. DNA Repair (Amst) 2016, 43, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Pillaire, M.J.; Betous, R.; Hoffmann, J.S. Role of DNA polymerase kappa in the maintenance of genomic stability. Mol. Cell. Oncol. 2014, 1, e29902. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Washington, M.T. Translesion Synthesis: Insights into the Selection and Switching of DNA Polymerases. Genes 2017, 8, 24. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, W.; Ogura, N.; Watashi, K.; Wakita, T. Host factor PRPF31 is involved in cccDNA production in HBV-replicating cells. Biochem. Biophys. Res. Commun. 2017, 482, 638–644. [Google Scholar] [CrossRef] [PubMed]
- Baumert, T.F.; Verrier, E.R.; Nassal, M.; Chung, R.T.; Zeisel, M.B. Host-targeting agents for treatment of hepatitis B virus infection. Curr. Opin. Virol. 2015, 14, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.T.; Guo, H. Metabolism and function of hepatitis B virus cccDNA: Implications for the development of cccDNA-targeting antiviral therapeutics. Antiviral Res. 2015, 122, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Testoni, B.; Durantel, D.; Zoulim, F. Novel targets for hepatitis B virus therapy. Liver Int. 2017, 37 (Suppl. 1), 33–39. [Google Scholar] [CrossRef] [PubMed]
- Faure-Dupuy, S.; Lucifora, J.; Durantel, D. Interplay between the hepatitis B virus and innate immunity: From an understanding to the development of therapeutic concepts. Viruses 2017, 9, 95. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, J.; Chen, J.; Li, Y.; Wang, W.; Du, X.; Song, W.; Zhang, W.; Lin, L.; Yuan, Z. Hepatitis B virus polymerase disrupts K63-linked ubiquitination of STING to block innate cytosolic DNA-sensing pathways. J. Virol. 2015, 89, 2287–2300. [Google Scholar] [CrossRef] [PubMed]
- Luangsay, S.; Gruffaz, M.; Isorce, N.; Testoni, B.; Michelet, M.; Faure-Dupuy, S.; Maadadi, S.; Ait-Goughoulte, M.; Parent, R.; Rivoire, M.; et al. Early inhibition of hepatocyte innate responses by hepatitis B virus. J. Hepatol. 2015, 63, 1314–1322. [Google Scholar] [CrossRef] [PubMed]









© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schreiner, S.; Nassal, M. A Role for the Host DNA Damage Response in Hepatitis B Virus cccDNA Formation—and Beyond? Viruses 2017, 9, 125. https://doi.org/10.3390/v9050125
Schreiner S, Nassal M. A Role for the Host DNA Damage Response in Hepatitis B Virus cccDNA Formation—and Beyond? Viruses. 2017; 9(5):125. https://doi.org/10.3390/v9050125
Chicago/Turabian StyleSchreiner, Sabrina, and Michael Nassal. 2017. "A Role for the Host DNA Damage Response in Hepatitis B Virus cccDNA Formation—and Beyond?" Viruses 9, no. 5: 125. https://doi.org/10.3390/v9050125
APA StyleSchreiner, S., & Nassal, M. (2017). A Role for the Host DNA Damage Response in Hepatitis B Virus cccDNA Formation—and Beyond? Viruses, 9(5), 125. https://doi.org/10.3390/v9050125